

The Lipophilic Purine Nucleoside—Tdp1 Inhibitor—Enhances DNA Damage Induced by Topotecan In Vitro and Potentiates the Antitumor Effect of Topotecan In Vivo
 , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Tdp1 Inhibition
2.3. The Effect of Tdp1 Inhibitors on Cells’ Viability
2.4. Type of Inhibition of Tdp1 Enzyme Reaction for the Most Potent Compounds
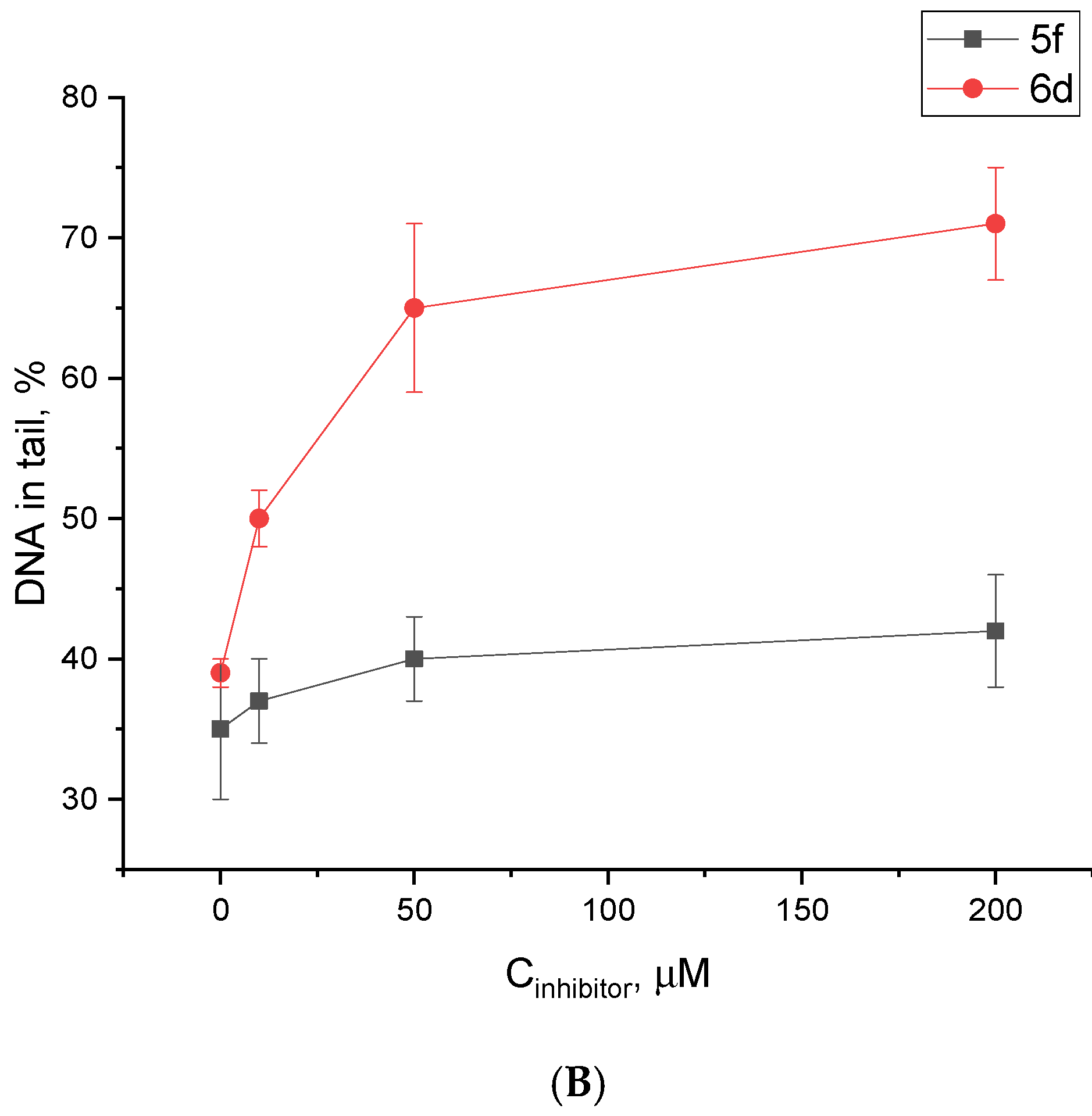
2.5. Influence of the Most Potent Compounds on the Accumulation of DNA Damage by the Alkaline Comet Assay
2.6. The Influence of Lead Compounds on the Action of Topotecan In Vivo
3. Materials and Methods
3.1. Chemistry
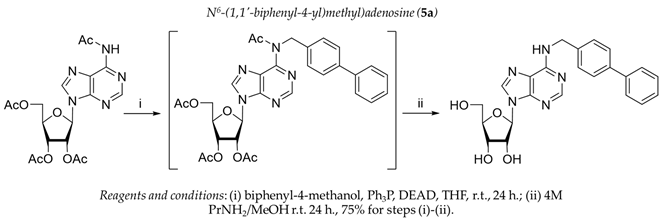
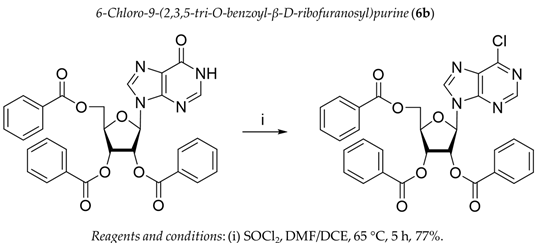

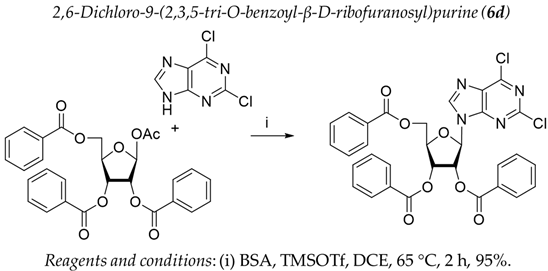
3.2. Biology
3.2.1. Real-Time Detection of Tdp1 Activity
3.2.2. Investigation of PARP1/2 Activity by Inclusion of α-[32P]-NAD+ in Poly(ADP-ribose)
3.2.3. Alkaline Comet Assay
3.2.4. Cell Culture Cytotoxicity Assay
3.3. Experiments In Vivo
3.3.1. Lab Animals
3.3.2. Tumor Models
3.3.3. Treatment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bezborodova, O.A.; Nemtsova, E.R.; Karmakova, T.A.; Venediktova, J.V.; Pankratov, A.A.; Alekseenko, I.V.; Pleshkan, V.V.; Zinoveva, M.V.; Monastyrskaya, G.S.; Sverdlov, E.D.; et al. Modern trends in the development of antitumor gene and cell therapy. RPMJ 2019, 6, 65. [Google Scholar]
- Lorusso, D.; Pietragalla, A.; Mainenti, S.; Masciullo, V.; Di Vagno, G.; Scambia, G. Review role of topotecan in gynaecological cancers: Current indications and perspectives. Crit. Rev. Oncol. Hematol. 2010, 74, 163–174. [Google Scholar] [CrossRef]
- O’Brien, M.; Eckardt, J.; Ramlau, R. Recent advances with topotecan in the treatment of lung cancer. Oncologist 2007, 12, 1194–1204. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; Huang, S.Y.; Gao, R.; Das, B.B.; Murai, J.; Marchand, C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair. 2014, 19, 114–129. [Google Scholar] [CrossRef] [Green Version]
- Pourquier, P.; Pilon, A.A.; Kohlhagen, G.; Mazumder, A.; Sharma, A.; Pommier, Y. Trapping of mammalian topoisomerase I and recombinations induced by damaged DNA containing nicks or gaps. Importance of DNA end phosphorylation and camptothecin effects. J. Biol. Chem. 1997, 272, 26441–26447. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Liu, L.F. Processing of topoisomerase I cleavable complexes into DNA damage by transcription. Nucleic Acids Res. 1997, 25, 4181–4186. [Google Scholar] [CrossRef] [Green Version]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Regairaz, M.; Seiler, J.A.; Agama, K.K.; Doroshow, J.H.; Pommier, Y. Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 2011, 39, 3607–3620. [Google Scholar] [CrossRef] [Green Version]
- Sacho, E.J.; Maizels, N. DNA repair factor MRE11/RAD50 cleaves 3’-phosphotyrosyl bonds and resects DNA to repair damage caused by topoisomerase 1 poisons. J. Biol. Chem. 2011, 286, 44945–44951. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Spitz, G.S.; Veturi, U.; Lach, F.P.; Auerbach, A.D.; Smogorzewska, A. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood 2013, 121, 54–63. [Google Scholar] [CrossRef]
- Nakamura, K.; Kogame, T.; Oshiumi, H.; Shinohara, A.; Sumitomo, Y.; Agama, K.; Pommier, Y.; Tsutsui, K.M.; Tsutsui, K.; Hartsuiker, E.; et al. Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLoS Genet. 2010, 6, e1000828. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.W.; Burgin, A.B., Jr.; Huizenga, B.N.; Robertson, C.A.; Yao, K.C.; Nash, H.A. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl. Acad. Sci. USA 1996, 93, 11534–11539. [Google Scholar] [CrossRef] [Green Version]
- Hirano, R.; Interthal, H.; Huang, C.; Nakamura, T.; Deguchi, K.; Choi, K.; Bhattacharjee, M.; Arimura, K.; Umehara, F.; Izumo, S.; et al. Spinocerebellar ataxia with axonal neuropathy: Consequence of a Tdp1 recessive neomorphic mutation? EMBO J. 2007, 26, 4732–4743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; van Waardenburg, R.C.A.M.; Babaoglu, K.; Price, A.C.; Nitiss, K.C.; Nitiss, J.L.; Bjornsti, M.-A.; White, S.W. Mutation of a conserved active site residue converts tyrosyl-DNA phosphodiesterase I into a DNA topoisomerase I-dependent poison. J. Mol. Biol. 2007, 372, 1070–1081. [Google Scholar] [CrossRef] [PubMed]
- Interthal, H.; Chen, H.J.; Kehl-Fie, T.E.; Zotzmann, J.; Leppard, J.B.; Champoux, J.J. SCAN1 mutant Tdp1 accumulates the enzyme–DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 2005, 24, 2224–2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katyal, S.; Khamisy, S.F.; Russell, H.R.; Li, Y.; Ju, L.; Caldecott, K.W.; McKinnon, P.J. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 2007, 26, 4720–4731. [Google Scholar] [CrossRef] [Green Version]
- El-Khamisy, S.F.; Katyal, S.; Patel, P.; Ju, L.; McKinnon, P.J.; Caldecott, K.W. Synergistic decrease of DNA single-strand break repair rates in mouse neural cells lacking both Tdp1 and aprataxin. DNA Repair. 2009, 8, 760–766. [Google Scholar] [CrossRef] [Green Version]
- Barthelmes, H.U.; Habermeyer, M.; Christensen, M.O.; Mielke, C.; Interthal, H.; Pouliot, J.J.; Boege, F.; Marko, D. TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II. J. Biol. Chem. 2004, 279, 55618–55625. [Google Scholar] [CrossRef] [Green Version]
- Nivens, M.C.; Felder, T.; Galloway, A.H.; Pena, M.M.; Pouliot, J.J.; Spencer, H.T. Engineered resistance to camptothecin and antifolates by retroviral coexpression of tyrosyl DNA phosphodiesterase-I and thymidylate synthase. Cancer Chemother. Pharmacol. 2004, 53, 107–115. [Google Scholar] [CrossRef]
- Alagoz, M.; Gilbert, D.C.; El-Khamisy, S.; Chalmers, A.J. DNA repair and resistance to topoisomerase I inhibitors: Mechanisms, biomarkers and therapeutic targets. Curr. Med. Chem. 2012, 19, 3874–3885. [Google Scholar] [CrossRef]
- Perego, P.; Cossa, G.; Tinelli, S.; Corna, E.; Carenini, N.; Gatti, L.; De Cesare, M.; Ciusani, E.; Zunino, F.; Luison, E.; et al. Role of tyrosyl-DNA phosphodiesterase 1 and inter-players in regulation of tumor cell sensitivity to topoisomerase I inhibition. Biochem. Pharmacol. 2012, 83, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Meisenberg, C.; Ward, S.E.; Schmid, P.; El-Khamisy, S.F. TDP1/TOP1 ratio as a promising indicator for the response of small cell lung cancer to topotecan. J. Cancer Sci. Ther. 2014, 6, 258–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczor, C.A.; Torres, R.A.; Lewis, W. The role of transporters in the toxicity of nucleoside and nucleotide analogs. Expert Opin. Drug Metab. Toxicol. 2012, 8, 665–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efremova, A.S.; Zakharenko, A.L.; Shram, S.I.; Kulikova, I.V.; Drenichev, M.S.; Sukhanova, M.V.; Khodyreva, S.N.; Myasoedov, N.F.; Lavrik, O.I.; Mikhailov, S.N.; et al. Disaccharide pyrimidine nucleosides and their derivatives: A novel group of cell-penetrating inhibitors of poly (ADP-ribose) polymerase 1. Nucleosides Nucleotides Nucleic Acids 2013, 32, 510–528. [Google Scholar] [CrossRef]
- King, A.E.; Ackley, M.A.; Cass, C.E.; Young, J.D.; Baldwin, S.A. Nucleoside transporters: From scavengers to novel therapeutic targets. Trends Pharmacol. Sci. 2006, 27, 416–425. [Google Scholar] [CrossRef]
- Komarova, A.O.; Drenichev, M.S.; Dyrkheeva, N.S.; Kulikova, I.V.; Oslovsky, V.E.; Zakharova, O.D.; Zakharenko, A.L.; Mikhailov, S.N.; Lavrik, O.I. Novel group of tyrosyl-DNA-phosphodiesterase 1 inhibitors based on disaccharide nucleosides as drug prototypes for anti-cancer therapy. J. Enzyme Inhib. Med. Chem. 2018, 33, 1415–1429. [Google Scholar] [CrossRef] [Green Version]
- Sherstyuk, Y.V.; Ivanisenko, N.V.; Zakharenko, A.L.; Sukhanova, M.V.; Peshkov, R.Y.; Eltsov, I.V.; Kutuzov, M.M.; Kurgina, T.A.; Belousova, E.A.; Ivanisenko, V.A.; et al. Design, Synthesis and Molecular Modeling Study of Conjugates of ADP and Morpholino Nucleosides as A Novel Class of Inhibitors of PARP-1, PARP-2 and PARP-3. Int. J. Mol. Sci. 2019, 21, 214. [Google Scholar] [CrossRef] [Green Version]
- Amé, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef]
- Pazzaglia, S.; Pioli, C. Multifaceted role of PARP-1 in DNA repair and inflammation: Pathological and therapeutic implications in cancer and non-cancer diseases. Cells 2019, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Shall, S. Proceedings: Experimental manipulation of the specific activity of poly(ADP-ribose) polymerase. Biochem. J. 1975, 77, 2. [Google Scholar] [CrossRef]
- Purnell, M.R.; Whish, W.J. Novel inhibitors of poly(ADP-ribose) synthetase. Biochem. J. 1980, 185, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Terada, M.; Fujiki, H.; Marks, P.A.; Sugimura, T. Induction of erythroid differentiation of murine erythroleukemia cells by nicotinamide and related compounds. Proc. Natl. Acad. Sci. USA 1979, 76, 6411–6414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Huang, S.-Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharenko, A.L.; Drenichev, M.S.; Dyrkheeva, N.S.; Ivanov, G.A.; Oslovsky, V.E.; Ilina, E.S.; Chernyshova, I.A.; Lavrik, O.I.; Mikhailov, S.N. Inhibition of tyrosyl-DNA phosphodiesterase 1 by lipophilic pyrimidine nucleosides. Molecules 2020, 25, 3694. [Google Scholar] [CrossRef]
- Dyrkheeva, N.S.; Chernyshova, I.A.; Ivanov, G.A.; Porozov, Y.B.; Zenchenko, A.A.; Oslovsky, V.E.; Zakharenko, A.L.; Nasyrova, D.I.; Likhatskaya, G.N.; Mikhailov, S.N.; et al. In Vitro and In Silico studies of human tyrosyl-dna phosphodiesterase 1 (Tdp1) inhibition by stereoisomeric forms of lipophilic nucleosides: The role of carbohydrate stereochemistry in ligand-enzyme interactions. Molecules 2022, 27, 2433. [Google Scholar] [CrossRef]
- Drenichev, M.S.; Oslovsky, V.E.; Sun, L.; Tijsma, A.; Kurochkin, N.N.; Tararov, V.I.; Chizhov, A.O.; Neyts, J.; Pannecouque, C.; Leyssen, P.; et al. Modification of the length and structure of the linker of N(6)-benzyladenosine modulates its selective antiviral activity against enterovirus 71. Eur. J. Med. Chem. 2016, 111, 84–94. [Google Scholar] [CrossRef]
- Orlov, A.A.; Drenichev, M.S.; Oslovsky, V.E.; Kurochkin, N.N.; Solyev, P.N.; Kozlovskaya, L.I.; Palyulin, V.A.; Karganova, G.G.; Mikhailov, S.N.; Osolodkin, D.I.; et al. New tools in nucleoside toolbox of tick-borne encephalitis virus reproduction inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 1267–1273. [Google Scholar] [CrossRef]
- Drenichev, M.S.; Oslovsky, V.E.; Tararov, V.I.; Mikhailov, S.N. Synthesis of N6 -Substituted Adenosines as Cytokinin Nucleosides. Curr. Protoc. Nucleic Acid. Chem. 2018, 72, 14.15.1–14.15.16. [Google Scholar] [CrossRef]
- Zhong, M.; Nowak, I.; Robins, M.J. 6-(2-Alkylimidazol-1-yl)purines undergo regiospecific glycosylation at N9. Org. Lett. 2005, 7, 4601–4603. [Google Scholar] [CrossRef]
- Sniady, A.; Bedore, M.W.; Jamison, T.F. One-flow, multistep synthesis of nucleosides by Brønsted acid-catalyzed glycosylation. Angew. Chem. Int. Ed. Engl. 2011, 50, 2155–2158. [Google Scholar] [CrossRef] [Green Version]
- Dumbre, S.G.; Jang, M.Y.; Herdewijn, P. Synthesis of α-L-threose nucleoside phosphonates via regioselective sugar protection. J. Org. Chem. 2013, 78, 7137–7144. [Google Scholar] [CrossRef] [PubMed]
- Vorbrüggen, H.; Ruh-Pohlenz, C. Handbook of Nucleoside Synthesis; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Zakharenko, A.L.; Khomenko, T.M.; Zhukova, S.G.; Koval, O.; Zakharova, O.; Anarbaev, R.; Lebedeva, N.; Korchagina, D.; Komarova, N.; Vasiliev, V.; et al. Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety. Bioorg. Med. Chem. 2015, 23, 2044–2052. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, A.; Klöcking, H.P. In vitro toxicity of surfactants in U937 cells: Cell membrane integrity and mitochondrial function. Exp. Toxicol. Pathol. 1998, 50, 472. [Google Scholar] [CrossRef] [PubMed]
- Munkuev, A.A.; Dyrkheeva, N.S.; Kornienko, T.E.; Ilina, E.S.; Ivankin, D.I.; Suslov, E.V.; Korchagina, D.V.; Gatilov, Y.V.; Zakharenko, A.L.; Malakhova, A.A.; et al. Adamantane-Monoterpenoid Conjugates Linked via Heterocyclic Linkers Enhance the Cytotoxic Effect of Topotecan. Molecules 2022, 27, 3374. [Google Scholar] [CrossRef] [PubMed]
- Takashima, H.; Boerkoel, C.F.; John, J.; Saifi, G.M.; Salih, M.A.; Armstrong, D.; Mao, Y.; Quiocho, F.A.; Roa, B.B.; Nakagawa, M.; et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet. 2002, 32, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Ostling, O.; Johanson, K.J. Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochem. Biophys. Res. Commun. 1984, 123, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Speit, G.; Hartmann, A. The comet assay: A sensitive genotoxicity test for the detection of DNA damage and repair. Methods Mol. Biol. 2006, 314, 275–286. [Google Scholar]
- Tice, R.R.; Strauss, G.H. The single cell gel electrophoresis/comet assay: A potential tool for detecting radiation-induced DNA damage in humans. Stem Cells. 1995, 13, 207–214. [Google Scholar]
- Collins, A.R. The comet assay for DNA damage and repair: Principles, applications, and limitations. Mol. Biotechnol. 2004, 26, 249–261. [Google Scholar] [CrossRef]
- Acar, A. In vivo toxicological assessment of diquat dibromide: Cytotoxic, genotoxic, and biochemical approach. Environ. Sci. Pollut. Res. Int. 2021, 28, 47550–47561. [Google Scholar] [CrossRef]
- Sanz-Serrano, J.; Vettorazzi, A.; Muruzabal, D.; López de Cerain, A.; Azqueta, A. In vitro genotoxicity assessment of functional ingredients: DHA, rutin and α-tocopherol. Food Chem. Toxicol. 2021, 153, 112237. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988, 175, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tice, R.R.; Agurell, E.; Anderson, D.; Burlinson, B.; Hartmann, A.; Kobayashi, H.; Miyamae, Y.; Rojas, E.; Ryu, J.-C.; Sasaki, Y.F. Single cell gel/comet assay: Guidelines for in vitro and in vivo genetic toxicology testing. Environ. Mol. Mutagen. 2000, 35, 206–221. [Google Scholar] [CrossRef]
- Klein, G.; Klein, E. The transformation of a solid transplantable mouse carcinoma into an “ascites tumor”. Cancer Res. 1951, 11, 466–469. [Google Scholar]
- Patt, H.M.; Blackford, M.E. Quantitative studies of the growth response of the Krebs ascites tumor. Cancer Res. 1954, 14, 391–396. [Google Scholar]
- Zakharenko, A.L.; Dyrkheeva, N.S.; Lavrik, O.I. Dual DNA topoisomerase 1 and tyrosyl-DNA phosphodiesterase 1 inhibition for improved anticancer activity. Med. Res. Rev. 2019, 39, 1427–1441. [Google Scholar] [CrossRef] [PubMed]
- Zakharenko, A.L.; Luzina, O.A.; Sokolov, D.N.; Kaledin, V.I.; Nikolin, V.P.; Popova, N.A.; Patel, J.; Zakharova, O.D.; Chepanova, A.A.; Zafar, A. Novel tyrosyl-DNA phosphodiesterase 1 inhibitors enhance the therapeutic impact of topotecan on in vivo tumor models. Eur. J. Med. Chem. 2019, 161, 581–593. [Google Scholar] [CrossRef]
- Khomenko, T.M.; Zakharenko, A.L.; Chepanova, A.A.; Ilina, E.S.; Zakharova, O.D.; Kaledin, V.I.; Nikolin, V.P.; Popova, N.A.; Korchagina, D.V.; Reynisson, J.; et al. Promising New Inhibitors of Tyrosyl-DNA Phosphodiesterase I (Tdp 1) Combining 4-Arylcoumarin and Monoterpenoid Moieties as Components of Complex Antitumor Therapy. Int. J. Mol. Sci. 2019, 21, 126. [Google Scholar] [CrossRef] [Green Version]
- Nikolin, V.P.; Popova, N.A.; Kaledin, V.I.; Luzina, O.A.; Zakharenko, A.L.; Salakhutdinov, N.F.; Lavrik, O.I. The influence of an enamine usnic acid derivative (a tyrosyl-DNA phosphodiesterase 1 inhibitor) on the therapeutic effect of topotecan against transplanted tumors in vivo. Clin. Exp. Metastasis 2021, 38, 431–440. [Google Scholar] [CrossRef]
- Tararov, V.I.; Kolyachkina, S.V.; Alexeev, C.S.; Mikhailov, S.N. N6-Acetyl-2′,3′,5′-tri-O-acetyladenosine; A convenient, ‘missed out’ substrate for regioselective N6-alkylations. Synthesis 2011, 15, 2483–2489. [Google Scholar]
- Yu Lin, H.; Xiang, L.; Ming, L. Synthesis and biological activity of novel 6-substituted purine derivatives. J. Mex. Chem. Soc. 2010, 54, 74–78. [Google Scholar]
- Prasad, A.K.; Kumar, V.; Malhotra, S.; Ravikumar, V.T.; Sanghvi, Y.S.; Parmar, V.S. ‘Green’ methodology for efficient and selective benzoylation of nucleosides using benzoyl cyanide in an ionic liquid. Bioorg. Med. Chem. 2005, 13, 4467–4472. [Google Scholar] [CrossRef] [PubMed]
- Dyrkheeva, N.S.; Anarbaev, R.O.; Lebedeva, N.A.; Kuprushkin, M.; Kuznetsova, A.; Kuznetsov, N.; Rechkunova, N.; Lavrik, O. Human Tyrosyl-DNA phosphodiesterase 1 possesses transphosphooligonucleotidation activity with primary alcohols. Front. Cell Dev. Biol. 2020, 8, 604732. [Google Scholar] [CrossRef] [PubMed]
- Amé, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Höger, T.; Ménissier-de Murcia, J.; de Murcia, G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef] [Green Version]
- Yushok, W.D.; Mallalieu, L.J.; Batt, W.G. Properties of Krebs 2 ascites carcinoma cells: Weight, size, specific gravity, and protein content. J. Frankl. Inst. 1956, 262, 507–509. [Google Scholar] [CrossRef]
- Parsons, D.F.; Marko, M.; Braun, S.J.; Wansor, K.J. Ascites tumor invasion of mouse peritoneum studied by high-voltage electron microscope stereoscopy. Cancer Res. 1982, 42, 4574–4583. [Google Scholar]
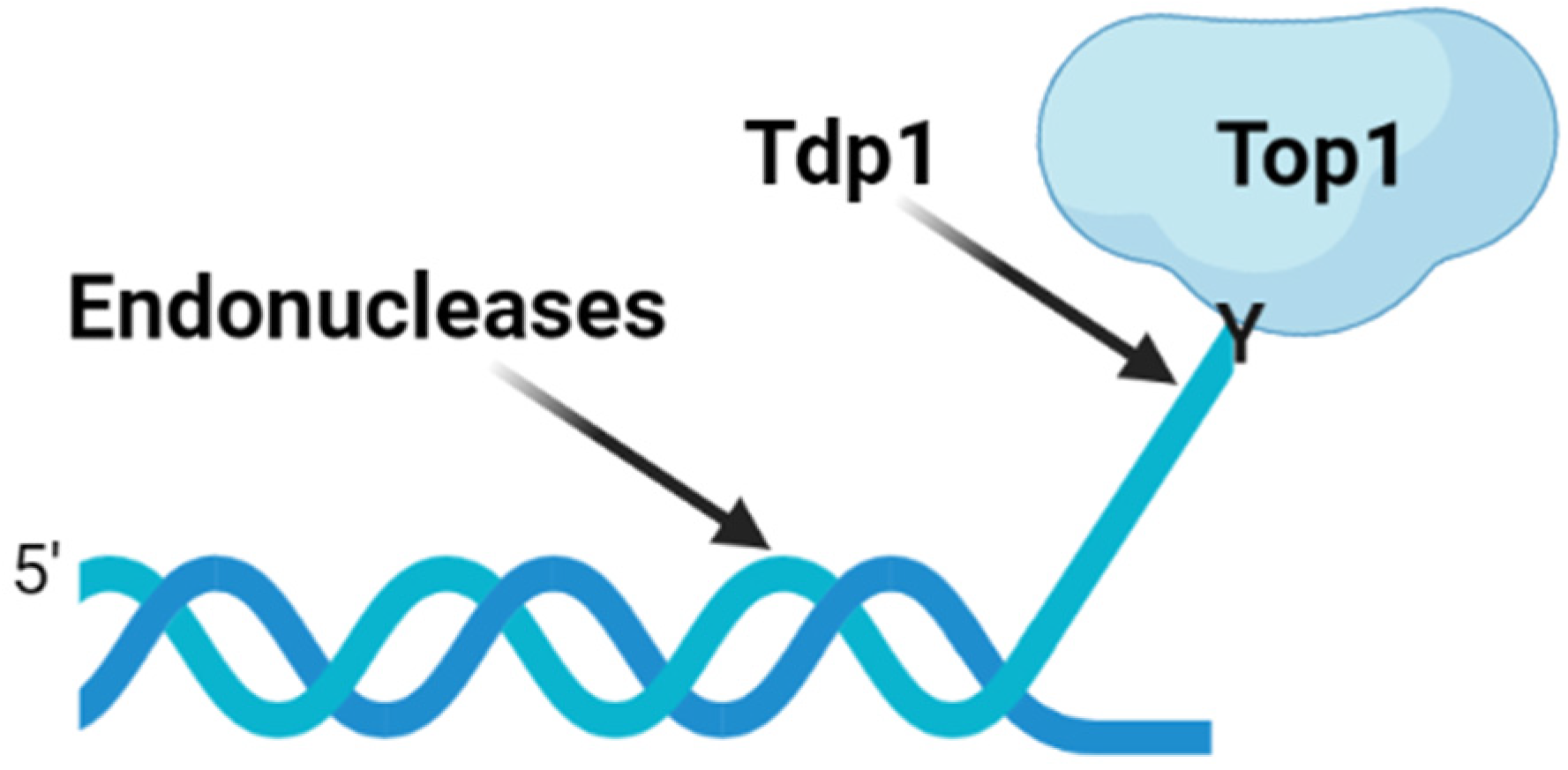





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Structure | Tdp1 IC50, μM | CC50, μM | Enhancement of Tpc Cytotoxicity by 10 μM Compounds, Times |
|---|---|---|---|---|
| Natural compounds | ||||
| 1 |  | >50 | nd * | nd * |
| 2 |  | >50 | nd | nd |
| 3 | 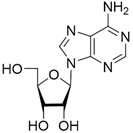 | >50 | nd | nd |
| 4 |  | >50 | nd | nd |
| Semisynthetic compounds | ||||
| 5a |  | >50 | >100 | 2.5 |
| 5b | 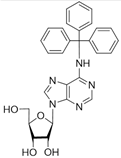 | >50 | 59.6 ± 0.6 | 1.7 |
| 5c |  | 22 ± 3 | 58 ± 2 | 2.2 |
| 5d |  | >50 | 56 ± 10 | 3.1 |
| 5f | 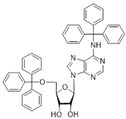 | 0.30 ± 0.06 | >100 | 1.7 |
| 6a |  ** ** | 7 ± 1 | 58 ± 5 | 1.8 |
| 6b |  | 2.0 ± 0.6 | 56 ± 26 | 1.9 |
| 6c | 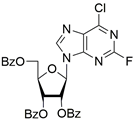 | 1.0 ± 0.2 | 35 ± 20 | 6.5 |
| 6d | 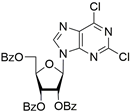 | 0.82 ± 0.02 | 33 ± 15 | 4.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chernyshova, I.A.; Zakharenko, A.L.; Kurochkin, N.N.; Dyrkheeva, N.S.; Kornienko, T.E.; Popova, N.A.; Nikolin, V.P.; Ilina, E.S.; Zharkov, T.D.; Kupryushkin, M.S.; et al. The Lipophilic Purine Nucleoside—Tdp1 Inhibitor—Enhances DNA Damage Induced by Topotecan In Vitro and Potentiates the Antitumor Effect of Topotecan In Vivo. Molecules 2023, 28, 323. https://doi.org/10.3390/molecules28010323
Chernyshova IA, Zakharenko AL, Kurochkin NN, Dyrkheeva NS, Kornienko TE, Popova NA, Nikolin VP, Ilina ES, Zharkov TD, Kupryushkin MS, et al. The Lipophilic Purine Nucleoside—Tdp1 Inhibitor—Enhances DNA Damage Induced by Topotecan In Vitro and Potentiates the Antitumor Effect of Topotecan In Vivo. Molecules. 2023; 28(1):323. https://doi.org/10.3390/molecules28010323
Chicago/Turabian StyleChernyshova, Irina A., Aleksandra L. Zakharenko, Nikolay N. Kurochkin, Nadezhda S. Dyrkheeva, Tatyana E. Kornienko, Nelly A. Popova, Valeriy P. Nikolin, Ekaterina S. Ilina, Timofey D. Zharkov, Maxim S. Kupryushkin, and et al. 2023. "The Lipophilic Purine Nucleoside—Tdp1 Inhibitor—Enhances DNA Damage Induced by Topotecan In Vitro and Potentiates the Antitumor Effect of Topotecan In Vivo" Molecules 28, no. 1: 323. https://doi.org/10.3390/molecules28010323