
Mechanism and Selectivity of Electrochemical Reduction of CO2 on Metalloporphyrin Catalysts from DFT Studies

Abstract
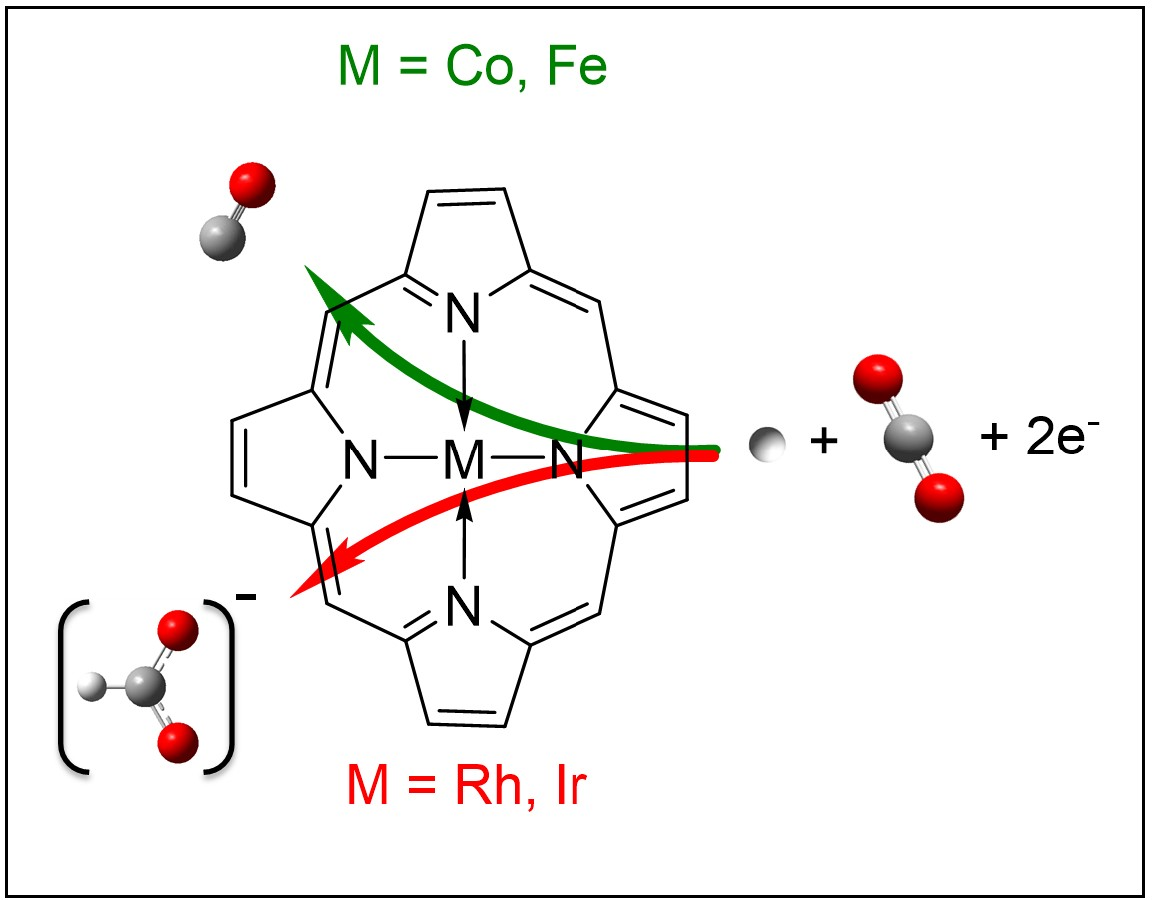
:
1. Introduction
2. Results
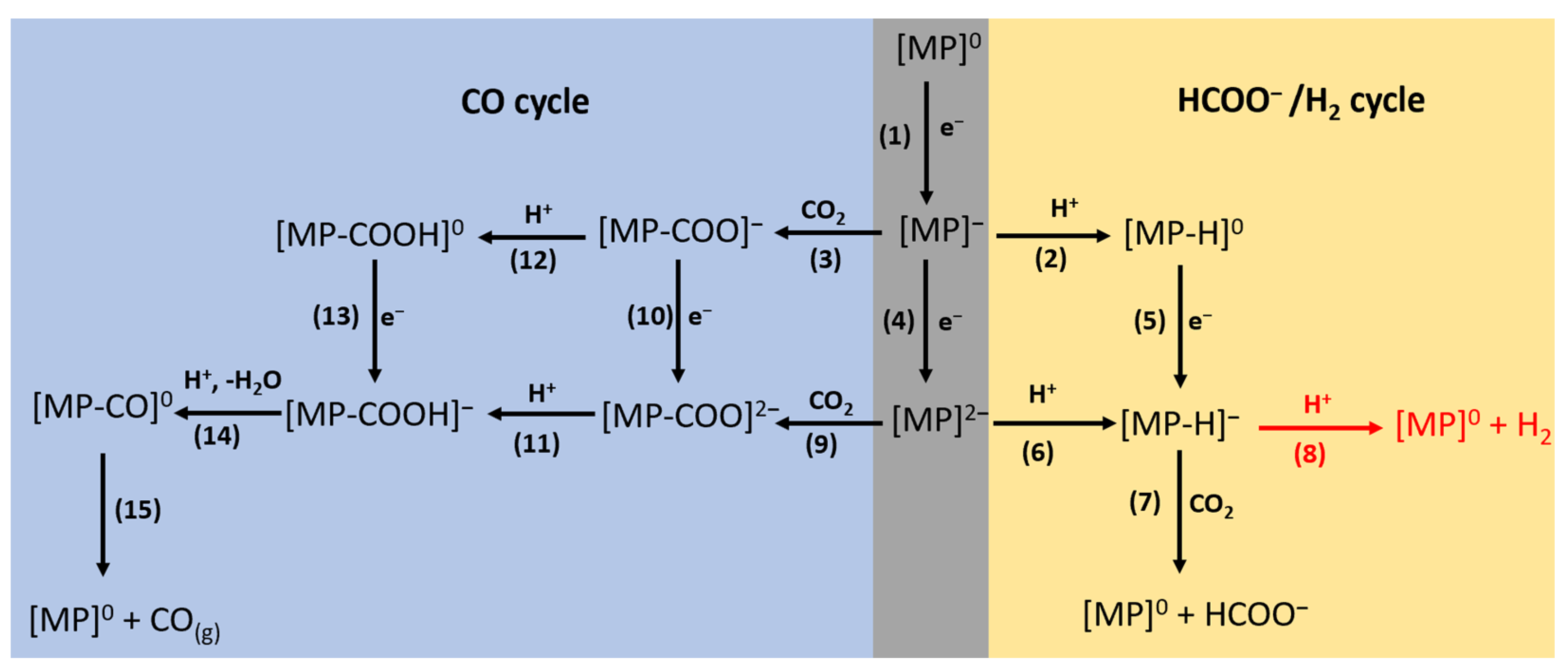
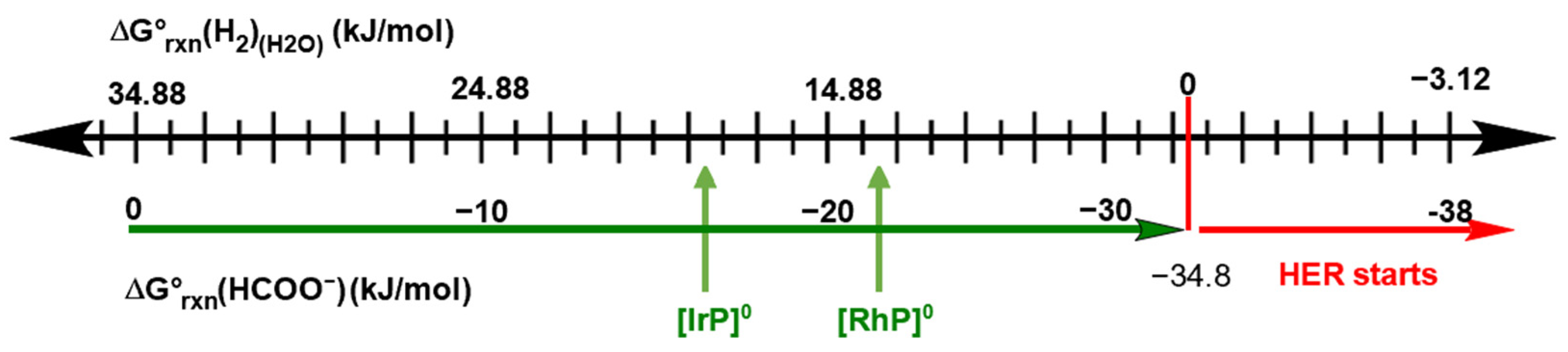
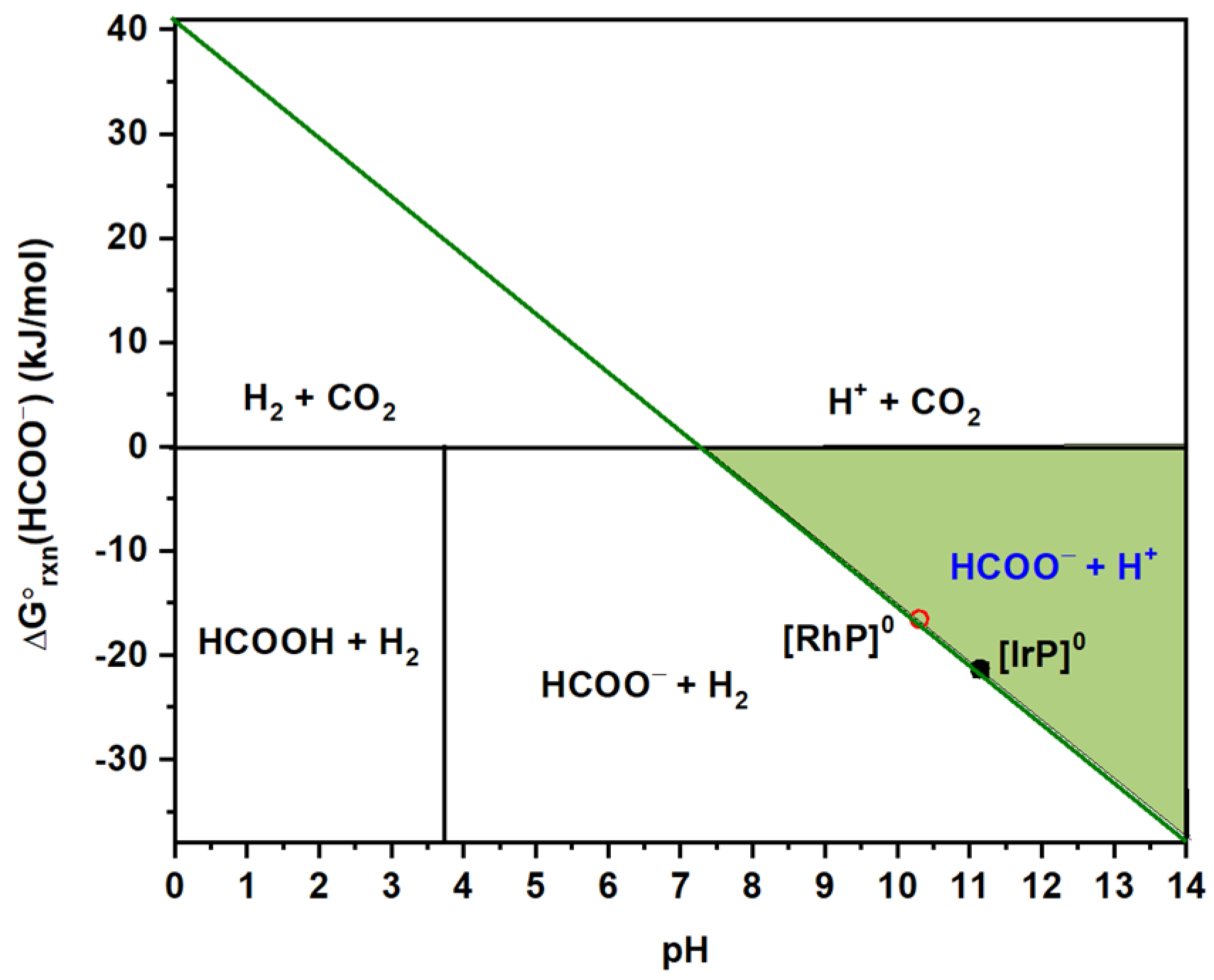
2.1. HCOO−/H2 Cycle
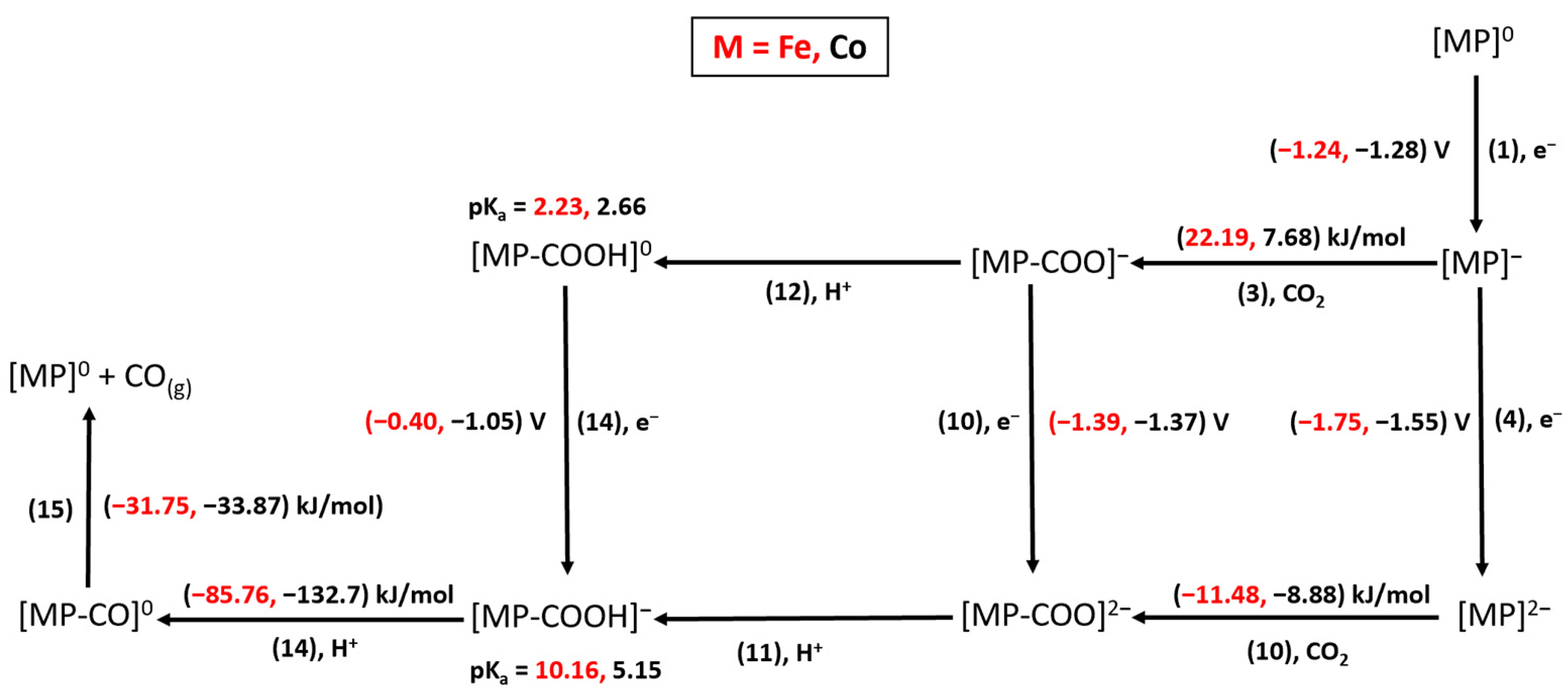
2.2. CO Cycle
3. Discussion
4. Computational Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Rogelj, J.; Den Elzen, M.; Höhne, N.; Fransen, T.; Fekete, H.; Winkler, H.; Schaeffer, R.; Sha, F.; Riahi, K.; Meinshausen, M. Paris Agreement climate proposals need a boost to keep warming well below 2 C. Nature 2016, 534, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpa, D.; Sarno, M. Single-Atom Catalysts for the Electro-Reduction of CO2 to Syngas with a Tunable CO/H2 Ratio: A Review. Catalysts 2022, 12, 275. [Google Scholar] [CrossRef]
- Lin, J.; Yan, S.; Zhang, C.; Hu, Q.; Cheng, Z. Electroreduction of CO2 toward High Current Density. Processes 2022, 10, 826. [Google Scholar] [CrossRef]
- Domingo-Tafalla, B.; Martínez-Ferrero, E.; Franco, F.; Palomares-Gil, E. Applications of Carbon Dots for the Photocatalytic and Electrocatalytic Reduction of CO2. Molecules 2022, 27, 1081. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.A.S.; Yahya, W.Z.N.; Bustam, M.A.; Kibria, M.G. Elucidation of the Roles of Ionic Liquid in CO2 Electrochemical Reduction to Value-Added Chemicals and Fuels. Molecules 2021, 26, 6962. [Google Scholar] [CrossRef]
- Kauffman, D.R.; Thakkar, J.; Siva, R.; Matranga, C.; Ohodnicki, P.R.; Zeng, C.; Jin, R. Efficient Electrochemical CO2 Conversion Powered by Renewable Energy. ACS Appl. Mater. Interfaces 2015, 7, 15626–15632. [Google Scholar] [CrossRef]
- Muis, Z.A.; Hashim, H.; Manan, Z.; Taha, F.; Douglas, P. Optimal planning of renewable energy-integrated electricity generation schemes with CO2 reduction target. Renew. Energy 2010, 35, 2562–2570. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the valorization of exhaust carbon: From CO2 to chemicals, materials, and fuels. Technological use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef]
- Ma, S.; Kenis, P.J. Electrochemical conversion of CO2 to useful chemicals: Current status, remaining challenges, and future opportunities. Curr. Opin. Chem. Eng. 2013, 2, 191–199. [Google Scholar]
- Wu, J.; Zhou, X.-D. Catalytic conversion of CO2 to value added fuels: Current status, challenges, and future directions. Chin. J. Catal. 2016, 37, 999–1015. [Google Scholar] [CrossRef]
- Dai, H.; Zhao, H.; Chen, S.; Jiang, B. A Microwave-Assisted Boudouard Reaction: A Highly Effective Reduction of the Greenhouse Gas CO2 to Useful CO Feedstock with Semi-Coke. Molecules 2021, 26, 1507. [Google Scholar] [CrossRef] [PubMed]
- Bulushev, D.A.; Ross, J.R. Towards sustainable production of formic acid. ChemSusChem 2018, 11, 821–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlow, J.M.; Yang, J.Y. Thermodynamic Considerations for Optimizing Selective CO2 Reduction by Molecular Catalysts. ACS Cent. Sci. 2019, 5, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, D.W.; Johnson, S.I.; Rheingold, A.L.; Ziller, J.W.; Goddard, W.A.; Nielsen, R.J.; Yang, J.Y. Reactivity of a Series of Isostructural Cobalt Pincer Complexes with CO2, CO, and H+. Inorg. Chem. 2014, 53, 13031–13041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, P.; Cheng, C.; Chen, Z.; Schauer, C.K.; Meyer, T.J.; Brookhart, M. Selective electrocatalytic reduction of CO2 to formate by water-stable iridium dihydride pincer complexes. J. Am. Chem. Soc. 2012, 134, 5500–5503. [Google Scholar] [CrossRef] [PubMed]
- Gafurov, Z.N.; Kantyukov, A.O.; Kagilev, A.A.; Kagileva, A.A.; Sakhapov, I.y.F.; Mikhailov, I.K.; Yakhvarov, D.G. Recent Advances in Chemistry of Unsymmetrical Phosphorus-Based Pincer Nickel Complexes: From Design to Catalytic Applications. Molecules 2021, 26, 4063. [Google Scholar] [CrossRef] [PubMed]
- Petersen, H.A.; Myren, T.H.T.; Luca, O.R. Redox-Active Manganese Pincers for Electrocatalytic CO2 Reduction. Inorganics 2020, 8, 62. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.-P.; Ruppert, R.; Sauvage, J.-P. Nickel(II)-cyclam: An extremely selective electrocatalyst for reduction of CO2 in water. J. Chem. Soc. Chem. Commun. 1984, 1315–1316. [Google Scholar] [CrossRef]
- Masood, Z.; Ge, Q. Electrochemical reduction of CO2 to CO and HCOO− using metal–cyclam complex catalysts: Predicting selectivity and limiting potential from DFT. Dalton Trans. 2021, 50, 11446–11457. [Google Scholar] [CrossRef]
- Behnke, S.L.; Manesis, A.C.; Shafaat, H.S. Spectroelectrochemical investigations of nickel cyclam indicate different reaction mechanisms for electrocatalytic CO2 and H+ reduction. Dalton Trans. 2018, 47, 15206–15216. [Google Scholar] [CrossRef] [PubMed]
- Froehlich, J.D.; Kubiak, C.P. Homogeneous CO2 reduction by Ni (cyclam) at a glassy carbon electrode. Inorg. Chem. 2012, 51, 3932–3934. [Google Scholar] [CrossRef] [PubMed]
- Ikeyama, S.; Amao, Y. The effect of the functional ionic group of the viologen derivative on visible-light driven CO2 reduction to formic acid with the system consisting of water-soluble zinc porphyrin and formate dehydrogenase. Photochem. Photobiol. Sci. 2018, 17, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.K.; Baeg, J.-O.; Oh, G.H.; Park, N.-J.; Kong, K.-j.; Kim, J.; Hwang, D.W.; Biswas, S.K. A photocatalyst–enzyme coupled artificial photosynthesis system for solar energy in production of formic acid from CO2. J. Am. Chem. Soc. 2012, 134, 11455–11461. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, W.; Wang, X.; Zhang, X.; Chen, H.; Hu, H.; Liu, L.; Ye, J.; Wang, D. Enhanced Photocatalytic CO2 Reduction over TiO2 Using Metalloporphyrin as the Cocatalyst. Catalysts 2020, 10, 654. [Google Scholar] [CrossRef]
- Mele, G.; Annese, C.; D’Accolti, L.; De Riccardis, A.; Fusco, C.; Palmisano, L.; Scarlino, A.; Vasapollo, G. Photoreduction of Carbon Dioxide to Formic Acid in Aqueous Suspension: A Comparison between Phthalocyanine/TiO2 and Porphyrin/TiO2 Catalysed Processes. Molecules 2015, 20, 396–415. [Google Scholar] [CrossRef] [Green Version]
- Birdja, Y.Y.; Shen, J.; Koper, M.T. Influence of the metal center of metalloprotoporphyrins on the electrocatalytic CO2 reduction to formic acid. Catal. Today 2017, 288, 37–47. [Google Scholar] [CrossRef]
- Taheri, A.; Thompson, E.J.; Fettinger, J.C.; Berben, L.A. An Iron Electrocatalyst for Selective Reduction of CO2 to Formate in Water: Including Thermochemical Insights. ACS Catal. 2015, 5, 7140–7151. [Google Scholar] [CrossRef]
- Taheri, A.; Berben, L.A. Tailoring electrocatalysts for selective CO2 or H+ reduction: Iron carbonyl clusters as a case study. Inorg. Chem. 2015, 55, 378–385. [Google Scholar] [CrossRef]
- Loewen, N.D.; Neelakantan, T.V.; Berben, L.A. Renewable formate from C–H bond formation with CO2: Using iron carbonyl clusters as electrocatalysts. Acc. Chem. Res. 2017, 50, 2362–2370. [Google Scholar] [CrossRef]
- Liu, J.-H.; Yang, L.-M.; Ganz, E. Efficient and Selective Electroreduction of CO2 by Single-Atom Catalyst Two-Dimensional TM–Pc Monolayers. ACS Sustain. Chem. Eng. 2018, 6, 15494–15502. [Google Scholar] [CrossRef]
- Furuya, N.; Matsui, K. Electroreduction of carbon dioxide on gas-diffusion electrodes modified by metal phthalocyanines. J. Electroanal. Chem. Interf. Electrochem. 1989, 271, 181–191. [Google Scholar] [CrossRef]
- Chacón, P.; Hernández-Lima, J.G.; Bazán-Jiménez, A.; García-Revilla, M.A. Modeling Adsorption and Optical Properties for the Design of CO2 Photocatalytic Metal-Organic Frameworks. Molecules 2021, 26, 3060. [Google Scholar] [CrossRef] [PubMed]
- Al-Omari, A.A.; Yamani, Z.H.; Nguyen, H.L. Electrocatalytic CO2 Reduction: From Homogeneous Catalysts to Heterogeneous-Based Reticular Chemistry. Molecules 2018, 23, 2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costentin, C.; Savéant, J.-M. Towards an intelligent design of molecular electrocatalysts. Nat. Rev. Chem. 2017, 1, 0087. [Google Scholar] [CrossRef]
- Costentin, C.; Savéant, J.-M. Homogeneous Molecular Catalysis of Electrochemical Reactions: Manipulating Intrinsic and Operational Factors for Catalyst Improvement. J. Am. Chem. Soc. 2018, 140, 16669–16675. [Google Scholar] [CrossRef]
- Costentin, C.; Savéant, J.-M. Homogeneous molecular catalysis of electrochemical reactions: Catalyst benchmarking and optimization strategies. J. Am. Chem. Soc. 2017, 139, 8245–8250. [Google Scholar] [CrossRef]
- Song, J.; Klein, E.L.; Neese, F.; Ye, S. The mechanism of homogeneous CO2 reduction by Ni (cyclam): Product selectivity, concerted proton–electron transfer and C–O bond cleavage. Inorg. Chem. 2014, 53, 7500–7507. [Google Scholar] [CrossRef]
- Ceballos, B.M.; Yang, J.Y. Directing the reactivity of metal hydrides for selective CO2 reduction. Proc. Natl. Acad. Sci. USA 2018, 115, 12686–12691. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.L.; Li, J.C.; Gao, W.; Jiang, Q. Cobalt-porphine catalyzed CO2 electro-reduction: A novel protonation mechanism. Phys. Chem. Chem. Phys. 2017, 19, 15067–15072. [Google Scholar] [CrossRef]
- Friães, S.; Realista, S.; Gomes, C.S.B.; Martinho, P.N.; Royo, B. Click-Derived Triazoles and Triazolylidenes of Manganese for Electrocatalytic Reduction of CO2. Molecules 2021, 26, 6325. [Google Scholar] [CrossRef]
- Li, J.; Zhang, S.; Wang, J.; Yin, X.; Han, Z.; Chen, G.; Zhang, D.; Wang, M. Electrocatalytic CO2 Reduction and H2 Evolution by a Copper (II) Complex with Redox-Active Ligand. Molecules 2022, 27, 1399. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.-M.; Tatin, A. Efficient and selective molecular catalyst for the CO2-to-CO electrochemical conversion in water. Proc. Natl. Acad. Sci. USA 2015, 112, 6882–6886. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Kortlever, R.; Kas, R.; Birdja, Y.Y.; Diaz-Morales, O.; Kwon, Y.; Ledezma-Yanez, I.; Schouten, K.J.P.; Mul, G.; Koper, M.T. Electrocatalytic reduction of carbon dioxide to carbon monoxide and methane at an immobilized cobalt protoporphyrin. Nat. Commun. 2015, 6, 8177. [Google Scholar] [CrossRef] [Green Version]
- de Groot, M.T.; Koper, M.T.M. Redox transitions of chromium, manganese, iron, cobalt and nickel protoporphyrins in aqueous solution. Phys. Chem. Chem. Phys. 2008, 10, 1023–1031. [Google Scholar] [CrossRef]
- Mohamed, E.A.; Zahran, Z.N.; Naruta, Y. Efficient electrocatalytic CO2 reduction with a molecular cofacial iron porphyrin dimer. Chem. Commun. 2015, 51, 16900–16903. [Google Scholar] [CrossRef]
- Ait Ahsaine, H.; Zbair, M.; BaQais, A.; Arab, M. CO2 Electroreduction over Metallic Oxide, Carbon-Based, and Molecular Catalysts: A Mini-Review of the Current Advances. Catalysts 2022, 12, 450. [Google Scholar] [CrossRef]
- Connelly, S.J.; Wiedner, E.S.; Appel, A.M. Predicting the reactivity of hydride donors in water: Thermodynamic constants for hydrogen. Dalton Trans. 2015, 44, 5933–5938. [Google Scholar] [CrossRef]
- Tsay, C.; Livesay, B.N.; Ruelas, S.; Yang, J.Y. Solvation Effects on Transition Metal Hydricity. J. Am. Chem. Soc. 2015, 137, 14114–14121. [Google Scholar] [CrossRef]
- Göttle, A.J.; Koper, M.T. Proton-coupled electron transfer in the electrocatalysis of CO2 reduction: Prediction of sequential vs. concerted pathways using DFT. Chem. Sci 2017, 8, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Miao, T.; Deng, Q.; Xu, Y.; Shi, H.; Huang, Y.; Fu, X. Accelerating Nickel-Based Molecular Construction via DFT Guidance for Advanced Photocatalytic Hydrogen Production. ACS Appl. Mater. Interfaces 2022, 14, 17486–17499. [Google Scholar] [CrossRef]
- Ma, Y.; Du, J.; Fang, Y.; Wang, X. Encapsulation of cobalt oxide into metal-organic frameworks for an improved photocatalytic CO2 reduction. ChemSusChem 2021, 14, 946–951. [Google Scholar] [CrossRef]
- Bhugun, I.; Lexa, D.; Savéant, J.-M. Homogeneous Catalysis of Electrochemical Hydrogen Evolution by Iron(0) Porphyrins. J. Am. Chem. Soc. 1996, 118, 3982–3983. [Google Scholar] [CrossRef]
- Ceballos, B.M.; Yang, J.Y. Highly Selective Electrocatalytic CO2 Reduction by [Pt(dmpe)2]2+ through Kinetic and Thermodynamic Control. Organometallics 2020, 39, 1491–1496. [Google Scholar] [CrossRef]
- Riplinger, C.; Sampson, M.D.; Ritzmann, A.M.; Kubiak, C.P.; Carter, E.A. Mechanistic Contrasts between Manganese and Rhenium Bipyridine Electrocatalysts for the Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2014, 136, 16285–16298. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.; Chabalowski, C.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154123. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16 Revision B. 01, 2016; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the first-row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules—A Quantum Theory; Clarendon Press: Oxford, UK, 1994; ISBN 9780198558651. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | First Reduction Potential (E°) | Second Reduction Potential |
|---|---|---|
| [FeP]0 | −1.24 | −1.74 |
| [CoP]0 | −1.28 | −1.55 |
| [RhP]0 | −0.02 | −1.64 |
| [IrP]0 | −0.26 | −1.71 |
| Catalyst | [MP-H]0 | [MP-H]− |
|---|---|---|
| [FeP]0 | 2.45 | 19.45 |
| [CoP]0 | 6.29 | 13.45 |
| [RhP]0 | 10.07 | 17.53 |
| [IrP]0 | 11.15 | 23.75 |
| Catalyst | [MP]0 | [MP]− | |
|---|---|---|---|
| [FeP]0 | Fe | 1.25 | 1.19 (−0.06) |
| P | −1.25 | −2.19 (−0.94) | |
| [CoP]0 | Co | 1.17 | 1.04 (−0.13) |
| P | −1.17 | −2.04 (−0.87) | |
| [RhP]0 | Rh | 0.89 | 0.49 (−0.64) |
| P | −0.89 | −1.49 (−0.36) | |
| [IrP]0 | Ir | 1.13 | 0.59 (−0.54) |
| P | −1.13 | −1.60 (−0.46) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masood, Z.; Ge, Q. Mechanism and Selectivity of Electrochemical Reduction of CO2 on Metalloporphyrin Catalysts from DFT Studies. Molecules 2023, 28, 375. https://doi.org/10.3390/molecules28010375
Masood Z, Ge Q. Mechanism and Selectivity of Electrochemical Reduction of CO2 on Metalloporphyrin Catalysts from DFT Studies. Molecules. 2023; 28(1):375. https://doi.org/10.3390/molecules28010375
Chicago/Turabian StyleMasood, Zaheer, and Qingfeng Ge. 2023. "Mechanism and Selectivity of Electrochemical Reduction of CO2 on Metalloporphyrin Catalysts from DFT Studies" Molecules 28, no. 1: 375. https://doi.org/10.3390/molecules28010375
APA StyleMasood, Z., & Ge, Q. (2023). Mechanism and Selectivity of Electrochemical Reduction of CO2 on Metalloporphyrin Catalysts from DFT Studies. Molecules, 28(1), 375. https://doi.org/10.3390/molecules28010375