

Branched Linkers for Site-Specific Fluorescent Labeling of Antibodies

 , , and
, , and
Abstract
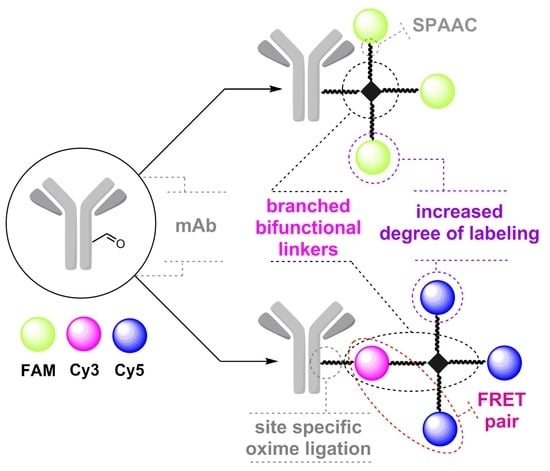
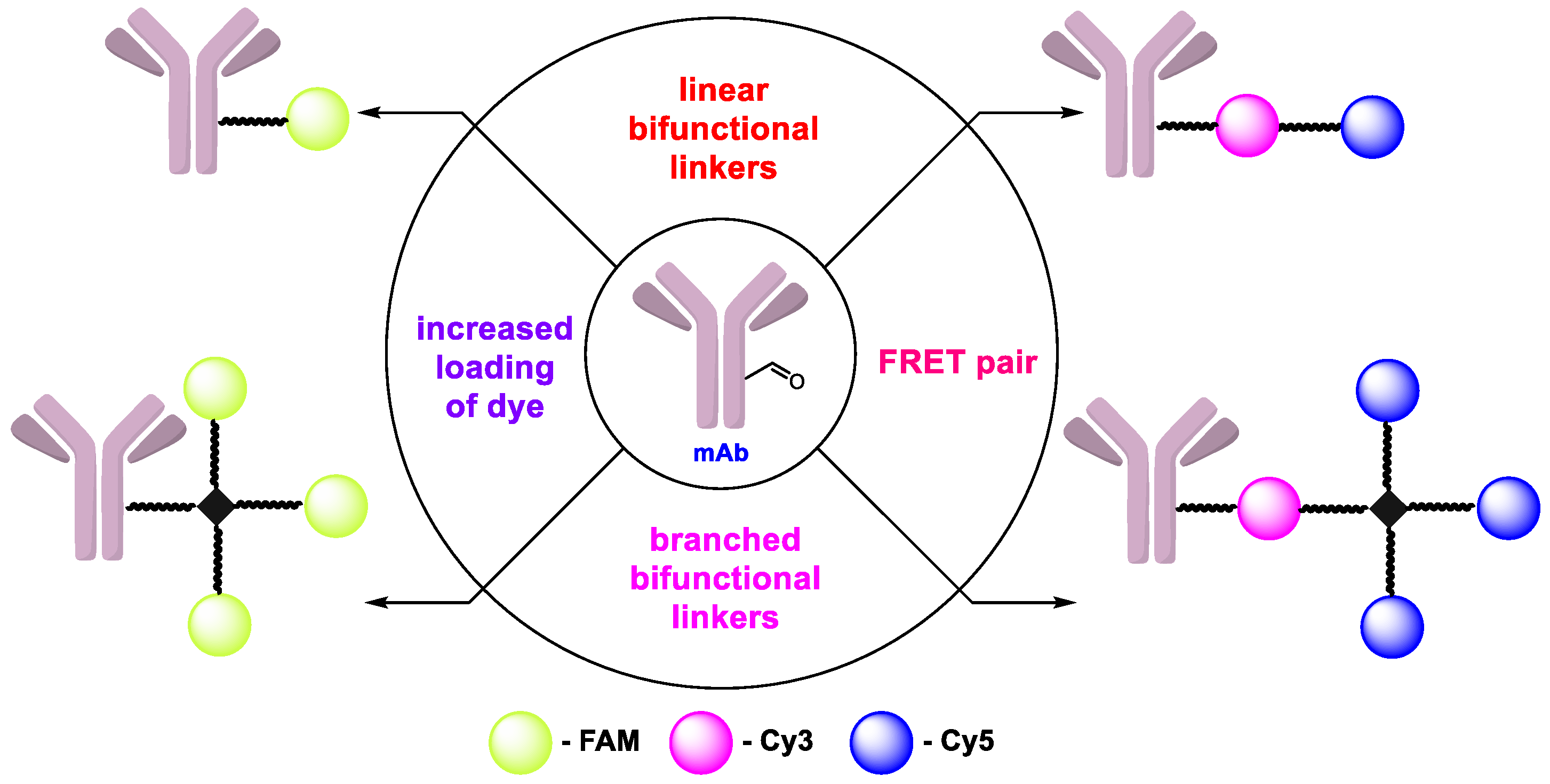
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Azide Linkers
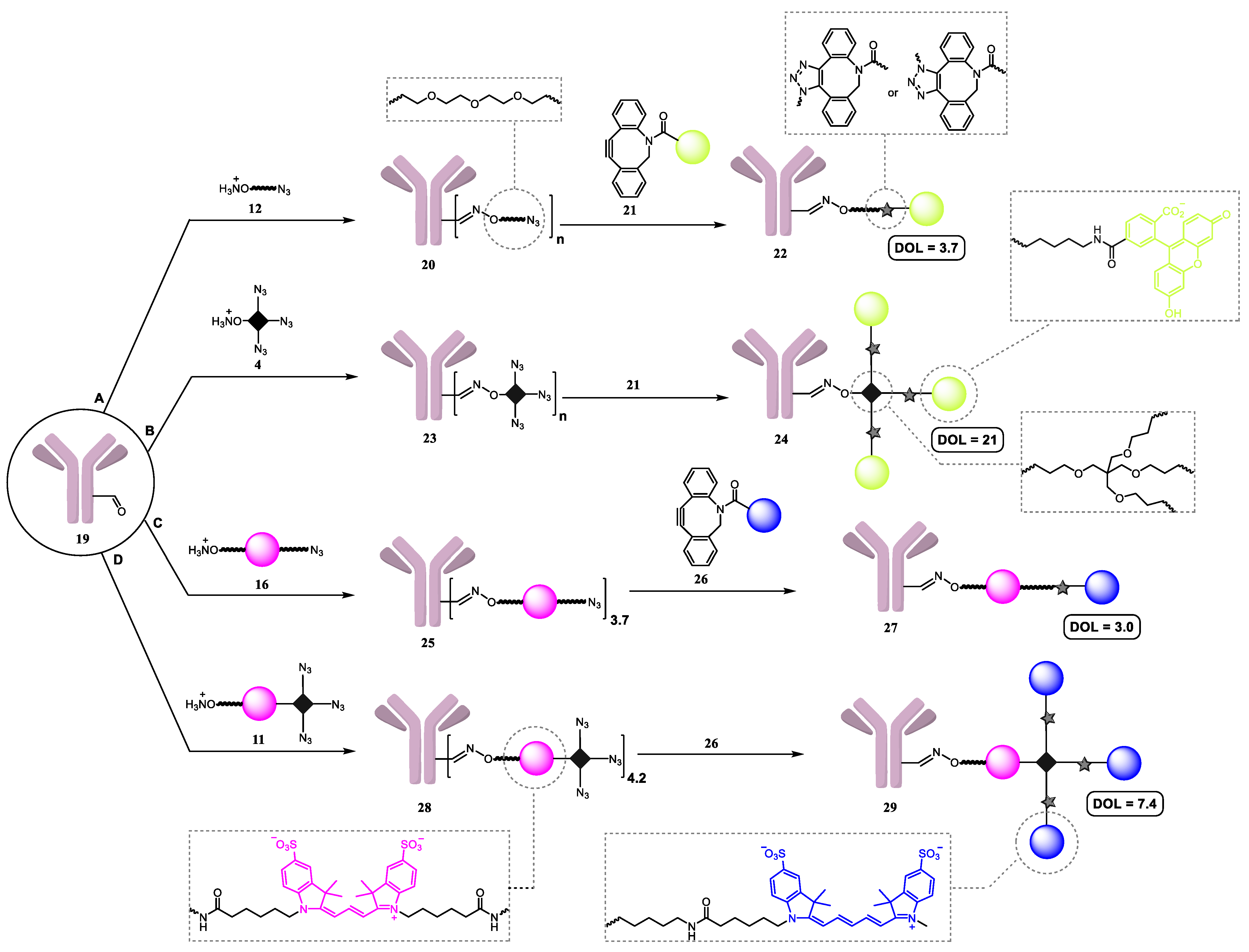
2.2. Synthesis of Antibody Conjugates
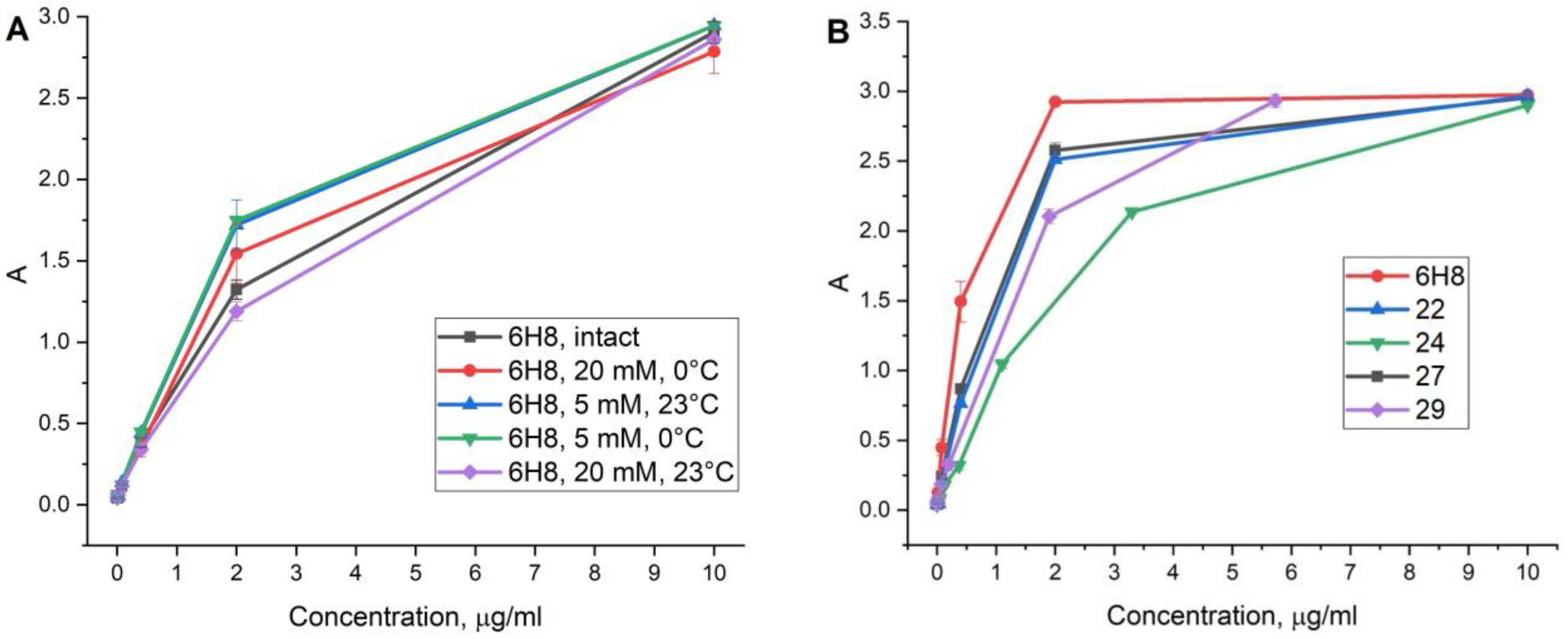
2.3. Degree of Labeling
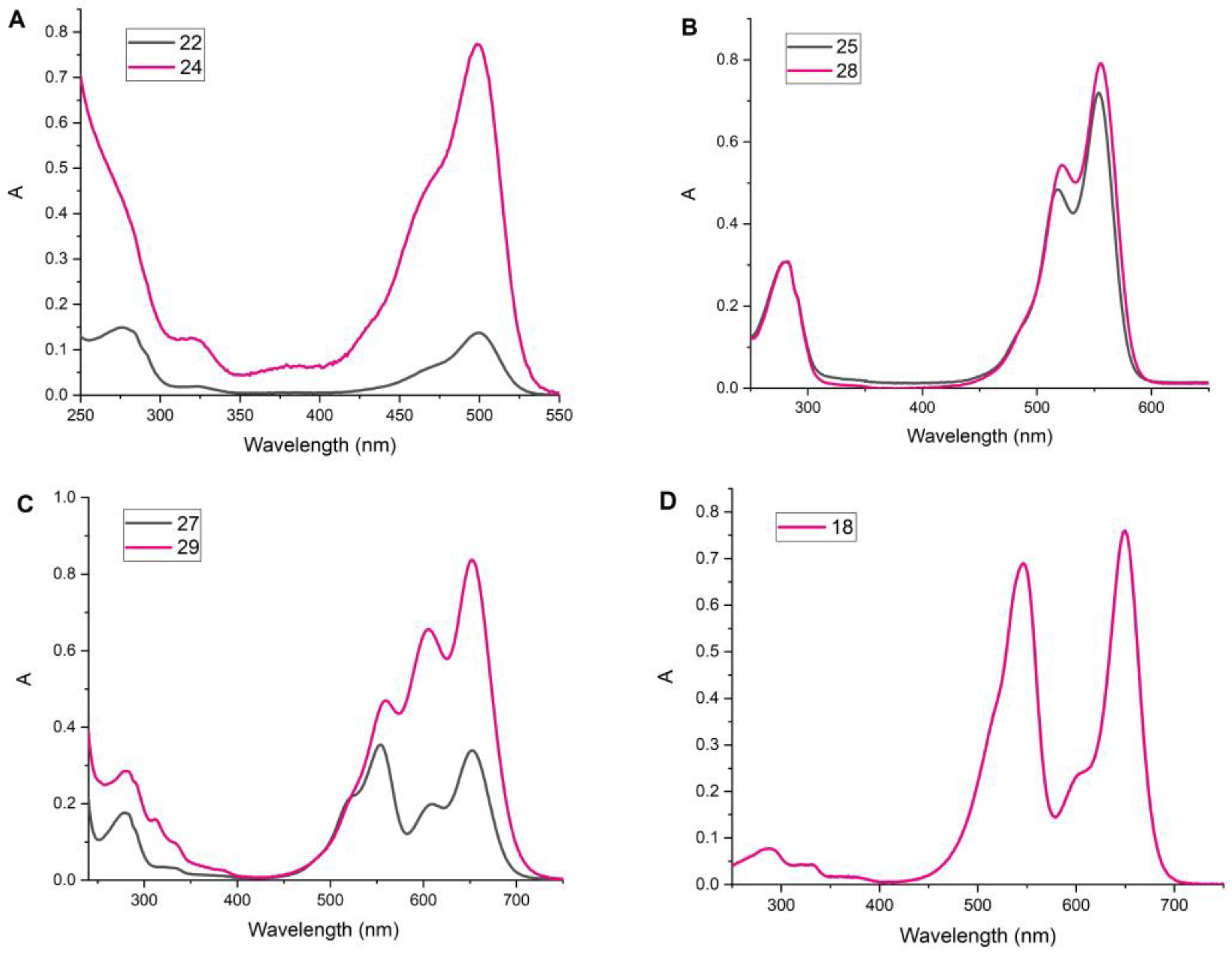
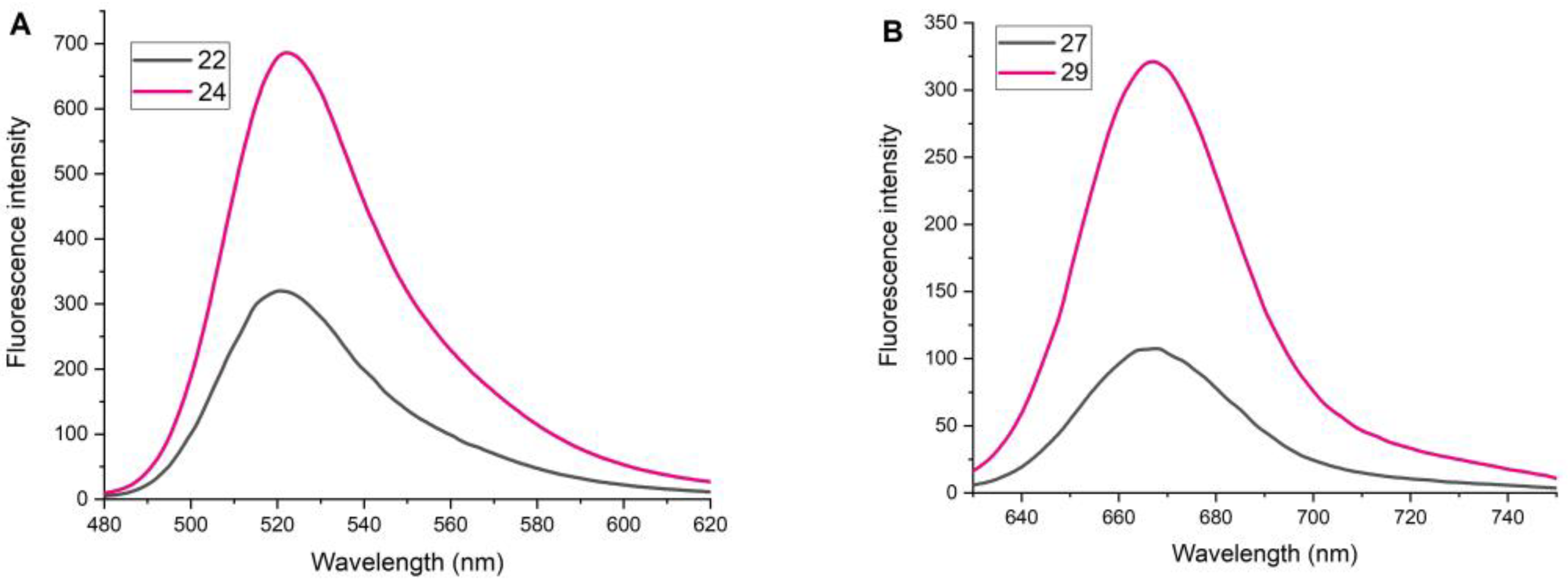
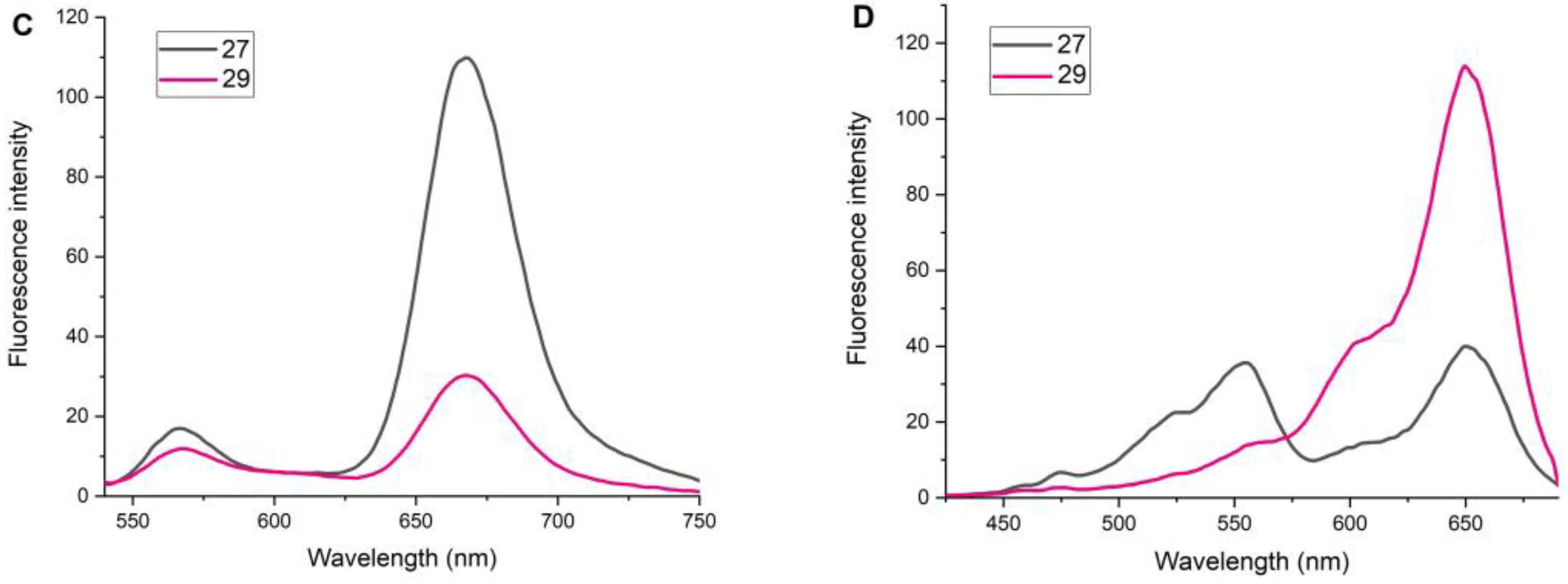
2.4. Study of Fluorescent Properties
2.5. Antibody Conjugate Affinity Determination
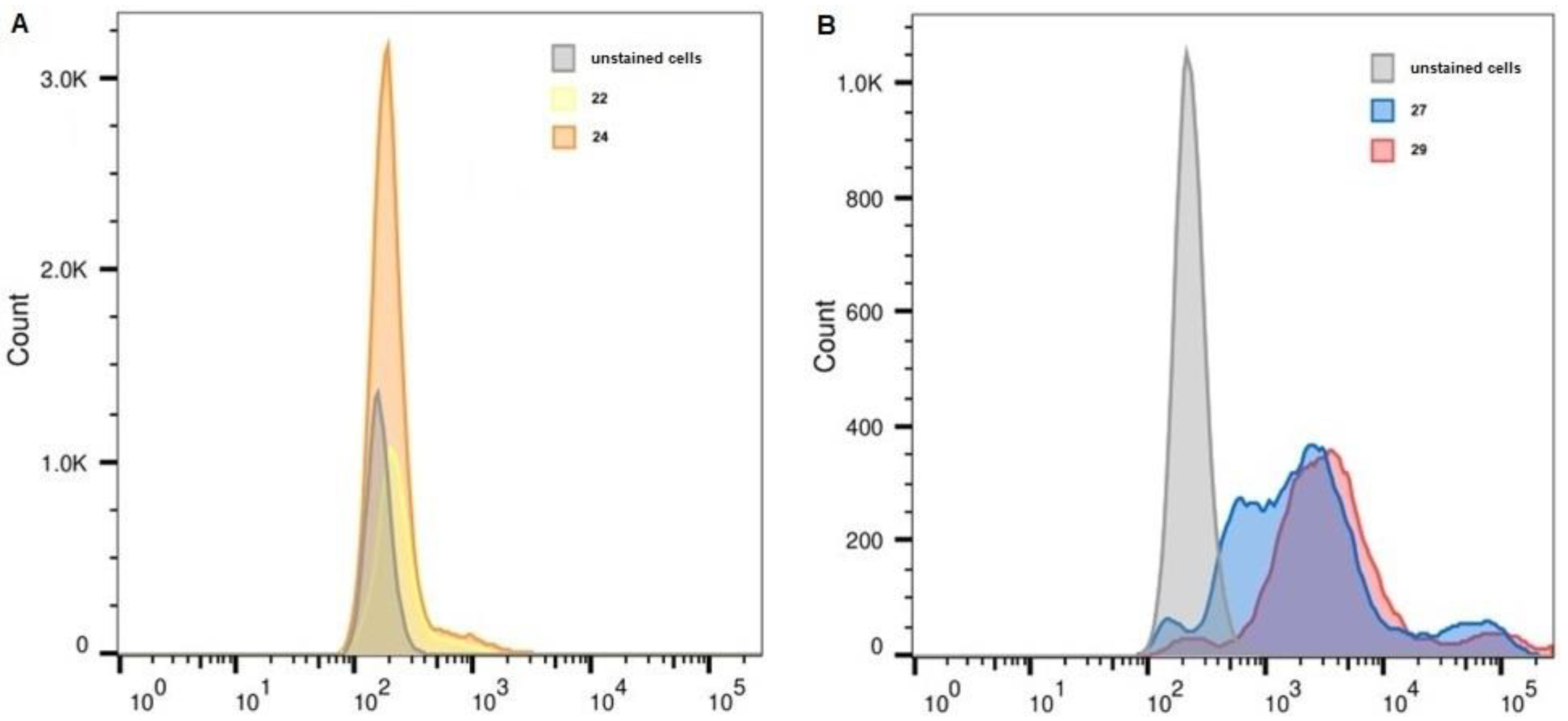
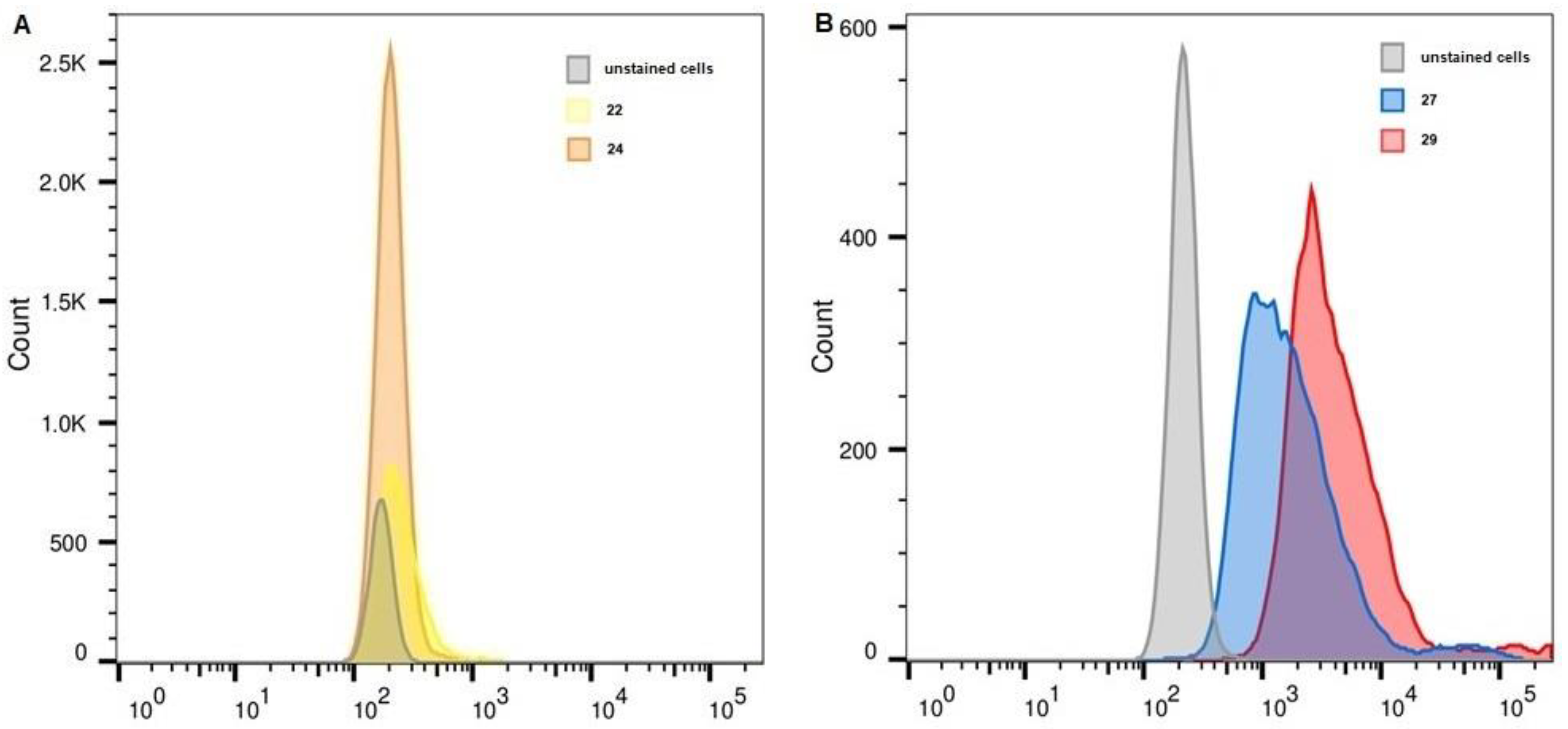
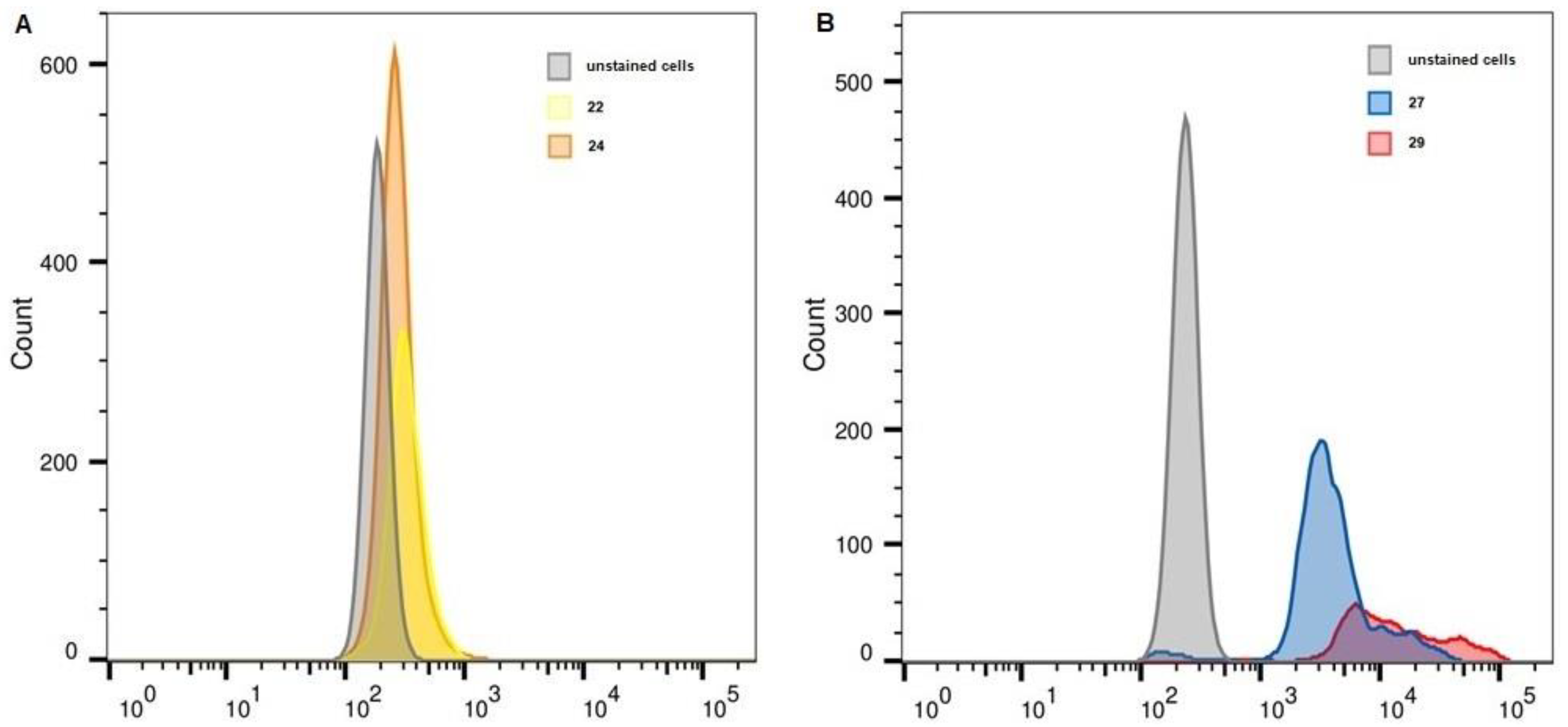
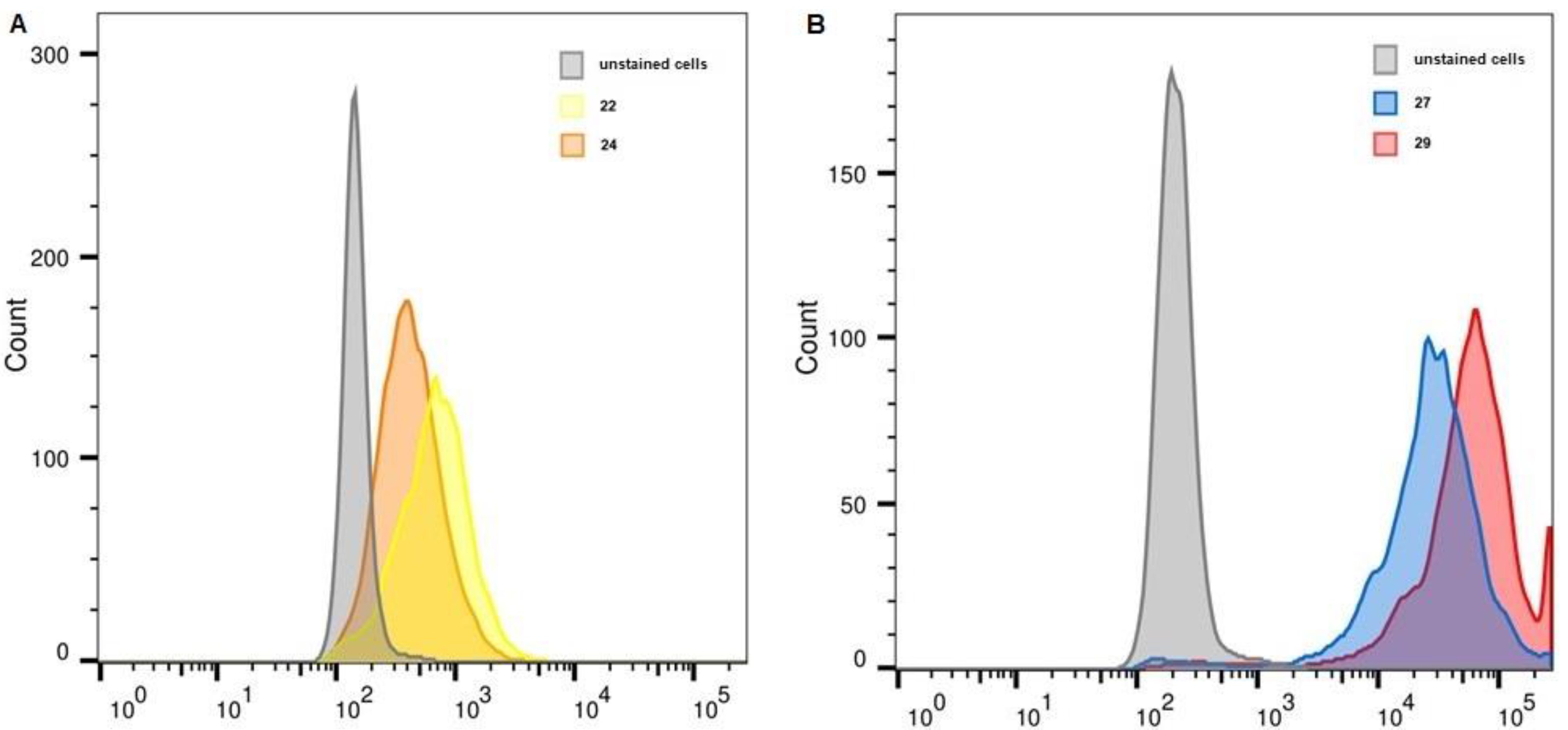
2.6. Flow Cytometry
3. Materials and Methods
3.1. General Methods
3.2. Synthetic Procedures
3.2.1. Mesylate 2
3.2.2. Imidate 3
3.2.3. Phthalimide 5
3.2.4. Amine 6
3.2.5. Imidate 9
3.2.6. Triazide 10
3.2.7. Monoazide 14
3.2.8. Imidate 15
3.2.9. Conjugate 18
3.2.10. General Procedure for Oxyamine Deprotection
3.3. Synthesis of Antibody Conjugates
3.3.1. General Procedure A
Conjugate 20
Conjugate 23
Conjugate 25
Conjugate 28
3.3.2. General Procedure B
Conjugate 22
Conjugate 24
Conjugate 27
Conjugate 29
3.4. Flow Cytometry
3.5. ELISA Assay
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- McKinnon, K.M. Flow cytometry: An overview. Curr. Prot. Immunol. 2018, 120, 5.1.1–5.1.11. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.-I. Immunocytochemistry: Theory and Practice, 1st ed.; CRC Press: Boca Raton, FL, USA, 2020; ISBN 978-1-00-306844-0. [Google Scholar]
- Vira, S.; Mekhedov, E.; Humphrey, G.; Blank, P.S. Fluorescent-labeled antibodies: Balancing functionality and degree of labeling. Anal. Biochem. 2010, 402, 146–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu-Shin, L.P.-Y.; Fung, A.; Malhotra, A.; Ratnaswamy, G. Evidence of disulfide bond scrambling during production of an antibody-drug conjugate. mAbs 2018, 10, 1190–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrens, C.R.; Liu, B. Methods for site-specific drug conjugation to antibodies. mAbs 2014, 6, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q. Site-specific antibody conjugation for ADC and beyond. Biomedicines 2017, 5, 64. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, Z.; Wang, Z.; Luo, F.; Guan, M.; Xu, M.; Li, Y.; Zhang, Y.; Wang, Z.; Wang, W. A simple and efficient method to generate dual site-specific conjugation ADCs with cysteine residue and an unnatural amino acid. Bioconjugate Chem. 2021, 32, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Marshall, M.J.E.; Cragg, M.S.; Crispin, M. Improving antibody-based cancer therapeutics through glycan engineering. BioDrugs 2017, 31, 151–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Shi, W.; Dong, Q.; Li, W.; Zhang, J.; Ren, X.; Tang, C.; Liu, B.; Song, Y.; Wu, Y.; et al. A traceless site-specific conjugation on native antibodies enables efficient one-step payload assembly. Angew. Chem. Int. Ed. 2022, 61, e202204132. [Google Scholar] [CrossRef]
- Shrestha, D.; Bagosi, A.; Szöllősi, J.; Jenei, A. Comparative study of the three different fluorophore antibody conjugation strategies. Anal. Bioanal. Chem. 2012, 404, 1449–1463. [Google Scholar] [CrossRef]
- Adumeau, P.; Sharma, S.K.; Brent, C.; Zeglis, B.M. Site-specifically labeled immunoconjugates for molecular imaging—Part 1: Cysteine residues and glycans. Mol. Imaging Biol. 2016, 18, 1–17. [Google Scholar] [CrossRef]
- Szabó, Á.; Szendi-Szatmári, T.; Ujlaky-Nagy, L.; Rádi, I.; Vereb, G.; Szöllősi, J.; Nagy, P. The effect of fluorophore conjugation on antibody affinity and the photophysical properties of dyes. Biophys. J. 2018, 114, 688–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kölmel, D.K.; Kool, E.T. Oximes and hydrazones in bioconjugation: Mechanism and catalysis. Chem. Rev. 2017, 117, 10358–10376. [Google Scholar] [CrossRef]
- Ponomarenko, A.I.; Brylev, V.A.; Sapozhnikova, K.A.; Ustinov, A.V.; Prokhorenko, I.A.; Zatsepin, T.S.; Korshun, V.A. Tetrahedral DNA conjugates from pentaerythritol-based polyazides. Tetrahedron 2016, 72, 2386–2391. [Google Scholar] [CrossRef]
- Sapozhnikova, K.A.; Misyurin, V.A.; Ryazantsev, D.Y.; Kokin, E.A.; Finashutina, Y.P.; Alexeeva, A.V.; Ivanov, I.A.; Kocharovskaya, M.V.; Tikhonova, N.A.; Popova, G.P.; et al. Sensitive immunofluorescent detection of the PRAME antigen using a practical antibody conjugation approach. Int. J. Mol. Sci. 2021, 22, 12845. [Google Scholar] [CrossRef] [PubMed]
- Fleminger, G.; Hadas, E.; Wolf, T.; Solomon, B. Oriented immobilization of periodate-oxidized monoclonal antibodies on amino and hydrazide derivatives of eupergit C. Appl. Biochem. Biotechnol. 1990, 23, 123–137. [Google Scholar] [CrossRef]
- Misyurin, V.A.; Finashutina, Y.P.; Turba, A.A.; Larina, M.V.; Solopova, O.N.; Lyzhko, N.A.; Kesaeva, L.A.; Kasatkina, N.N.; Aliev, T.K.; Misyurin, A.V.; et al. Epitope analysis of murine and chimeric monoclonal antibodies recognizing the cancer testis antigen PRAME. Dokl. Biochem. Biophys. 2020, 492, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Sapozhnikova, K.A.; Misyurin, A.V.; Pestov, N.B.; Meleshkina, E.G.; Oreshkov, S.D.; Ganzhula, E.P.; Mikhailova, A.S.; Korshun, V.A.; Misyurin, V.A.; Brylev, V.A. Detection of the PRAME protein on the surface of melanoma cells using a fluorescently labeled monoclonal antibody. Russ. J. Bioorg. Chem. 2021, 47, 1077–1085. [Google Scholar] [CrossRef]
- Field, M.G.; Durante, M.A.; Decatur, C.L.; Tarlan, B.; Oelschlager, K.M.; Stone, J.F.; Kuznetsov, J.; Bowcock, A.M.; Kurtenbach, S.; Harbour, J.W. Epigenetic reprogramming and aberrant expression of PRAME are associated with increased metastatic risk in class 1 and class 2 uveal melanomas. Oncotarget 2016, 7, 59209–59219. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-L.; Gokgoz, N.; Samman, B.; Andrulis, I.L.; Wunder, J.S.; Demicco, E.G. RNA expression profiling reveals PRAME, a potential immunotherapy target, is frequently expressed in solitary fibrous tumors. Mod. Pathol. 2021, 34, 951–960. [Google Scholar] [CrossRef]
- Hovander, D.; Allen, J.; Oda, D.; Moshiri, A.S. PRAME immunohistochemistry is useful in the diagnosis of oral malignant melanoma. Oral Oncol. 2022, 124, 105500. [Google Scholar] [CrossRef]
- Kirkey, D.C.; Loeb, A.; Castro, S.; McKay, C.N.; Perkins, L.; Pardo, L.; Leonti, A.R.; Tang, T.; Loken, M.R.; Eidenschink Brodersen, L.; et al. Therapeutic targeting PRAME with mTCRCAR T cells in acute myeloid leukemia. Blood Adv. 2022, bloodadvances.2022008304. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowski, M.; Chłopek, M.; Kruczak, A.; Ryś, J.; Lasota, J.; Miettinen, M. PRAME expression in cancer. A systematic immunohistochemical study of >5800 epithelial and nonepithelial tumors. Am. J. Surg. Pathol. 2022, 46, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- VanBrunt, M.P.; Shanebeck, K.; Caldwell, Z.; Johnson, J.; Thompson, P.; Martin, T.; Dong, H.; Li, G.; Xu, H.; D’Hooge, F.; et al. Genetically encoded azide containing amino acid in mammalian cells enables site-specific antibody–drug conjugates using click cycloaddition chemistry. Bioconjugate Chem. 2015, 26, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Finashutina, Y.P.; Misyurin, A.V.; Akhlynina, T.V.; Lyzhko, N.A.; Krutov, A.A.; Aksenova, E.V.; Misyurin, V.A.; Baryshnikov, A.Y. Production of purified human recombinant antigen PRAME and specific monoclonal antibodies. Russ. J. Biother. 2015, 14, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Schwabacher, A.W.; Lane, J.W.; Schiesher, M.W.; Leigh, K.M.; Johnson, C.W. Desymmetrization reactions: Efficient preparation of unsymmetrically substituted linker molecules. J. Org. Chem. 1998, 63, 1727–1729. [Google Scholar] [CrossRef]

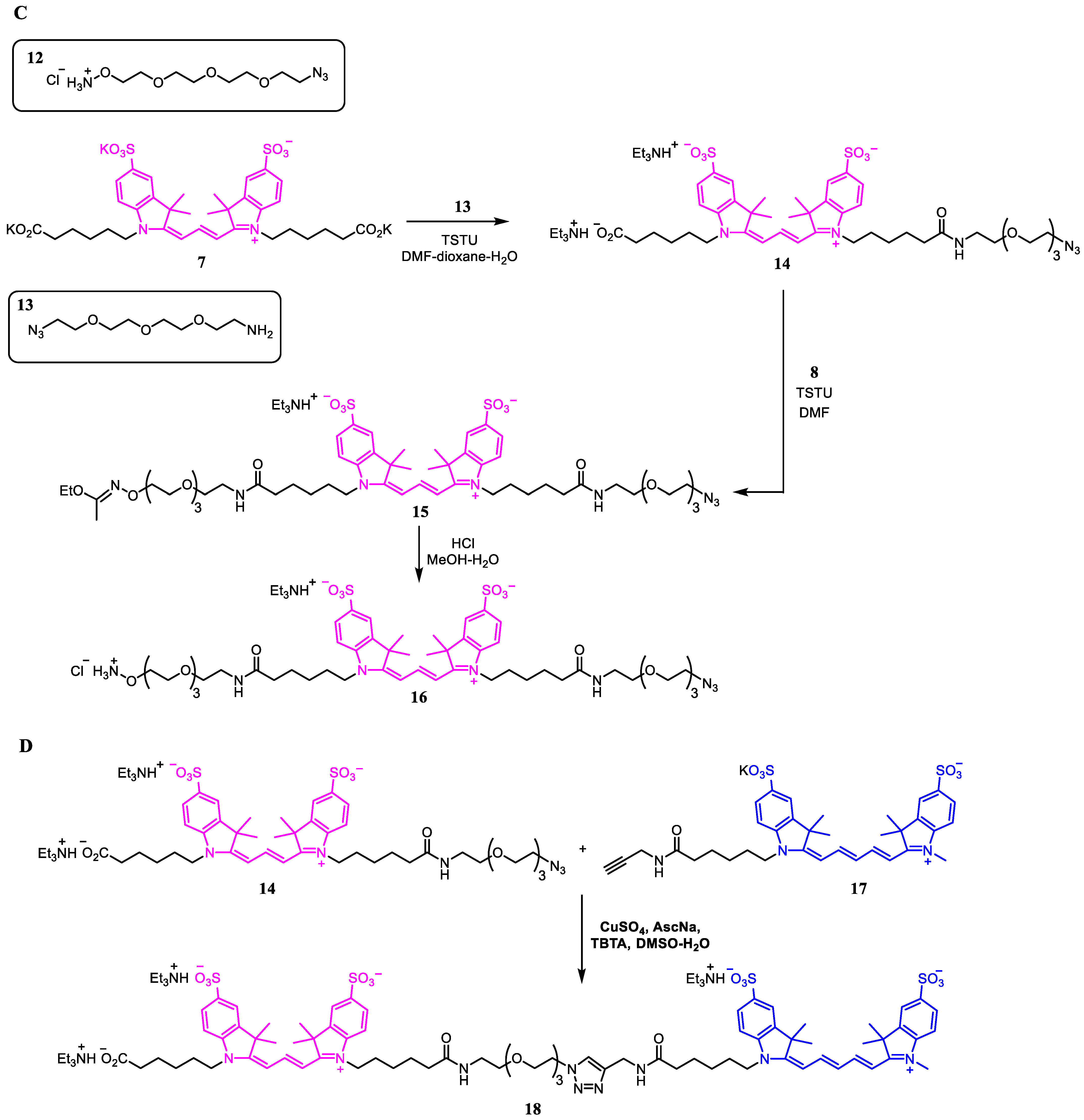

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ε280, M−1cm−1 | εmax long wave, M−1cm−1 | |
|---|---|---|
| 6H8 | 2.10 × 105 | - |
| FAM-DBCO | 2.17 × 104 | 7.40 × 104 |
| sCy3 | 9.72 × 103 | 1.62 × 105 |
| sCy5-DBCO | 1.08 × 104 | 2.71 × 105 |
| Unstained, FITC | 22 | 24 | Unstained, APC | 27 | 29 | |
|---|---|---|---|---|---|---|
| K-562 | 161 | 219 | 192 | 226 | 1956 | 3223 |
| THP-1 | 170 | 234 | 203 | 216 | 1447 | 3384 |
| MelP | 186 | 316 | 267 | 234 | 3521 | 10,477 |
| WI-38 PRAME+ | 148 | 676 | 404 | 208 | 27,267 | 56,987 |
| Dye | Extinction | Emission | Laser | Detector |
|---|---|---|---|---|
| FAM | 488 | 515 | 488 | 530/30 FITC |
| Cy5 | 643 | 667 | 640 | 675/30 APC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sapozhnikova, K.A.; Gulyak, E.L.; Misyurin, V.A.; Simonova, M.A.; Ryabukhina, E.V.; Alexeeva, A.V.; Tikhonova, N.A.; Lyzhko, N.A.; Popova, G.P.; Misyurin, A.V.; et al. Branched Linkers for Site-Specific Fluorescent Labeling of Antibodies. Molecules 2023, 28, 425. https://doi.org/10.3390/molecules28010425
Sapozhnikova KA, Gulyak EL, Misyurin VA, Simonova MA, Ryabukhina EV, Alexeeva AV, Tikhonova NA, Lyzhko NA, Popova GP, Misyurin AV, et al. Branched Linkers for Site-Specific Fluorescent Labeling of Antibodies. Molecules. 2023; 28(1):425. https://doi.org/10.3390/molecules28010425
Chicago/Turabian StyleSapozhnikova, Ksenia A., Evgeny L. Gulyak, Vsevolod A. Misyurin, Maria A. Simonova, Ekaterina V. Ryabukhina, Anastasiya V. Alexeeva, Nataliya A. Tikhonova, Natalia A. Lyzhko, Galina P. Popova, Andrey V. Misyurin, and et al. 2023. "Branched Linkers for Site-Specific Fluorescent Labeling of Antibodies" Molecules 28, no. 1: 425. https://doi.org/10.3390/molecules28010425
APA StyleSapozhnikova, K. A., Gulyak, E. L., Misyurin, V. A., Simonova, M. A., Ryabukhina, E. V., Alexeeva, A. V., Tikhonova, N. A., Lyzhko, N. A., Popova, G. P., Misyurin, A. V., Ustinov, A. V., Korshun, V. A., Alferova, V. A., Ryazantsev, D. Y., & Brylev, V. A. (2023). Branched Linkers for Site-Specific Fluorescent Labeling of Antibodies. Molecules, 28(1), 425. https://doi.org/10.3390/molecules28010425