
A Novel Multi-Target Mu/Delta Opioid Receptor Agonist, HAGD, Produced Potent Peripheral Antinociception with Limited Side Effects in Mice and Minimal Impact on Human Sperm Motility In Vitro

Abstract
:1. Introduction
2. Results
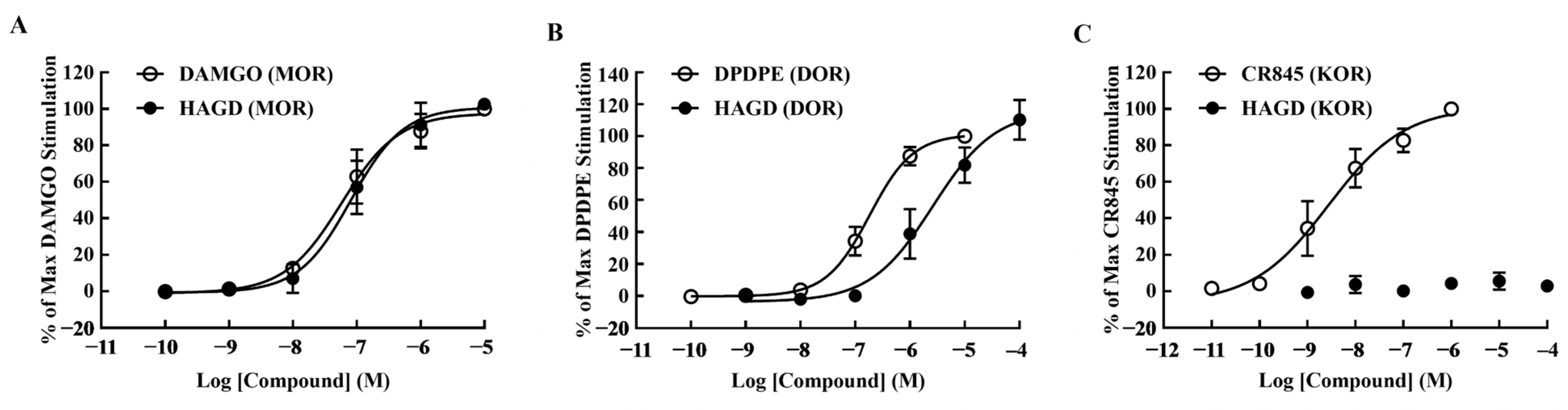
2.1. Calcium Mobilization Assays
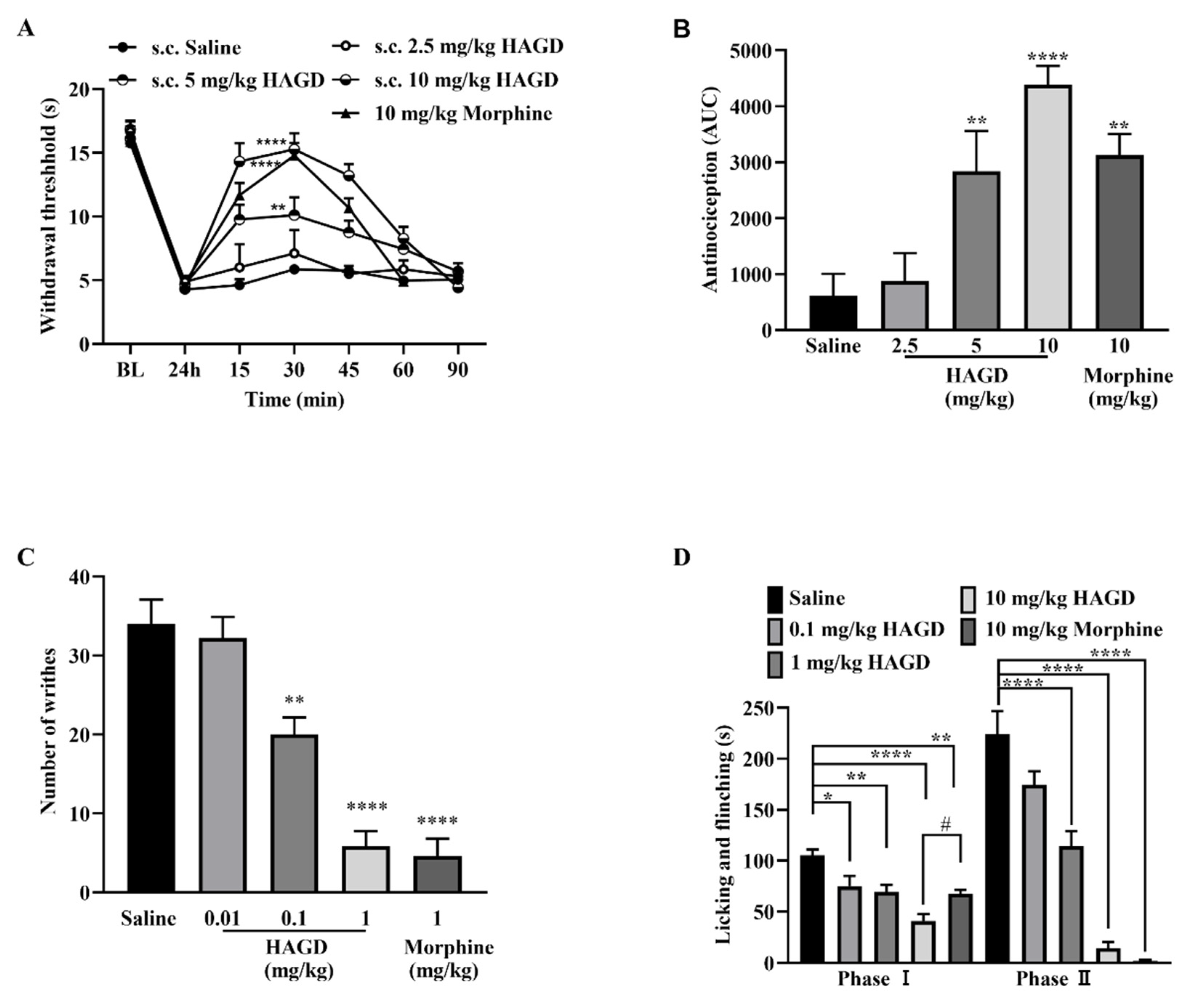
2.2. Antinociceptive Effects of Subcutaneous HAGD in the Tail-Flick Test
2.3. Antinociceptive Effects of Subcutaneous HAGD in Carrageenan-Induced Inflammatory Pain
2.4. Antinociceptive Effects of Subcutaneous HAGD in Acetic Acid Writhing Test
2.5. Antinociceptive Effects of Subcutaneous HAGD in the Formalin Test
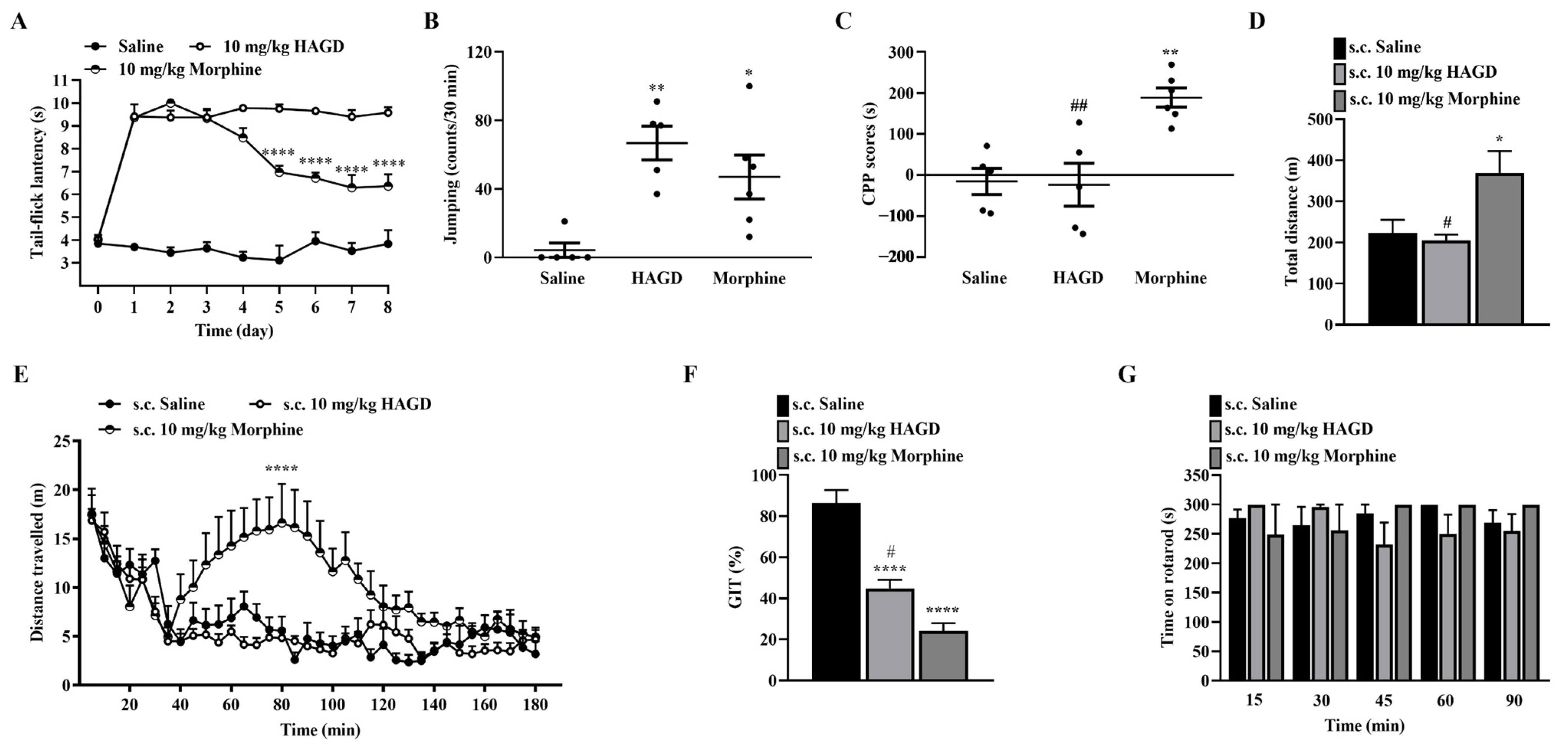
2.6. Side Effects Evaluation of Subcutaneous HAGD
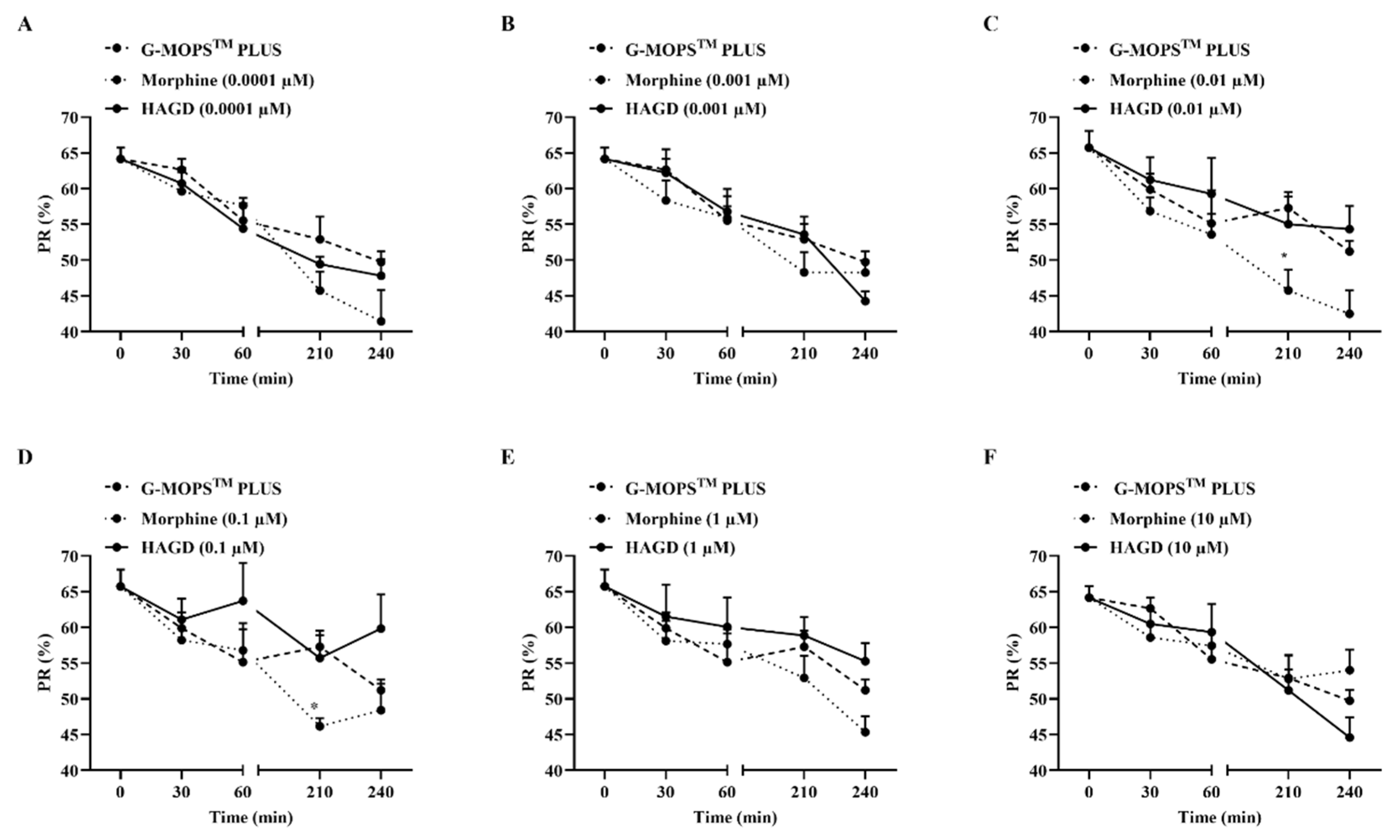
2.7. Effects on Sperm Motility of HAGD
3. Discussion
4. Materials and Methods
4.1. Drugs and Chemicals
4.2. Peptide Synthesis
4.3. Administration
4.4. Animals
4.5. Calcium Mobilization Assays
4.6. The Tail-Flick Test
4.7. Carrageenan-Induced Inflammatory Pain
4.8. Acetic Acid-Induced Writhing Test
4.9. Formalin Test
4.10. Antinociceptive Tolerance
4.11. Naloxone-Induced Withdrawal Response
4.12. Conditioned Place Preference
4.13. Acute Hyperlocomotion Test
4.14. Gastrointestinal Transit Test
4.15. Rotarod Test
4.16. Sperm Motility
4.16.1. Sperm Preparation
4.16.2. Incubation Medium
4.16.3. Motility Analysis
4.17. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Melnikova, I. Pain market. Nat. Rev. Drug Discov. 2010, 9, 589–590. [Google Scholar] [CrossRef] [PubMed]
- Hider-Mlynarz, K.; Cavalie, P.; Maison, P. Trends in analgesic consumption in France over the last 10 years and comparison of patterns across Europe. Br. J. Clin. Pharmacol. 2018, 84, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monje, B.; Giménez-Manzorro, Á.; Ortega-Navarro, C.; Herranz-Alonso, A.; Sanjurjo-Sáez, M. Trends in hospital consumption of analgesics after the implementation of a pain performance improvement plan. Braz. J. Anesthesiol. 2019, 69, 259–265. [Google Scholar] [CrossRef]
- Lutz, P.E.; Kieffer, B.L. Opioid receptors: Distinct roles in mood disorders. Trends Neurosci. 2013, 36, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Dinges, H.C.; Otto, S.; Stay, D.K.; Baumlein, S.; Waldmann, S.; Kranke, P.; Wulf, H.F.; Eberhart, L.H. Side Effect Rates of Opioids in Equianalgesic Doses via Intravenous Patient-Controlled Analgesia: A Systematic Review and Network Meta-analysis. Anesth. Analg. 2019, 129, 1153–1162. [Google Scholar] [CrossRef]
- Subiran, N.; Candenas, L.; Pinto, F.M.; Cejudo-Roman, A.; Agirregoitia, E.; Irazusta, J. Autocrine regulation of human sperm motility by the met-enkephalin opioid peptide. Fertil. Steril. 2012, 98, 617–625 e613. [Google Scholar] [CrossRef]
- Agirregoitia, E.; Valdivia, A.; Carracedo, A.; Casis, L.; Gil, J.; Subiran, N.; Ochoa, C.; Irazusta, J. Expression and localization of delta-, kappa-, and mu-opioid receptors in human spermatozoa and implications for sperm motility. J. Clin. Endocrinol. Metab. 2006, 91, 4969–4975. [Google Scholar] [CrossRef]
- Agirregoitia, E.; Subiran, N.; Valdivia, A.; Gil, J.; Zubero, J.; Irazusta, J. Regulation of human sperm motility by opioid receptors. Andrologia 2012, 44 (Suppl. 1), 578–585. [Google Scholar] [CrossRef]
- Zadina, J.E.; Hackler, L.; Ge, L.-J.; Kastin, A.J.J.N. A potent and selective endogenous agonist for the µ-opiate receptor. Nature 1997, 386, 499–502. [Google Scholar] [CrossRef]
- Czapla, M.A.; Gozal, D.; Alea, O.A.; Beckerman, R.C.; Zadina, J.E.J.A.j.o.r.; Medicine, C.C. Differential cardiorespiratory effects of endomorphin 1, endomorphin 2, DAMGO, and morphine. Am. J. Respir. Crit. Care Med. 2000, 162, 994–999. [Google Scholar] [CrossRef]
- Tömböly, C.; Péter, A.; Tóth, G.J.P. In vitro quantitative study of the degradation of endomorphins. Peptides 2002, 23, 1573–1580. [Google Scholar] [CrossRef]
- Machelska, H.; Celik, M.Ö. Advances in Achieving Opioid Analgesia Without Side Effects. Front Pharmacol. 2018, 9, 1388. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.K.; Smith, C.M.; Rahmatullah, M.; Nissapatorn, V.; Wilairatana, P.; Spetea, M.; Gueven, N.; Dietis, N. Opioid Analgesia and Opioid-Induced Adverse Effects: A Review. Pharmaceuticals 2021, 14, 1091. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, X.; Gao, X.; Gan, Y.; Liu, Y.; Zhao, X.; Hu, J.; Ma, X.; Wu, Y.; Ma, P.; et al. Original endomorphin-1 analogues exhibit good analgesic effects with minimal implications for human sperm motility. Bioorg. Med. Chem. Lett. 2017, 27, 2119–2123. [Google Scholar] [CrossRef]
- Mika, J.; Wawrzczak-Bargiela, A.; Osikowicz, M.; Makuch, W.; Przewlocka, B.J.B. Attenuation of morphine tolerance by minocycline and pentoxifylline in naive and neuropathic mice. Brain Behav. Immun. 2009, 23, 75–84. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, M.; Shi, X.; Zhang, R.; Chen, D.; Chen, Y.; Wang, Z.; Qiu, Y.; Zhang, T.; Xu, K.; et al. The multifunctional peptide DN-9 produced peripherally acting antinociception in inflammatory and neuropathic pain via mu- and kappa-opioid receptors. Br. J. Pharmacol. 2020, 177, 93–109. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, B.; Li, N.; Liu, H.; Shi, X.; Zhang, Q.; Shi, Y.; Xu, K.; Xiao, J.; Chen, D.; et al. Synthesis and Biological Characterization of Cyclic Disulfide-Containing Peptide Analogs of the Multifunctional Opioid/Neuropeptide FF Receptor Agonists That Produce Long-Lasting and Nontolerant Antinociception. J. Med. Chem. 2020, 63, 15709–15725. [Google Scholar] [CrossRef]
- Przewłocki, R.; Łabuz, D.; Mika, J.; Przewłocka, B.; Tomboly, C.; Toth, G. Pain inhibition by endomorphins. Ann. New York Acad. Sci. 1999, 897, 154–164. [Google Scholar] [CrossRef]
- Vicario, N.; Pasquinucci, L.; Spitale, F.M.; Chiechio, S.; Turnaturi, R.; Caraci, F.; Tibullo, D.; Avola, R.; Gulino, R.; Parenti, R.; et al. Simultaneous Activation of Mu and Delta Opioid Receptors Reduces Allodynia and Astrocytic Connexin 43 in an Animal Model of Neuropathic Pain. Mol. Neurobiol. 2019, 56, 7338–7354. [Google Scholar] [CrossRef]
- Pasquinucci, L.; Turnaturi, R.; Montenegro, L.; Caraci, F.; Chiechio, S.; Parenti, C. Simultaneous targeting of MOR/DOR: A useful strategy for inflammatory pain modulation. Eur. J. Pharmacol. 2019, 847, 97–102. [Google Scholar] [CrossRef]
- Pasquinucci, L.; Turnaturi, R.; Calo, G.; Pappalardo, F.; Ferrari, F.; Russo, G.; Arena, E.; Montenegro, L.; Chiechio, S.; Prezzavento, O.; et al. (2S)-N-2-methoxy-2-phenylethyl-6,7-benzomorphan compound (2S-LP2): Discovery of a biased mu/delta opioid receptor agonist. Eur. J. Med. Chem. 2019, 168, 189–198. [Google Scholar] [CrossRef]
- Gendron, L.; Mittal, N.; Beaudry, H.; Walwyn, W. Recent advances on the δ opioid receptor: From trafficking to function. Br. J. Pharmacol. 2015, 172, 403–419. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Huang, M.; Wang, W.; Zhang, Y.; Ma, X.; Luo, L.; Xu, X.; Xu, L.; Shi, H.; Xu, Y.; et al. Microglial TLR4-induced TAK1 phosphorylation and NLRP3 activation mediates neuroinflammation and contributes to chronic morphine-induced antinociceptive tolerance. Pharmacol. Res. 2021, 165, 105482. [Google Scholar] [CrossRef]
- Klenowski, P.; Morgan, M.; Bartlett, S.E. The role of δ-opioid receptors in learning and memory underlying the development of addiction. Br. J. Pharmacol. 2015, 172, 297–310. [Google Scholar] [CrossRef] [Green Version]
- Laurent, V.; Morse, A.K.; Balleine, B.W. The role of opioid processes in reward and decision-making. Br. J. Pharmacol. 2015, 172, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Funada, M.; Suzuki, T.; Narita, M.; Misawa, M.; Nagase, H. Modification of morphine-induced locomotor activity by pertussis toxin: Biochemical and behavioral studies in mice. Brain Res. 1993, 619, 163–172. [Google Scholar] [CrossRef]
- Siegfried, B.; Filibeck, U.; Gozzo, S.; Castellano, C. Lack of morphine-induced hyperactivity in C57BL/6 mice following striatal kainic acid lesions. Behav. Brain Res. 1982, 4, 389–399. [Google Scholar] [CrossRef]
- Günther, T.; Dasgupta, P.; Mann, A.; Miess, E.; Kliewer, A.; Fritzwanker, S.; Steinborn, R.; Schulz, S. Targeting multiple opioid receptors-improved analgesics with reduced side effects? Br. J. Pharmacol. 2018, 175, 2857–2868. [Google Scholar] [CrossRef] [Green Version]
- Varga, B.R.; Streicher, J.M.; Majumdar, S. Strategies towards safer opioid analgesics—A review of old and upcoming targets. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Ragni, G.; de LAURETIS, L.; BESTETTI, O.; SGHEDONI, D.; ARO, V.G.A. Gonadal function in male heroin and methadone addicts. Int. J. Androl. 1988, 11, 93–100. [Google Scholar] [CrossRef]
- Wu, Y.; Zhao, X.; Gan, Y.; Zhang, X.; Wei, H.; Wang, L.; Liang, X.; Gao, X.; Liu, Y.; Hu, J.; et al. Original endomorphin-1 analogues exhibit good analgesic effects. Bioorg. Med. Chem. Lett. 2017, 27, 1557–1560. [Google Scholar] [CrossRef]
- Wang, C.-l.; Guo, C.; Wang, Y.-q.; Zhou, Y.; Li, Q.; Ni, J.-m.; Wang, R. Synthesis and antinociceptive effects of endomorphin-1 analogs with C-terminal linked by oligoarginine. Peptides 2011, 32, 293–299. [Google Scholar] [CrossRef]
- Chang, M.; Peng, Y.-L.; Dong, S.-L.; Han, R.-W.; Li, W.; Yang, D.-J.; Chen, Q.; Wang, R. Structure-activity studies on different modifications of nociceptin/orphanin FQ: Identification of highly potent agonists and antagonists of its receptor. Regul. Pept. 2005, 130, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Guo, Y.; Zhang, M.; Zhang, R.; Chen, D.; Zhang, Q.; Xiao, J.; Xu, K.; Li, N.; Qiu, Y.; et al. Central and peripheral modulation of gastrointestinal transit in mice by DN-9, a multifunctional opioid/NPFF receptor agonist. Neurogastroenterol. Motil. 2020, 32, e13848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toll, L.; Berzetei-Gurske, I.P.; Polgar, W.E.; Brandt, S.R.; Adapa, I.D.; Rodriguez, L.; Schwartz, R.W.; Haggart, D.; O’Brien, A.; White, A.; et al. Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Res. Monogr. 1998, 178, 440–466. [Google Scholar]
- Lewanowitsch, T.; Irvine, R.J. Naloxone and its quaternary derivative, naloxone methiodide, have differing affinities for mu, delta, and kappa opioid receptors in mouse brain homogenates. Brain Res. 2003, 964, 302–305. [Google Scholar] [CrossRef]
- Raynor, K.; Kong, H.; Chen, Y.; Yasuda, K.; Yu, L.; Bell, G.I.; Reisine, T. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol. Pharmacol. 1994, 45, 330–334. [Google Scholar]
- Liu-Chen, L.Y.; Li, S.X.; Tallarida, R.J. Studies on kinetics of [3H]beta-funaltrexamine binding to mu opioid receptor. Mol. Pharmacol. 1990, 37, 243–250. [Google Scholar]
- Zhu, J.; Chen, C.; Xue, J.C.; Kunapuli, S.; DeRiel, J.K.; Liu-Chen, L.Y. Cloning of a human kappa opioid receptor from the brain. Life Sci. 1995, 56, PL201–PL207. [Google Scholar] [CrossRef]
- Miao, X.; Zhou, T.; Zhang, J.; Xu, J.; Guo, X.; Hu, H.; Zhang, X.; Hu, M.; Li, J.; Yang, W.; et al. Enhanced cell selectivity of hybrid peptides with potential antimicrobial activity and immunomodulatory effect. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129532. [Google Scholar] [CrossRef]
- Rizzi, A.; Malfacini, D.; Cerlesi, M.C.; Ruzza, C.; Marzola, E.; Bird, M.F.; Rowbotham, D.J.; Salvadori, S.; Guerrini, R.; Lambert, D.G.; et al. In vitro and in vivo pharmacological characterization of nociceptin/orphanin FQ tetrabranched derivatives. Br. J. Pharmacol. 2014, 171, 4138–4153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Xu, B.; Jiang, C.; Zhang, T.; Zhang, M.; Li, N.; Zhang, Q.; Xu, K.; Chen, D.; Xiao, J.; et al. Spinal DN-9, a Peptidic Multifunctional Opioid/Neuropeptide FF Agonist Produced Potent Nontolerance Forming Analgesia with Limited Side Effects. J. Pain 2020, 21, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xu, B.; Zhang, Q.; Chen, D.; Zhang, M.; Zhao, G.; Xu, K.; Xiao, J.; Zhu, H.; Niu, J.; et al. Spinal administration of the multi-functional opioid/neuropeptide FF agonist BN-9 produced potent antinociception without development of tolerance and opioid-induced hyperalgesia. Eur. J. Pharmacol. 2020, 880, 173169. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Han, Z.L.; Xu, B.; Zhang, M.N.; Zhang, T.; Shi, X.R.; Zhao, W.D.; Guo, Y.Y.; Zhang, Q.Q.; Fang, Q. Systemic administration of the bifunctional opioid/neuropeptide FF receptors agonist BN-9 produced peripheral antinociception in preclinical mouse models of pain. Eur. J. Pharmacol. 2018, 837, 53–63. [Google Scholar] [CrossRef]
- Lin, Y.T.; Kao, S.C.; Day, Y.J.; Chang, C.C.; Chen, J.C. Altered nociception and morphine tolerance in neuropeptide FF receptor type 2 over-expressing mice. Eur. J. Pain 2016, 20, 895–906. [Google Scholar] [CrossRef]
- Chen, C.; Xu, B.; Shi, X.; Zhang, M.; Zhang, Q.; Zhang, T.; Zhao, W.; Zhang, R.; Wang, Z.; Li, N.; et al. GpTx-1 and [Ala(5), Phe(6), Leu(26), Arg(28) ]GpTx-1, two peptide NaV 1.7 inhibitors: Analgesic and tolerance properties at the spinal level. Br. J. Pharmacol. 2018, 175, 3911–3927. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Xiao, J.; Xu, K.; Zhang, Q.; Chen, D.; Zhang, R.; Zhang, M.; Zhu, H.; Niu, J.; Zheng, T.; et al. VF-13, a chimeric peptide of VD-hemopressin(alpha) and neuropeptide VF, produces potent antinociception with reduced cannabinoid-related side effects. Neuropharmacology 2020, 175, 108178. [Google Scholar] [CrossRef]
- World Health Organization. WHO Laboratory Manual for the Examination and Processing of Human Semen, 5th ed.; Geneva WHO Press: Switzerland, Geneva, 2010. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | pEC50 (95% CI) | Emax ± S.E.M. | EC50 (nM) | R2 |
|---|---|---|---|---|
| DAMGO (MOR) | 7.24 (7.02–7.44) | 100.00 ± 0.00 | 58.01 | 0.98 |
| HAGD (MOR) | 7.09 (6.85–7.29) | 102.56 ± 1.58 | 81.69 | 0.97 |
| DPDPE (DOR) | 6.74 (6.61–6.85) | 100.00 ± 0.00 | 183.1 | 0.99 |
| HAGD (DOR) | 5.58 (5.06–5.9) | 110.36 ± 7.21 | 2628 | 0.97 |
| CR845 (KOR) | 8.56 (8.18–8.89) | 100.00 ± 0.00 | 2.79 | 0.95 |
| HAGD (KOR) | (-) | 2.95 ± 0.92 | (-) | (-) |
| Compound | Molecular Weight | Ki(MOR) (nM) | Ki(DOR) (nM) | Ki(KOR) (nM) | Agonist | Antagonist | Dose a (mg/kg, μmol/kg) |
|---|---|---|---|---|---|---|---|
| morphine [35] | 321.85 | 1.0 | 125.9 | 50.1 | MOR DOR KOR | 10, 31.07 | |
| HAGD [14] | 455 | 2.1 | 3800 | MOR DOR | 10, 21.98 | ||
| naloxone [36] | 399.87 | 1.9 | 3.1 | 32.6 | MOR DOR KOR | 1, 2.5 | |
| naloxone methiodide [36] | 469.31 | 28.9 | 1010 | 203.5 | MOR DOR KOR | 1, 2.13 | |
| β-funaltrexamine [37,38] | 581.01 | 0.3 | MOR b | 1, 1.72 | |||
| naltrindole [37] | 482.48 | 0.02 | DOR | 1, 2.07 | |||
| nor-binaltorphimine [39] | 842.81 | 0.00025 | KOR | 1, 1.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Yue, F.; Zhang, W.; Xu, B.; Wang, Y.; Zhang, X. A Novel Multi-Target Mu/Delta Opioid Receptor Agonist, HAGD, Produced Potent Peripheral Antinociception with Limited Side Effects in Mice and Minimal Impact on Human Sperm Motility In Vitro. Molecules 2023, 28, 427. https://doi.org/10.3390/molecules28010427
Li F, Yue F, Zhang W, Xu B, Wang Y, Zhang X. A Novel Multi-Target Mu/Delta Opioid Receptor Agonist, HAGD, Produced Potent Peripheral Antinociception with Limited Side Effects in Mice and Minimal Impact on Human Sperm Motility In Vitro. Molecules. 2023; 28(1):427. https://doi.org/10.3390/molecules28010427
Chicago/Turabian StyleLi, Fangfang, Feng Yue, Wei Zhang, Biao Xu, Yiqing Wang, and Xuehong Zhang. 2023. "A Novel Multi-Target Mu/Delta Opioid Receptor Agonist, HAGD, Produced Potent Peripheral Antinociception with Limited Side Effects in Mice and Minimal Impact on Human Sperm Motility In Vitro" Molecules 28, no. 1: 427. https://doi.org/10.3390/molecules28010427
APA StyleLi, F., Yue, F., Zhang, W., Xu, B., Wang, Y., & Zhang, X. (2023). A Novel Multi-Target Mu/Delta Opioid Receptor Agonist, HAGD, Produced Potent Peripheral Antinociception with Limited Side Effects in Mice and Minimal Impact on Human Sperm Motility In Vitro. Molecules, 28(1), 427. https://doi.org/10.3390/molecules28010427