
Effect of 3d Transition Metal Atom Intercalation Concentration on the Electronic and Magnetic Properties of Graphene/MoS2 Heterostructure: A First-Principles Study

Abstract
:1. Introduction
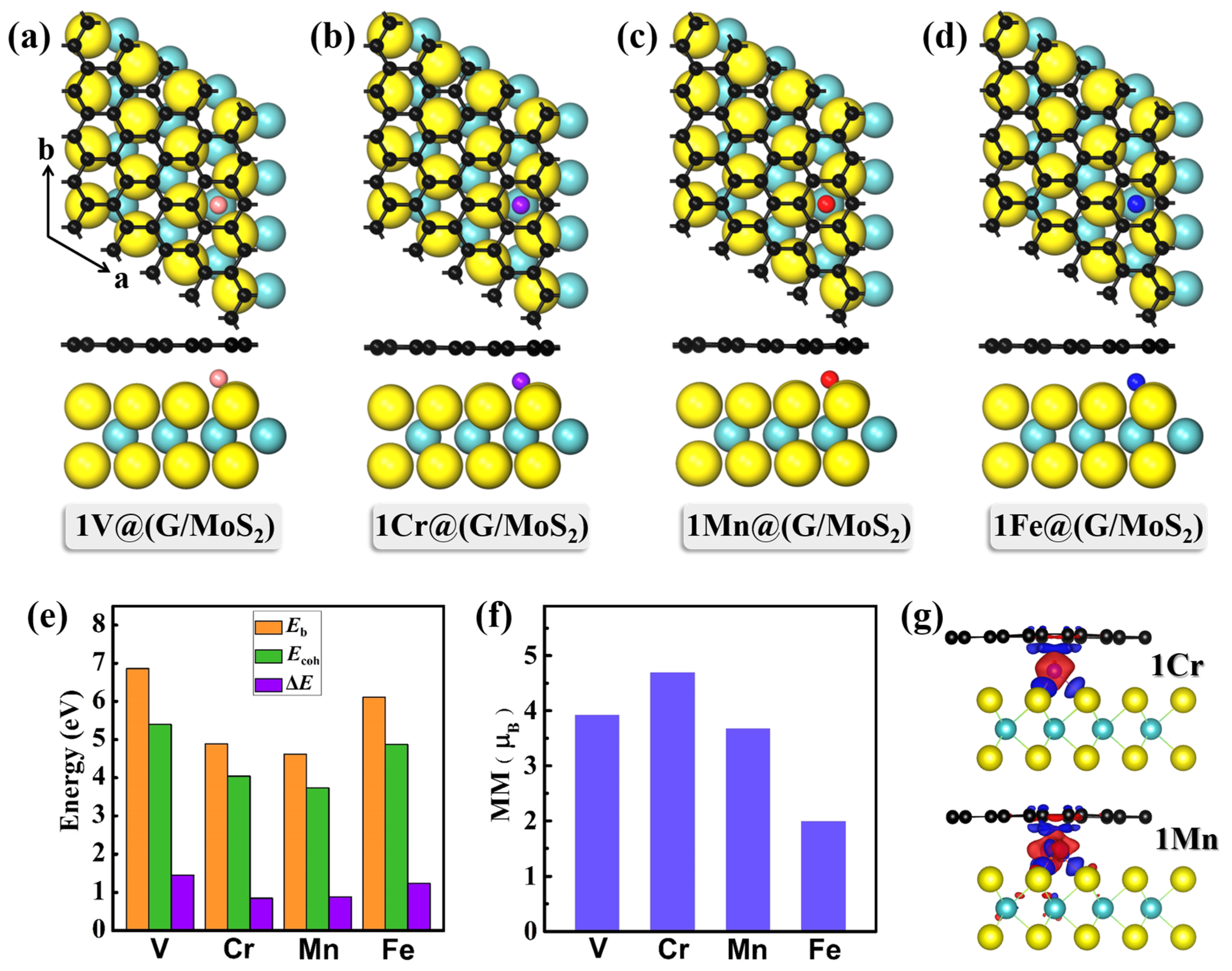
2. Results and Discussion
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Habib, M.R.; Liang, T.; Yu, X.; Pi, X.; Liu, Y.; Xu, M. A review of theoretical study of graphene chemical vapor deposition synthesis on metals: Nucleation, growth, and the role of hydrogen and oxygen. Rep. Prog. Phys. 2018, 81, 036501. [Google Scholar] [CrossRef] [PubMed]
- Han, G.H.; Duong, D.L.; Keum, D.H.; Yun, S.J.; Lee, Y.H. van der Waals metallic transition metal dichalcogenides. Chem. Rev. 2018, 118, 6297–6336. [Google Scholar] [CrossRef]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yu, Y.; Ye, G.J.; Ge, Q.; Ou, X.; Wu, H.; Feng, D.; Chen, X.H.; Zhang, Y. Black phosphorus field-effect transistors. Nat. Nanotechnol. 2014, 9, 372–377. [Google Scholar] [CrossRef] [Green Version]
- Abellán, G.; Wild, S.; Lloret, V.; Scheuschner, N.; Gillen, R.; Mundloch, U.; Maultzsch, J.; Varela, M.; Hauke, F.; Hirsch, A. Fundamental Insights into the Degradation and Stabilization of Thin Layer Black Phosphorus. J. Am. Chem. Soc. 2017, 139, 10432–10440. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Dai, Y.; Wei, W.; Niu, C.; Yu, L.; Huang, B. First-Principles Study of the Graphene@MoSe2 Heterobilayers. J. Phys. Chem. C 2011, 115, 20237–20241. [Google Scholar] [CrossRef]
- Hwang, B.; Hwang, J.; Yoon, J.K.; Lim, S.; Kim, S.; Lee, M.; Kwon, J.H.; Baek, H.; Sung, D.; Kim, G.; et al. Energy Bandgap and Edge States in an Epitaxially Grown Graphene/h-BN Heterostructure. Sci. Rep. 2016, 6, 31160. [Google Scholar] [CrossRef] [Green Version]
- Kochaev, A.; Katin, K.; Maslov, M.; Meftakhutdinov, R. AA-Stacked Borophene-Graphene Bilayer with Covalent Bonding: Ab Initio Investigation of Structural, Electronic and Elastic properties. J. Phys. Chem. Lett. 2020, 11, 5668–5673. [Google Scholar] [CrossRef]
- Geim, A.K.; Grigorieva, I.V. Van der Waals heterostructures. Nature 2013, 499, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Dean, C.R.; Young, A.F.; Cadden-Zimansky, P.; Wang, L.; Ren, H.; Watanabe, K.; Taniguchi, T.; Kim, P.; Hone, J.; Shepard, K.L. Multicomponent fractional quantum Hall effect in graphene. Nat. Phys. 2011, 7, 693–696. [Google Scholar] [CrossRef]
- Neupane, H.K.; Adhikari, N.P.; Hang, D.-R. Tuning Structural, Electronic, and Magnetic Properties of C Sites Vacancy Defects in Graphene/MoS2 van der Waals Heterostructure Materials: A First-Principles Study. Adv. Condens. Matter. Phys. 2020, 2020, 8850701. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, P.; Zu, H.; Gao, F.; Zu, X. Electronic Structures and Magnetic Properties of MoS2 Nanostructures: Atomic Defects, Nanoholes, Nanodots and Antidots. Phys. Chem. Chem. Phys. 2013, 15, 10385–10394. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Wang, D.; Ma, Z.; Liu, G.; Zhu, X.; Yan, M.; Fu, Y. The first-principles study on the halogen-doped graphene/MoS2 heterojunction. Solid State Commun. 2021, 334–335, 114366. [Google Scholar] [CrossRef]
- Shu, H.; Luo, P.F.; Liang, P.; Cao, D.; Chen, X.S. Layer-Dependent Dopant Stability and Magnetic Exchange Coupling of Iron-Doped MoS2 Nanosheets. ACS Appl. Mater. Interfaces 2015, 7, 7534–7541. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Chen, L.; Yang, M.; Mi, Z.; Zhang, M.; Gao, K.; Zhang, D.; Su, S.; Hou, W. Interface contact and modulated electronic properties by external vertical strains and electric fields in graphene/MoS2 heterostructure. Curr. Appl. Phys. 2022, 39, 331–338. [Google Scholar] [CrossRef]
- Ghorbani-Asl, M.; Bristowe, P.D.; Koziol, K.; Heine, T.; Kuc, A. Effect of compression on the electronic, optical and transport properties of MoS2/graphene-based junctions. 2D Mater. 2016, 3, 025018. [Google Scholar] [CrossRef]
- Manchanda, P.; Sharma, V.; Yu, H.; Sellmyer, D.J.; Skomski, R. Magnetism of Ta Dichalcogenide Monolayers Tuned by Strain and Hydrogenation. Appl. Phys. Lett. 2015, 107, 032402. [Google Scholar] [CrossRef] [Green Version]
- Larson, D.T.; Fampiou, I.; Kim, G.; Kaxiras, E. Lithium intercalation in graphene–MoS2 heterostructures. J. Phys. Chem. C 2018, 122, 24535–24541. [Google Scholar] [CrossRef]
- Jung, Y.; Zhou, Y.; Cha, J.J. Intercalation in Two-Dimensional Transition Metal Chalcogenides. Inorg. Chem. Front. 2016, 3, 452–463. [Google Scholar] [CrossRef]
- Wan, J.; Lacey, S.D.; Dai, J.; Bao, W.; Fuhrer, M.S.; Hu, L. Tuning two-dimensional nanomaterials by intercalation: Materials, properties and applications. Chem. Soc. Rev. 2016, 45, 6742–6765. [Google Scholar] [CrossRef]
- Stark, M.S.; Kuntz, K.L.; Martens, S.J.; Warren, S.C. Intercalation of Layered Materials from Bulk to 2D. Adv. Mater. 2019, 31, e1808213. [Google Scholar] [CrossRef]
- Ghosh, D.; Devi, P.; Kumar, P. Intercalation in two-dimensional transition metal chalcogenides: Interlayer engineering and applications. Prog. Energy 2022, 4, 022001. [Google Scholar] [CrossRef]
- Zhou, J.; Lin, Z.; Ren, H.; Duan, X.; Shakir, I.; Huang, Y.; Duan, X. Layered Intercalation Materials. Adv. Mater. 2021, 33, e2004557. [Google Scholar] [CrossRef] [PubMed]
- Kühne, M.; Börrnert, F.; Fecher, S.; Ghorbani-Asl, M.; Biskupek, J.; Samuelis, D.; Krasheninnikov, A.V.; Kaiser, U.; Smet, J.H. Reversible superdense ordering of lithium between two graphene sheets. Nature 2018, 564, 234–239. [Google Scholar] [CrossRef]
- Zhu, G.; Liu, J.; Zheng, Q.; Zhang, R.; Li, D.; Banerjee, D.; Cahill, D.G. Tuning thermal conductivity in molybdenum disulfide by electrochemical intercalation. Nat. Commun. 2016, 7, 13211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.S.; Ke, M.; Hu, Y. Ionic Intercalation in Two-Dimensional van der Waals Materials: In Situ Characterization and Electrochemical Control of the Anisotropic Thermal Conductivity of Black Phosphorus. Nano Lett. 2017, 17, 1431–1438. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, A.; Wu, X.; van de Groep, J.; Tang, P.; Li, S.; Liu, B.; Shi, F.; Wan, J.; Li, Q.; et al. Reversible and selective ion intercalation through the top surface of few-layer MoS2. Nat. Commun. 2018, 9, 5289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, H.K.; Abutaha, A.; Voiry, D.; Verzhbitskiy, I.; Cai, Y.; Zhang, G.; Liu, Y.; Wu, J.; Chhowalla, M.; Eda, G.; et al. Effects of Structural Phase Transition on Thermoelectric Performance in Lithium-Intercalated Molybdenum Disulfide (LixMoS2). ACS Appl. Mater. Interfaces 2019, 11, 12184–12189. [Google Scholar] [CrossRef] [PubMed]
- Profeta, G.; Calandra, M.; Mauri, F. Phonon-mediated superconductivity in graphene by lithium deposition. Nat. Phys. 2012, 8, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Xiong, F.; Wang, H.; Liu, X.; Sun, J.; Brongersma, M.; Pop, E.; Cui, Y. Li Intercalation in MoS2: In Situ Observation of Its Dynamics and Tuning Optical and Electrical Properties. Nano Lett. 2015, 15, 6777–6784. [Google Scholar] [CrossRef]
- Bediako, D.K.; Rezaee, M.; Yoo, H.; Larson, D.T.; Zhao, S.Y.F.; Taniguchi, T.; Watanabe, K.; Brower-Thomas, T.L.; Kaxiras, E.; Kim, P. Heterointerface effects in the electrointercalation of van der Waals heterostructures. Nature 2018, 558, 425–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhao, X.; Liu, Y. Ab Initio Study of Structural, Electronic, and Magnetic Properties of Transition Metal Atoms Intercalated AA-Stacked Bilayer Graphene. J. Phys. Chem. C 2016, 120, 22710–22717. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, W.; Dai, J.; Liu, Y. Role of embedded 3d transition metal atoms on the electronic and magnetic properties of defective bilayer graphene. Carbon 2017, 118, 376–383. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Ma, L.; Zhao, X.; Yao, X. Modulating the electronic and magnetic properties of bilayer borophene via transition metal atoms intercalation: From metal to half metal and semiconductor. Nanotechnology 2018, 29, 305706. [Google Scholar] [CrossRef]
- Zhang, X.; Bao, Z.; Ye, X.; Xu, W.; Wang, Q.; Liu, Y. Half-metallic properties of 3d transition metal atom-intercalated graphene@MS2 (M = W, Mo) hybrid structures. Nanoscale 2017, 9, 11231–11238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, Y.; Gao, W.; Lin, Y.; Zhao, X.; Wang, Q.; Yao, X.; He, M.; Ye, X.; Liu, Y. Sizable bandgaps of graphene in 3d transition metal intercalated defective graphene/WSe2 heterostructures. RSC Adv. 2019, 9, 18157–18164. [Google Scholar] [CrossRef] [Green Version]
- Krasheninnikov, A.V.; Lehtinen, P.O.; Foster, A.S.; Pyykko, P.; Nieminen, R.M. Embedding transition-metal atoms in graphene: Structure, bonding, and magnetism. Phys. Rev. Lett. 2009, 102, 126807. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-Q.; Chen, W.-G.; Zhu, Z.-L.; Jia, Y. Electronic and Magnetic Properties Modulated by Adsorption of 3d Transition Metal Atoms in Monolayer and Bilayer MoS2 Sheets. Acta Metall. Sin. 2015, 28, 793. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 2013, 46, 1740. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Q.; Wang, J. Electronic Structure of Twisted Bilayers of Graphene/MoS2 and MoS2/MoS2. J. Phys. Chem. C 2015, 119, 4752. [Google Scholar] [CrossRef]
- Jin, C.; Rasmussen, F.A.; Thygesen, K.S. Tuning the Schottky Barrier at the Graphene/MoS2 Interface by Electron Doping: Density Functional Theory and Many-Body Calculations. J. Phys. Chem. C 2015, 119, 19928. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bučko, T.; Hafner, J.; Lebègue, S.; Ángyán, J.G. Improved Description of the Structure of Molecular and Layered Crystals: Ab Initio DFT Calculations with van der Waals Corrections. J. Phys. Chem. A 2010, 114, 11814–11824. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liechtenstein, A.; Anisimov, V.I.; Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 1995, 52, R5467. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Wang, L.; Sun, Y.; Li, X.; Sun, J.; Wang, B.; He, M.; Zhang, X. Two-dimensional transition metal triborides: Monolayers with robust intrinsic magnetism and high spin stability. Phys. Rev. B 2022, 105, 214421. [Google Scholar] [CrossRef]
- Dou, M.; Li, H.; Yao, Q.; Wang, J.; Liu, Y.; Wu, F. Room-temperature ferromagnetism in two-dimensional transition metal borides: A first-principles investigation. Phys. Chem. Chem. Phys. 2021, 23, 10615. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1TM@(G/MoS2) | 2TM@(G/MoS2) | 3TM@(G/MoS2) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| d | Eb | ΔQ1/ΔQ2 | MM | d | Eb | ΔQ1/ΔQ2 | MM | d | Eb | ΔQ1/ΔQ2 | MM | |
| V | 3.20 | 6.86 | 0.46/0.66 | 3.93 | 3.53 | 6.03 | 0.43/0.64 | 5.81 | 3.59 | 5.70 | 0.42/0.64 | 9.64 |
| Cr | 3.27 | 4.89 | 0.41/0.65 | 4.70 | 3.74 | 4.08 | 0.36/0.55 | 10.51 | 3.80 | 3.79 | 0.33/0.58 | 14.73 |
| Mn | 3.19 | 4.62 | 0.38/0.57 | 3.69 | 3.52 | 4.02 | 0.30/0.49 | 6.83 | 3.64 | 3.72 | 0.24/0.50 | 10.63 |
| Fe | 3.29 | 6.11 | 0.12/0.56 | 2.00 | 3.52 | 5.23 | 0.26/0.41 | 5.12 | 3.57 | 5.15 | 0.21/0.42 | 8.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, F.; Wang, Z.; He, J.; Li, Z.; Meng, L.; Zhang, X. Effect of 3d Transition Metal Atom Intercalation Concentration on the Electronic and Magnetic Properties of Graphene/MoS2 Heterostructure: A First-Principles Study. Molecules 2023, 28, 509. https://doi.org/10.3390/molecules28020509
Wu F, Wang Z, He J, Li Z, Meng L, Zhang X. Effect of 3d Transition Metal Atom Intercalation Concentration on the Electronic and Magnetic Properties of Graphene/MoS2 Heterostructure: A First-Principles Study. Molecules. 2023; 28(2):509. https://doi.org/10.3390/molecules28020509
Chicago/Turabian StyleWu, Feng, Zijin Wang, Jiaqi He, Zhenzhe Li, Lijuan Meng, and Xiuyun Zhang. 2023. "Effect of 3d Transition Metal Atom Intercalation Concentration on the Electronic and Magnetic Properties of Graphene/MoS2 Heterostructure: A First-Principles Study" Molecules 28, no. 2: 509. https://doi.org/10.3390/molecules28020509