
N,N′-Diaryldihydrophenazines as a Sustainable and Cost-Effective Alternative to Precious Metal Complexes in the Photoredox-Catalyzed Alkylation of Aryl Alkyl Ketones

Abstract
:
1. Introduction
2. Results and Discussion
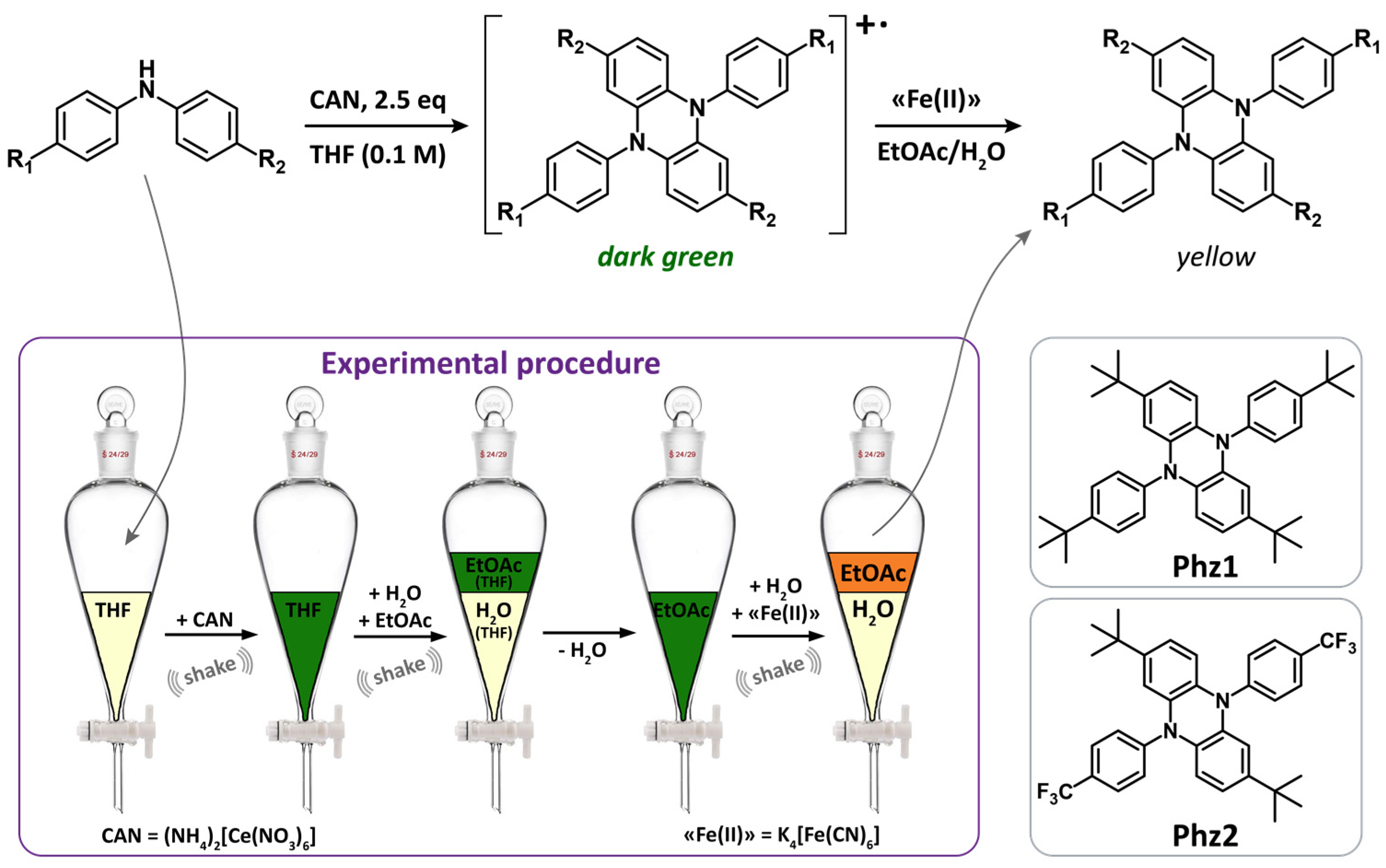
2.1. Synthesis and Characterization
2.2. Absorption and Emission Spectra
2.3. ESR Data
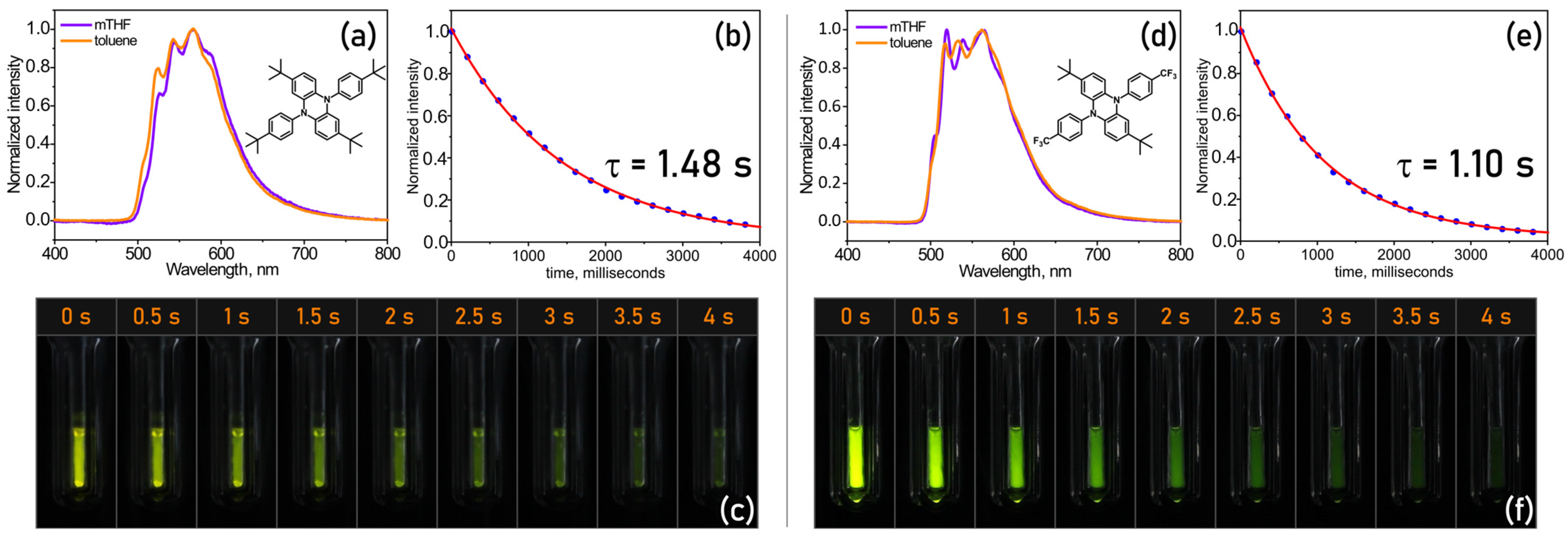
2.4. Phosphorescence Spectra
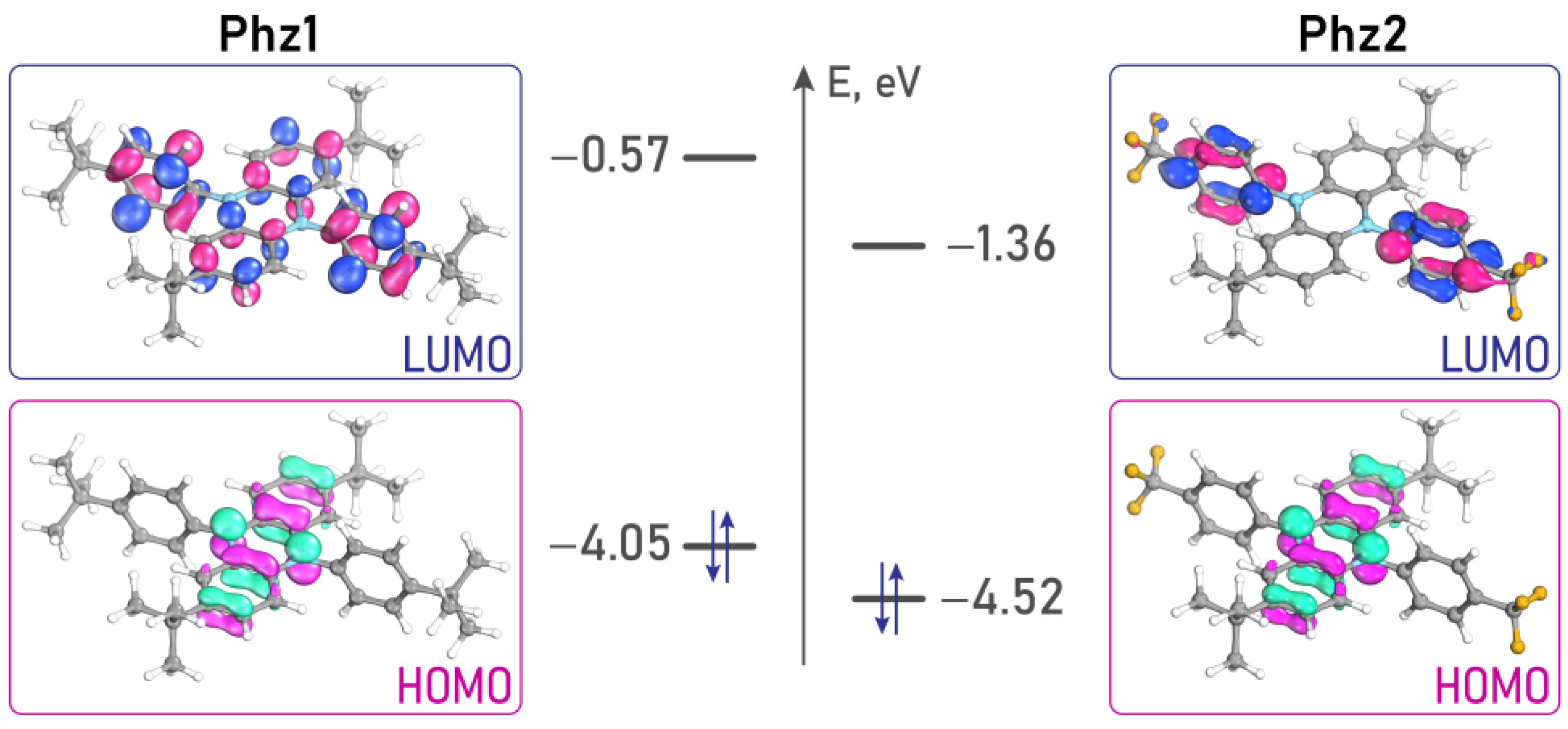
2.5. Frontier Orbital Localization
2.6. Redox Behavior
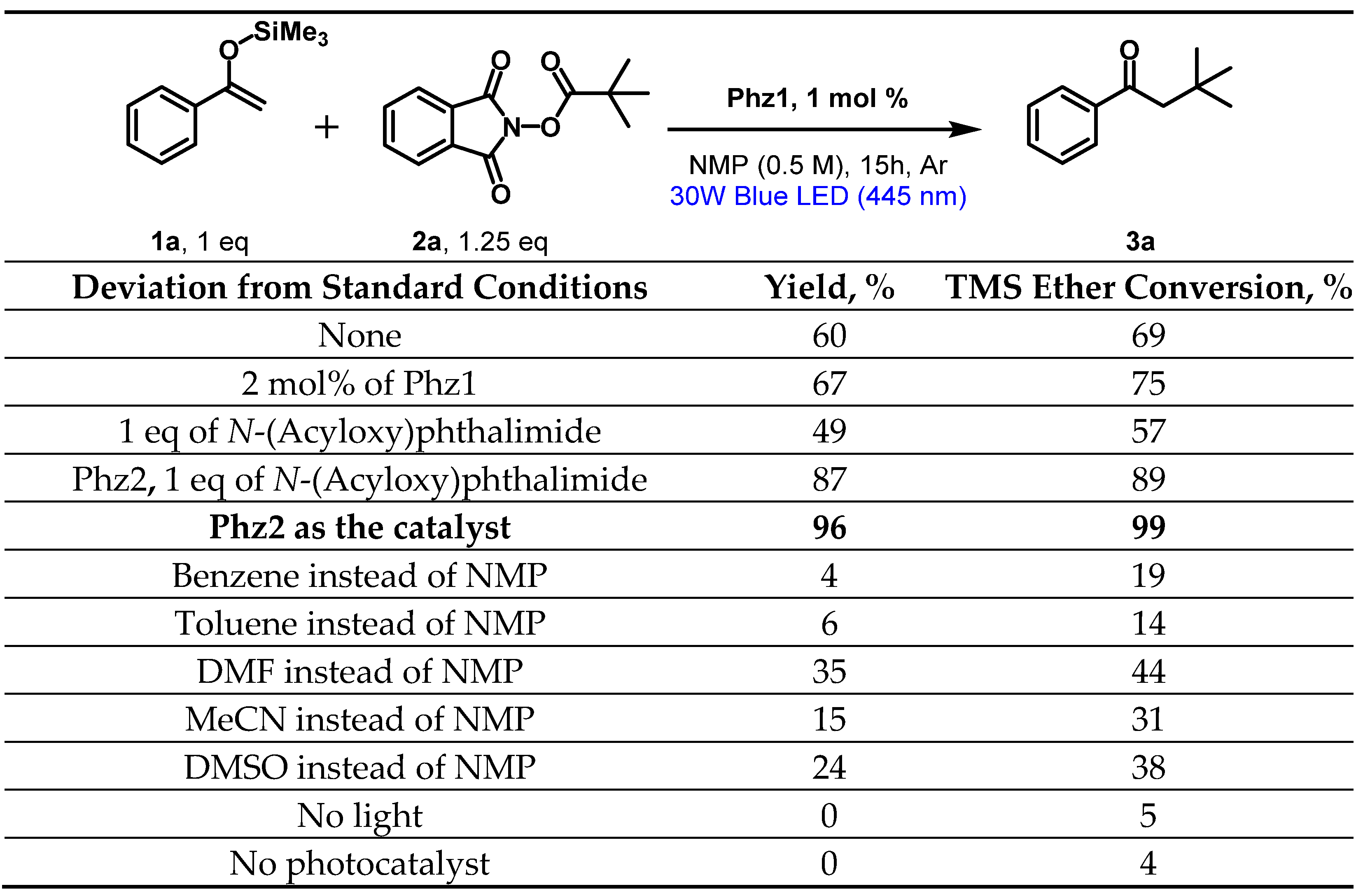
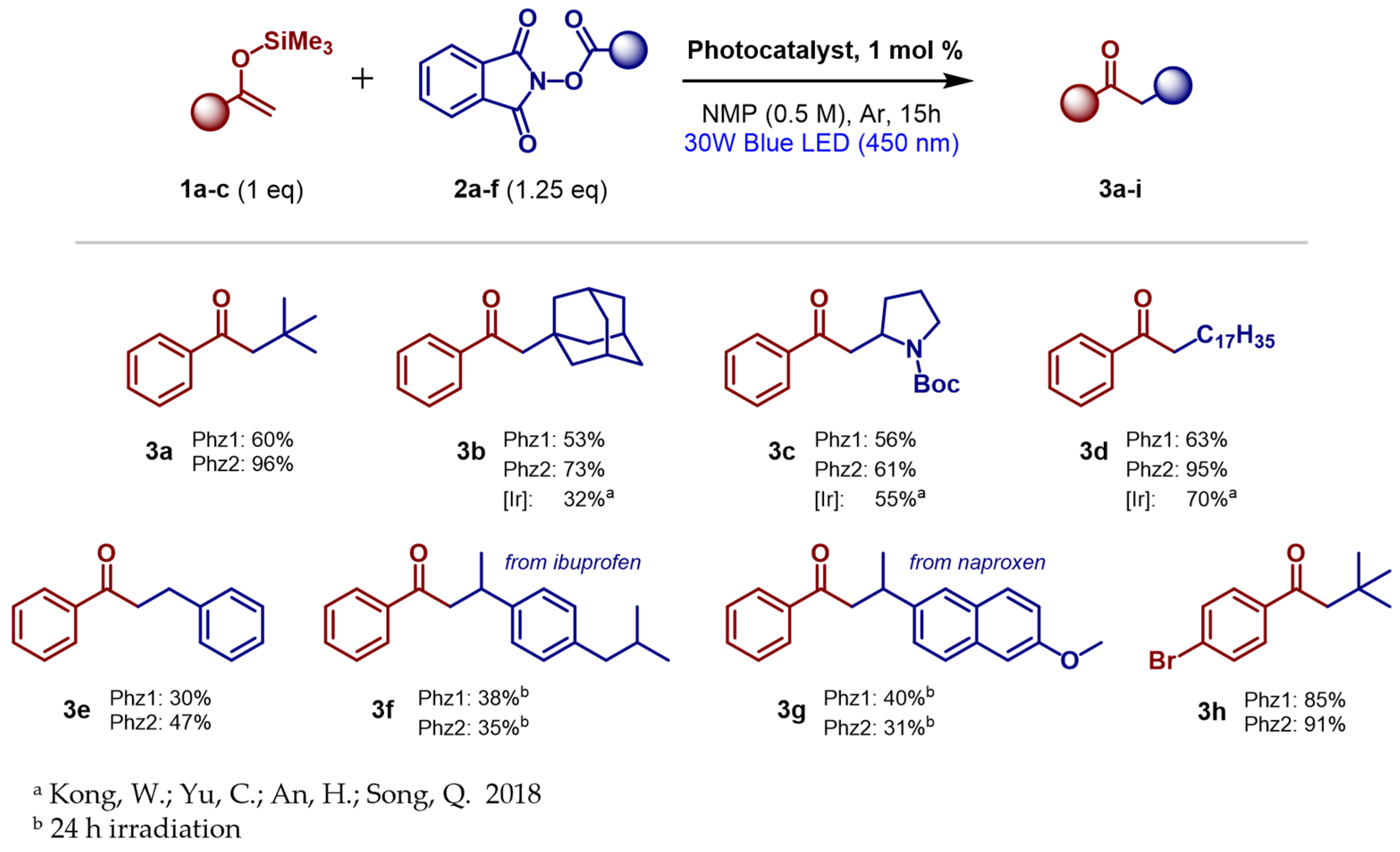
2.7. Testing in Photoredox-Catalyzed Decarboxylative Alkylation of Silyl Enol Ethers
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Computational Details
4.3. Synthesis of Diaryldihydrophenazines
4.4. Photoredox Alkylation of TMS Enol Ethers
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McAtee, R.C.; McClain, E.J.; Stephenson, C.R.J. Illuminating Photoredox Catalysis. Trends Chem. 2019, 1, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Yi, H.; Lei, A. Synthetic Applications of Photoredox Catalysis with Visible Light. Org. Biomol. Chem. 2013, 11, 2387–2403. [Google Scholar] [CrossRef]
- Xuan, J.; Xiao, W.J. Visible-Light Photoredox Catalysis. Angew. Chemie Int. Ed. 2012, 51, 6828–6838. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, D.; Fagnoni, M.; Albini, A. Photoorganocatalysis. What For? Chem. Soc. Rev. 2012, 42, 97–113. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef]
- Speckmeier, E.; Fischer, T.G.; Zeitler, K. A Toolbox Approach to Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor-Acceptor Cyanoarenes. J. Am. Chem. Soc. 2018, 140, 15353–15365. [Google Scholar] [CrossRef]
- Lee, Y.; Kwon, M.S. Emerging Organic Photoredox Catalysts for Organic Transformations. European J. Org. Chem. 2020, 2020, 6028–6043. [Google Scholar] [CrossRef]
- Vega-Peñaloza, A.; Mateos, J.; Companyó, X.; Escudero-Casao, M.; Dell’Amico, L. A Rational Approach to Organo-Photocatalysis: Novel Designs and Structure-Property Relationships. Angew. Chemie Int. Ed. 2021, 60, 1082–1097. [Google Scholar] [CrossRef]
- Bobo, M.V.; Kuchta, J.J.; Vannucci, A.K. Recent Advancements in the Development of Molecular Organic Photocatalysts. Org. Biomol. Chem. 2021, 19, 4816–4834. [Google Scholar] [CrossRef] [PubMed]
- Discekici, E.H.; Treat, N.J.; Poelma, S.O.; Mattson, K.M.; Hudson, Z.M.; Luo, Y.; Hawker, C.J.; De Alaniz, J.R. A Highly Reducing Metal-Free Photoredox Catalyst: Design and Application in Radical Dehalogenations. Chem. Commun. 2015, 51, 11705–11708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Jiao, M.J.; Feng, Z.T.; Wang, X.Z.; Xu, G.Q.; Xu, P.F. Design, Synthesis, and Application of Highly Reducing Organic Visible-Light Photocatalysts. Org. Lett. 2018, 20, 5700–5704. [Google Scholar] [CrossRef] [PubMed]
- Treat, N.J.; Sprafke, H.; Kramer, J.W.; Clark, P.G.; Barton, B.E.; Read De Alaniz, J.; Fors, B.P.; Hawker, C.J. Metal-Free Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2014, 136, 16096–16101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitskiy, O.A.; Dulov, D.A.; Nikitin, O.M.; Bogdanov, A.V.; Eremin, D.B.; Paseshnichenko, K.A.; Magdesieva, T.V. Competitive Routes for Electrochemical Oxidation of Substituted Diarylamines: The Guidelines. ChemElectroChem 2018, 5, 3391–3410. [Google Scholar] [CrossRef]
- Du, Y.; Pearson, R.M.; Lim, C.H.; Sartor, S.M.; Ryan, M.D.; Yang, H.; Damrauer, N.H.; Miyake, G.M. Strongly Reducing, Visible-Light Organic Photoredox Catalysts as Sustainable Alternatives to Precious Metals. Chem. A Eur. J. 2017, 23, 10962–10968. [Google Scholar] [CrossRef]
- Tlahuext-Aca, A.; Candish, L.; Garza-Sanchez, R.A.; Glorius, F. Decarboxylative Olefination of Activated Aliphatic Acids Enabled by Dual Organophotoredox/Copper Catalysis. ACS Catal. 2018, 8, 1715–1719. [Google Scholar] [CrossRef]
- Lim, C.H.; Ryan, M.D.; McCarthy, B.G.; Theriot, J.C.; Sartor, S.M.; Damrauer, N.H.; Musgrave, C.B.; Miyake, G.M. Intramolecular Charge Transfer and Ion Pairing in N, N-Diaryl Dihydrophenazine Photoredox Catalysts for Efficient Organocatalyzed Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2017, 139, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.D.; Theriot, J.C.; Lim, C.H.; Yang, H.; Lockwood, A.G.; Garrison, N.G.; Lincoln, S.R.; Musgrave, C.B.; Miyake, G.M. Solvent Effects on the Intramolecular Charge Transfer Character of N,N-Diaryl Dihydrophenazine Catalysts for Organocatalyzed Atom Transfer Radical Polymerization. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 3017–3027. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.P.; Federico, C.R.; Lim, C.H.; Miyake, G.M. Photoinduced Organocatalyzed Atom Transfer Radical Polymerization Using Low Ppm Catalyst Loading. Macromolecules 2019, 52, 747–754. [Google Scholar] [CrossRef]
- Theriot, J.C.; Lim, C.H.; Yang, H.; Ryan, M.D.; Musgrave, C.B.; Miyake, G.M. Organocatalyzed Atom Transfer Radical Polymerization Driven by Visible Light. Science 2016, 352, 1082–1086. [Google Scholar] [CrossRef]
- Singh, V.K.; Yu, C.; Badgujar, S.; Kim, Y.; Kwon, Y.; Kim, D.; Lee, J.; Akhter, T.; Thangavel, G.; Park, L.S.; et al. Highly Efficient Organic Photocatalysts Discovered via a Computer-Aided-Design Strategy for Visible-Light-Driven Atom Transfer Radical Polymerization. Nat. Catal. 2018, 1, 794–804. [Google Scholar] [CrossRef]
- Kong, W.; Yu, C.; An, H.; Song, Q. Photoredox-Catalyzed Decarboxylative Alkylation of Silyl Enol Ethers to Synthesize Functionalized Aryl Alkyl Ketones. Org. Lett. 2018, 20, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Terada, E.; Kozaki, M.; Uchida, M.; Kikukawa, S.; Okada, K. Facile Synthesis of 5,10-Diaryl-5,10-Dihydrophenazines and Application to EL Devices. Org. Lett. 2003, 5, 373–376. [Google Scholar] [CrossRef]
- Prass, B.; Von Borczyskowski, C.; Steidl, P.; Stehlik, D. Characterization of the Excited States of Photoreactive Dihydrophenazine Doped in Molecular Crystals. J. Phys. Chem. 1987, 91, 2298–2303. [Google Scholar] [CrossRef]
- Clarke, R.H. Magnetic Resonance Studies of Optical Spin Polarization in Triplet State Anthracene. Chem. Phys. Lett. 1970, 6, 413–416. [Google Scholar] [CrossRef]
- Hutchison, C.A.; Mangum, B.W. Paramagnetic Resonance Absorption in Naphthalene in Its Phosphorescent State. J. Chem. Phys. 2004, 34, 908. [Google Scholar] [CrossRef]
- Yang, Z.; Mao, Z.; Xie, Z.; Zhang, Y.; Liu, S.; Zhao, J.; Xu, J.; Chi, Z.; Aldred, M.P. Recent Advances in Organic Thermally Activated Delayed Fluorescence Materials. Chem. Soc. Rev. 2017, 46, 915–1016. [Google Scholar] [CrossRef] [PubMed]
- Dias, F.B.; Santos, J.; Graves, D.R.; Data, P.; Nobuyasu, R.S.; Fox, M.A.; Batsanov, A.S.; Palmeira, T.; Berberan-Santos, M.N.; Bryce, M.R.; et al. The Role of Local Triplet Excited States and D-A Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices. Adv. Sci. 2016, 3, 1600080. [Google Scholar] [CrossRef] [Green Version]
- Hofbeck, T.; Yersin, H. The Triplet State of Fac-Ir(Ppy)3. Inorg. Chem. 2010, 49, 9290–9299. [Google Scholar] [CrossRef]
- Endo, A.; Sato, K.; Yoshimura, K.; Kai, T.; Kawada, A.; Miyazaki, H.; Adachi, C. Efficient Up-Conversion of Triplet Excitons into a Singlet State and Its Application for Organic Light Emitting Diodes. Appl. Phys. Lett. 2011, 98, 083302. [Google Scholar] [CrossRef]
- Fang, F.; Zhu, L.; Li, M.; Song, Y.; Sun, M.; Zhao, D.; Zhang, J. Thermally Activated Delayed Fluorescence Material: An Emerging Class of Metal-Free Luminophores for Biomedical Applications. Adv. Sci. 2021, 8, 2102970. [Google Scholar] [CrossRef]
- Shi, Y.Z.; Wu, H.; Wang, K.; Yu, J.; Ou, X.M.; Zhang, X.H. Recent Progress in Thermally Activated Delayed Fluorescence Emitters for Nondoped Organic Light-Emitting Diodes. Chem. Sci. 2022, 13, 3625–3651. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Wang, Y.; Xu, Y.; Chen, Z.; Chen, G.; Liang, S.H. Ru-Photoredox-Catalyzed Decarboxylative Oxygenation of Aliphatic Carboxylic Acids through N-(Acyloxy)Phthalimide. Org. Lett. 2018, 20, 4824–4827. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Tan, Z.; Zhang, H.; Liu, J.; Zhang, S.; Wang, Z. Photoredox Catalysis Enabled Alkylation of Alkenyl Carboxylic Acids with N-(Acyloxy)Phthalimide via Dual Decarboxylation. Chem. Commun. 2017, 53, 10719–10722. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.M.; Shang, R.; Fu, M.C.; Fu, Y. Photoredox-Catalysed Decarboxylative Alkylation of N-Heteroarenes with N-(Acyloxy)Phthalimides. Chem. A Eur. J. 2017, 23, 2537–2541. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.M.B.; Pinto, A.R.T.; Domingues, M.M.; Serrano, I.; Heras, M.; Bardaji, E.R.; Tavares, I.; Castanho, M.A. Chemical Conjugation of the Neuropeptide Kyotorphin and Ibuprofen Enhances Brain Targeting and Analgesia. Mol. Pharm. 2011, 8, 1929–1940. [Google Scholar] [CrossRef]
- Biswas, G.; Kim, W.; Kim, K.T.; Cho, J.; Jeong, D.; Song, K.H.; Im, J. Synthesis of Ibuprofen Conjugated Molecular Transporter Capable of Enhanced Brain Penetration. J. Chem. 2017, 2017, 4746158. [Google Scholar] [CrossRef] [Green Version]
- Bryden, M.A.; Zysman-Colman, E. Organic Thermally Activated Delayed Fluorescence (TADF) Compounds Used in Photocatalysis. Chem. Soc. Rev. 2021, 50, 7587–7680. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Grimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent Structures and Interactions by Density Functional Theory with Small Atomic Orbital Basis Sets. J. Chem. Phys. 2015, 143, 054107. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phz1 | Phz2 | NaphtPhz [15] | Ir(ppy)3 [29] | |
|---|---|---|---|---|
| λmax(abs), nm | 375 | 370 | 343 | 377 |
| ε(λmax) | 4400 | 3800 | 5950 | 13100 |
| τ(TADF, 298 K), μs | <10 | 65 | 4.3 | 2.1 |
| EST(exp), eV | 0.22 | 0.11 | - | ~0 |
| EST(calc), eV | 0.22 ± 0.04 | 0.01 | - | - |
| τ(phos, 77 K, toluene) | 1.5 sec | 1.0 sec | - | ~10 sec |
| Photoluminescence quantum yield PL, % | 0.06 | 0.03 | - | 1 |
| Quantum yield of triplet state formation, T, % | 0.05 | 0.02 | 0.02 | 1 |
| E(Ox), V vs. Ag/AgCl | 0.24 | 0.37 | 0.25 | 0.81 |
| E(Ox)*T1, V vs. Ag/AgCl | −2.5 | −2.31 | −1.65 | −1.69 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dulov, D.A.; Bogdanov, A.V.; Dorofeev, S.G.; Magdesieva, T.V. N,N′-Diaryldihydrophenazines as a Sustainable and Cost-Effective Alternative to Precious Metal Complexes in the Photoredox-Catalyzed Alkylation of Aryl Alkyl Ketones. Molecules 2023, 28, 221. https://doi.org/10.3390/molecules28010221
Dulov DA, Bogdanov AV, Dorofeev SG, Magdesieva TV. N,N′-Diaryldihydrophenazines as a Sustainable and Cost-Effective Alternative to Precious Metal Complexes in the Photoredox-Catalyzed Alkylation of Aryl Alkyl Ketones. Molecules. 2023; 28(1):221. https://doi.org/10.3390/molecules28010221
Chicago/Turabian StyleDulov, Dmitry A., Alexey V. Bogdanov, Sergey G. Dorofeev, and Tatiana V. Magdesieva. 2023. "N,N′-Diaryldihydrophenazines as a Sustainable and Cost-Effective Alternative to Precious Metal Complexes in the Photoredox-Catalyzed Alkylation of Aryl Alkyl Ketones" Molecules 28, no. 1: 221. https://doi.org/10.3390/molecules28010221