
Ru-Controlled Thymine Tautomerization Frozen by a k1(O)-, k2(N,O)-Metallacycle: An Experimental and Theoretical Approach

 ,
,  , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
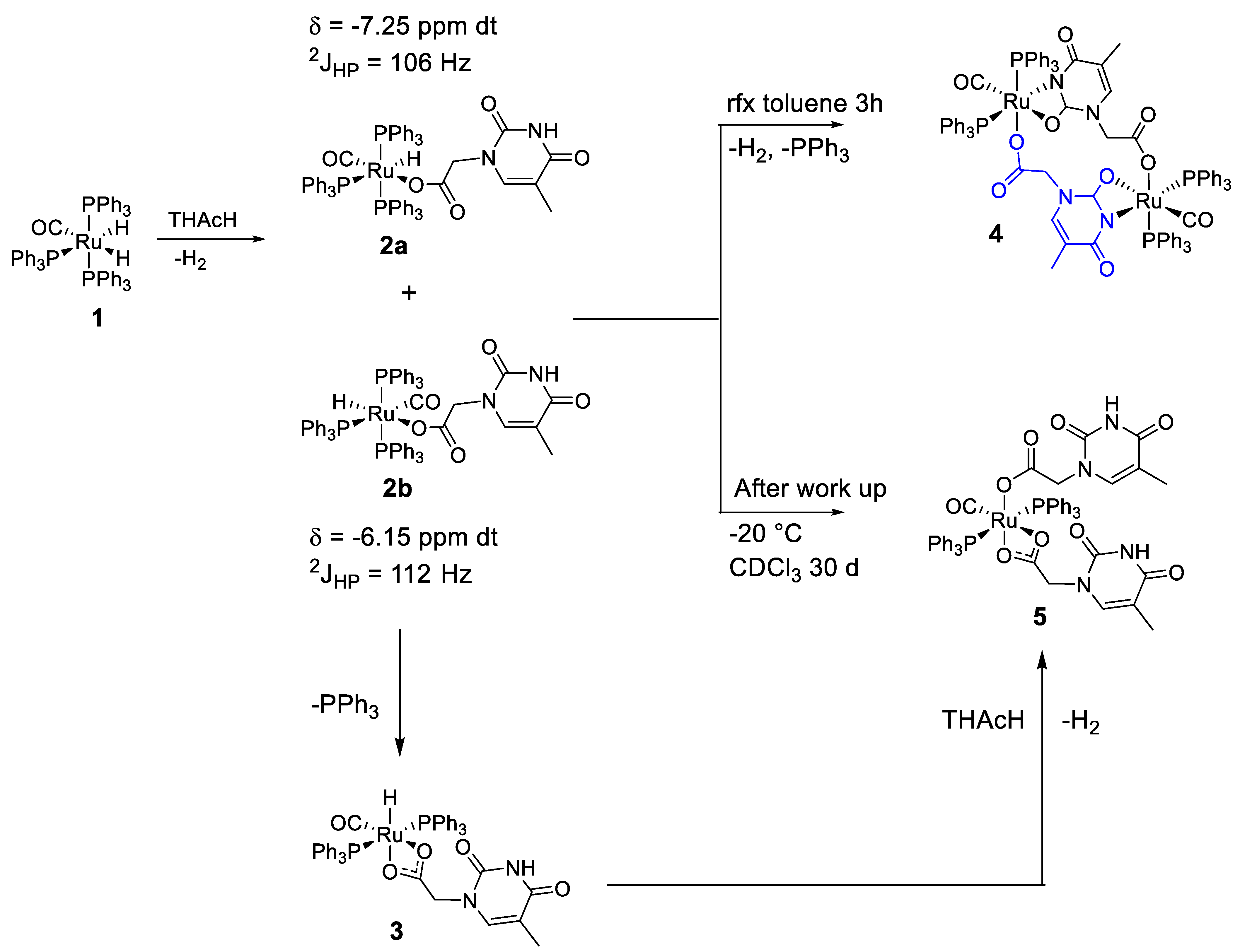
2.1. Synthesis of the Complexes cisP,P-k1(O), k2(N,O)-(Ru(CO)(PPh3)2(THAc)2) (5) + cisP,P- k1(O), k2(N,O)-(Ru(CO)(PPh3)2THAc)2 (4)
2.2. Characterization of 4 and 5
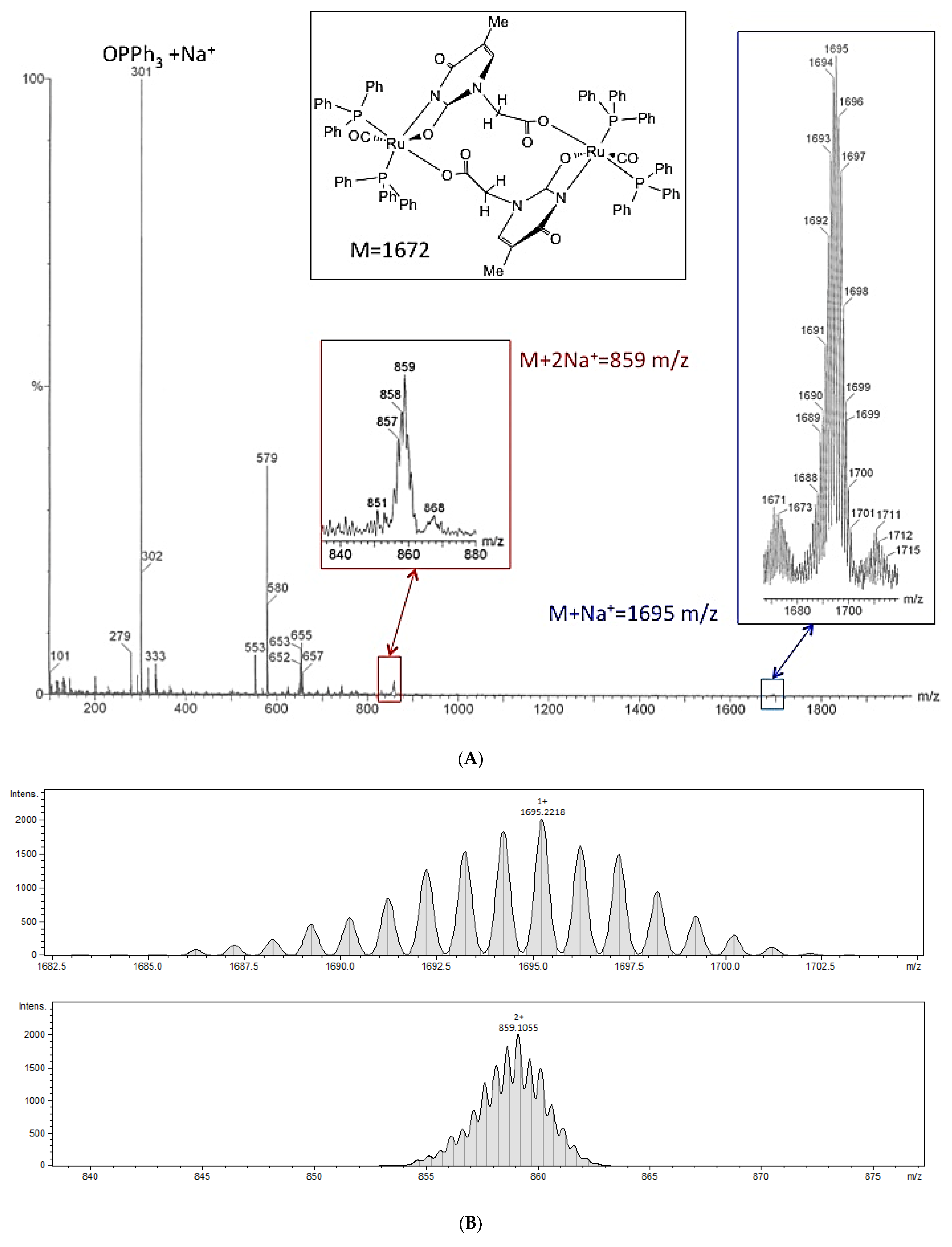
2.2.1. ESI-Ms of 4
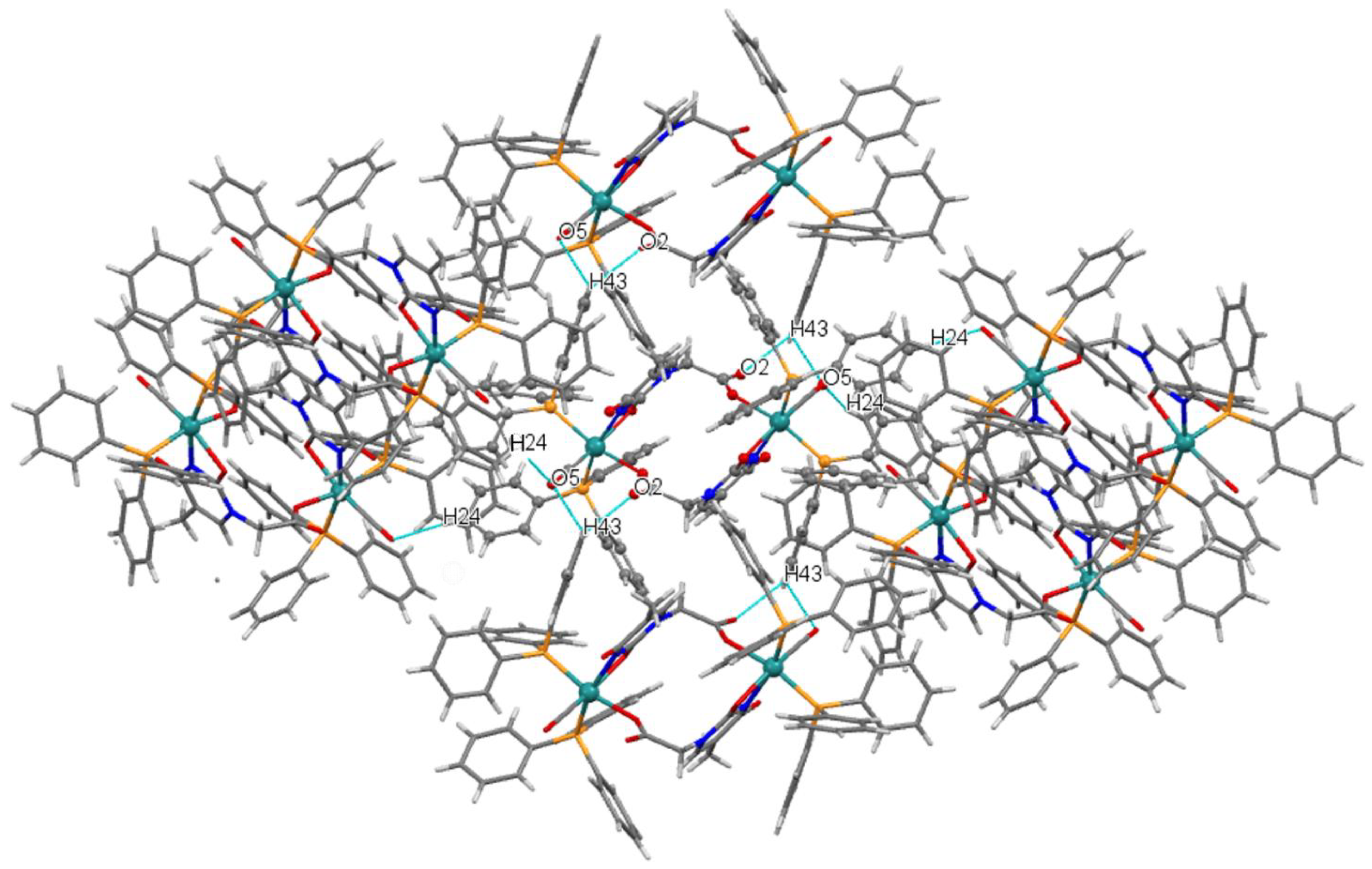
2.2.2. Description of the X-Ray Crystal Structure of 4
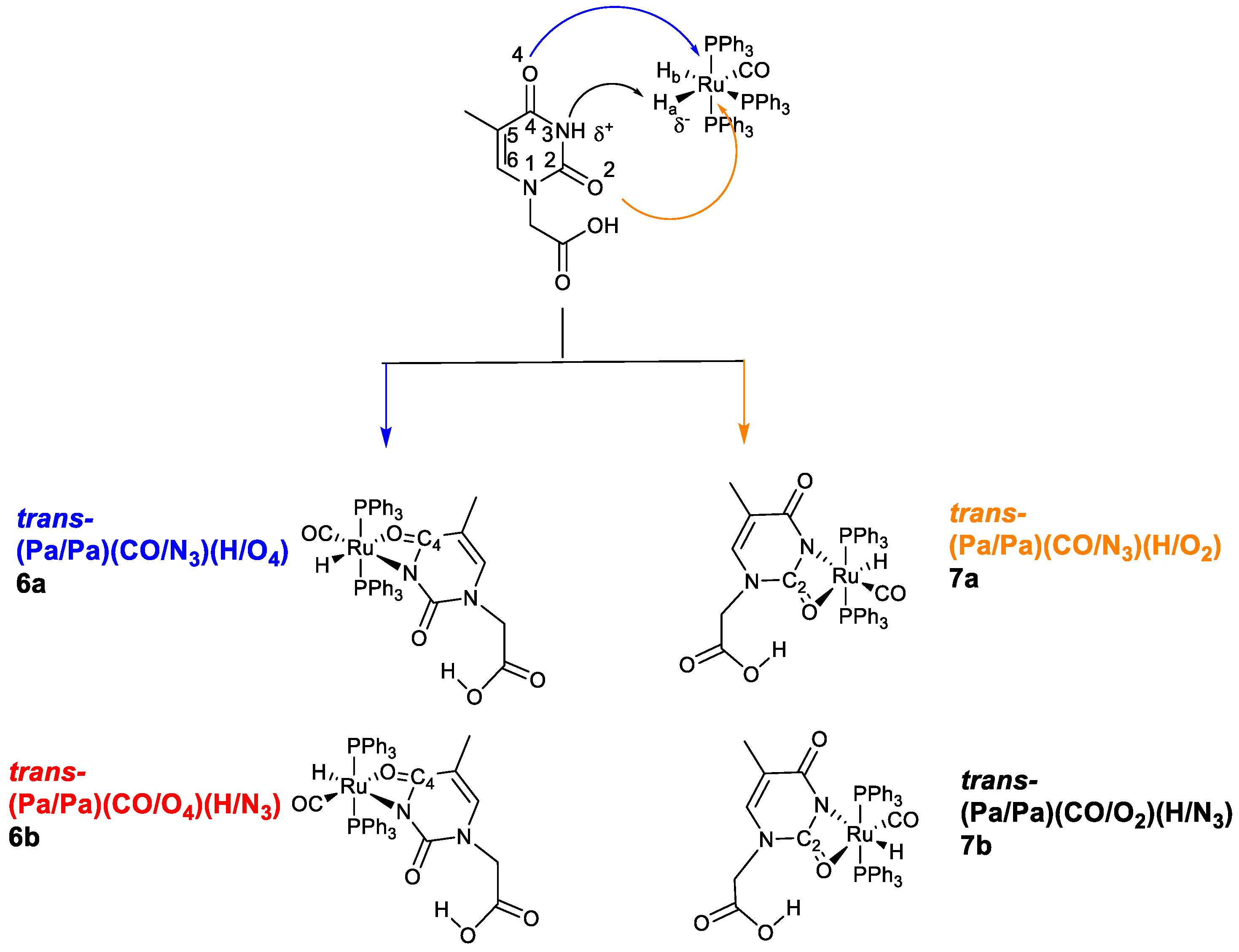
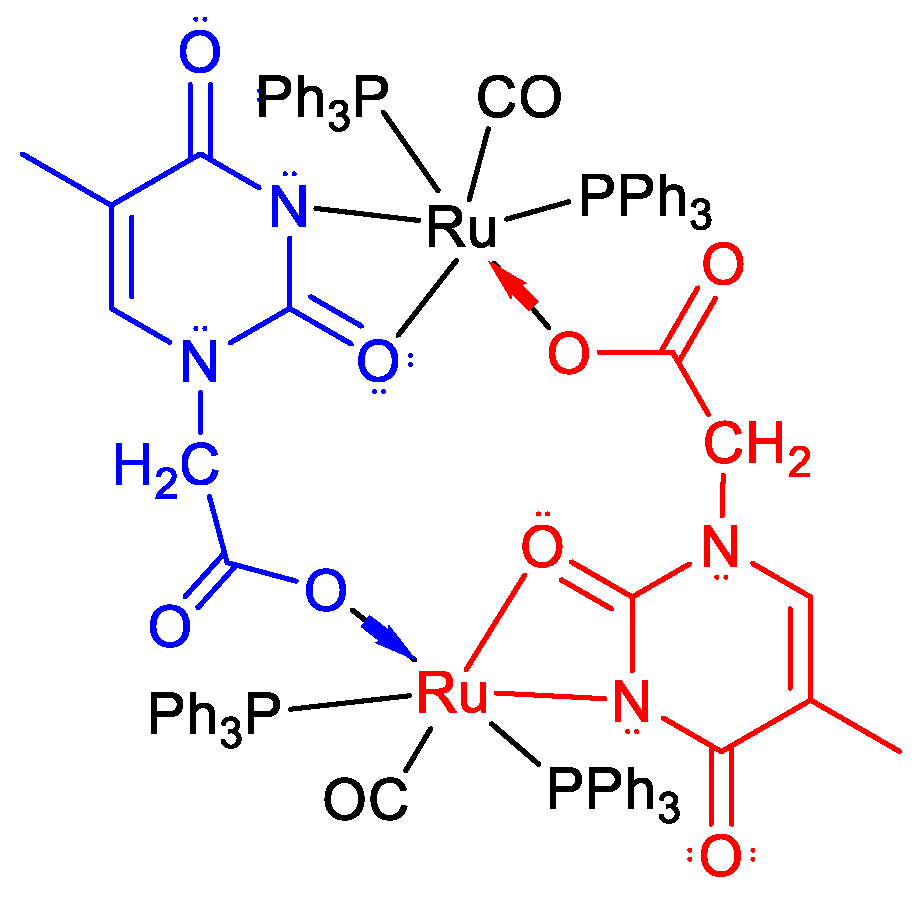
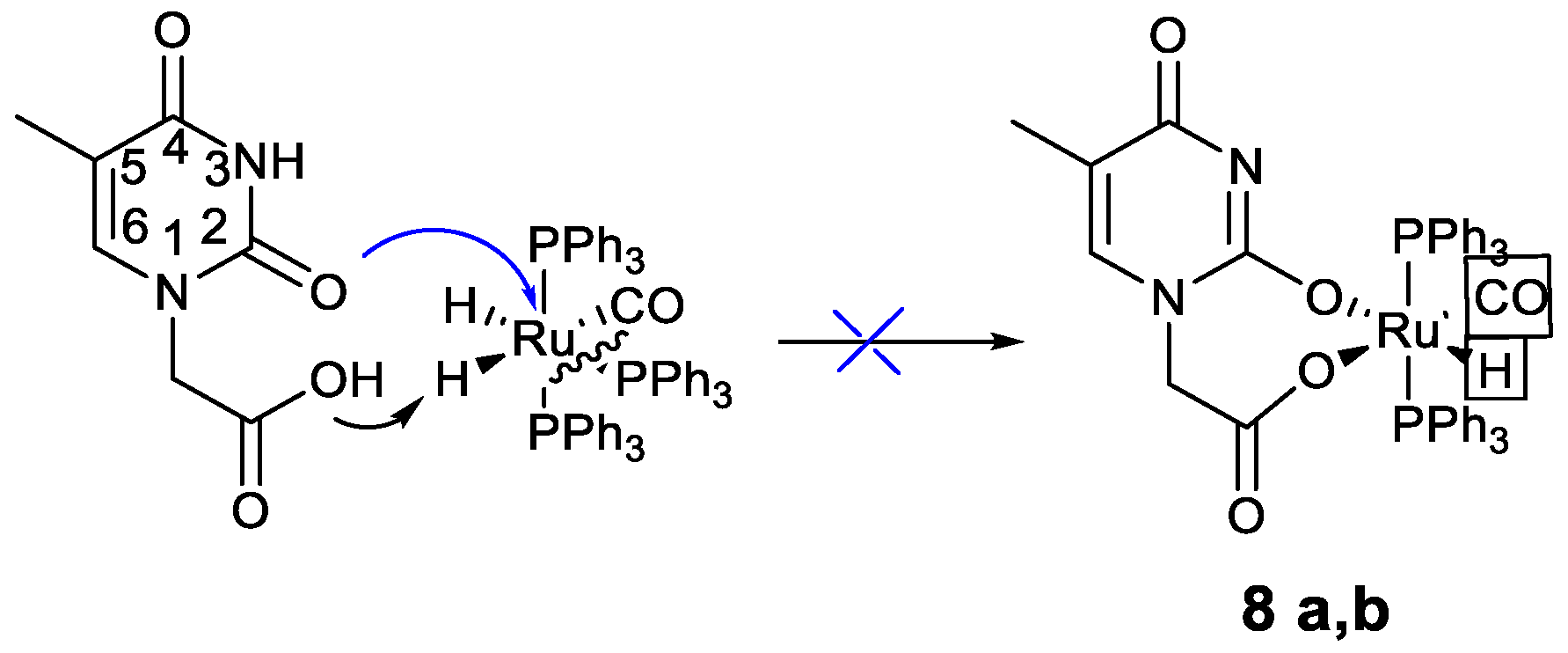
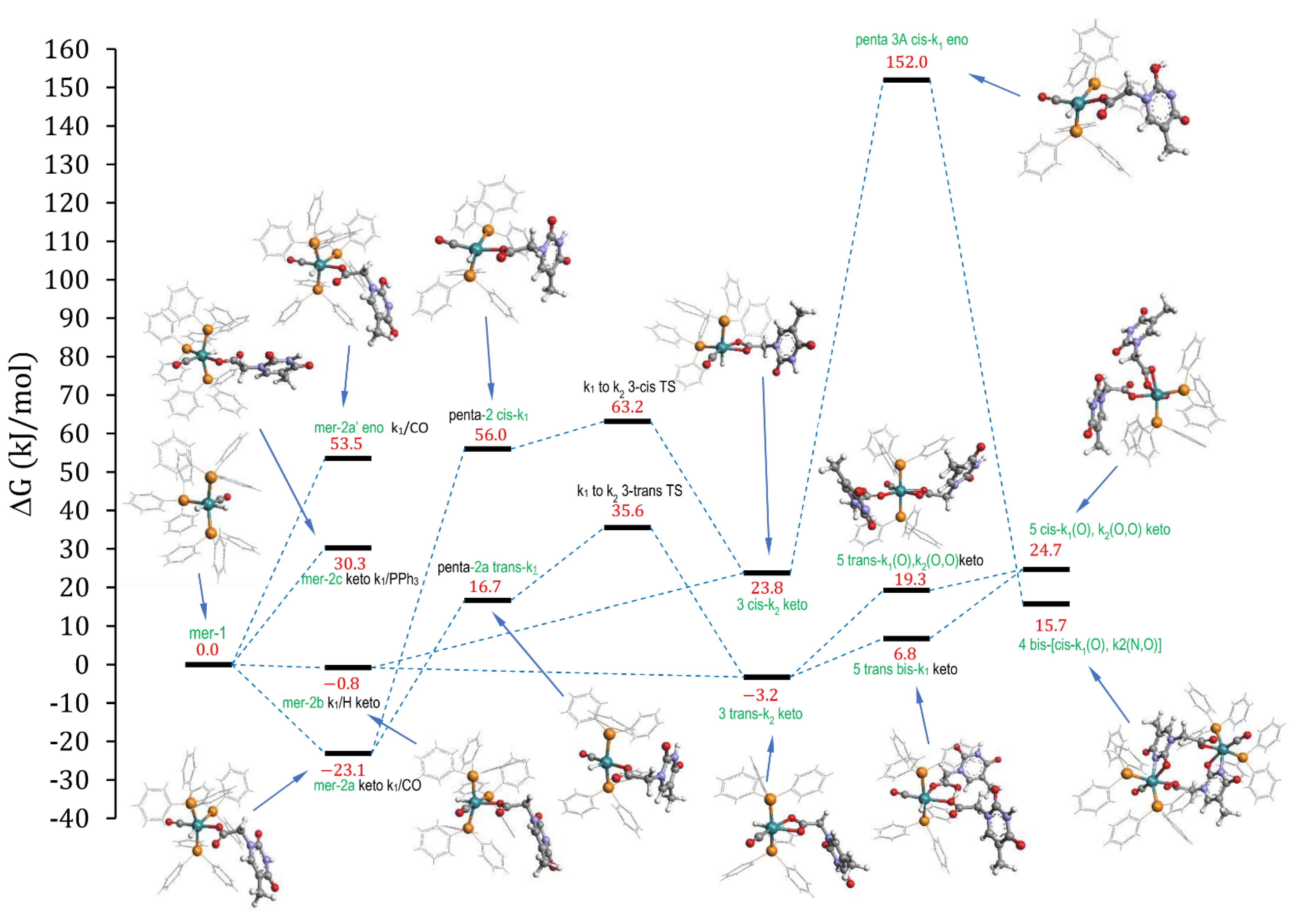
2.3. Proposed Reaction Path
- The rapid release of H2 from the reaction of the acidic NH-thymine ring and the Ru-H fragment;
- The dissociation of a PPh3 ligand through the action of the N-nucleophilic addition to the center of the Ru;
- Stereo-rearrangement of the phosphine ligands to achieve the less encumbered cisP,P conformation, which was ultimately exhibited by the Ru skeleton in the solid state.
- The poor solubility of the large molecule of 4 in chlorinated polar solvents, such as CH2Cl2, or CDCl3;
- The stronger nucleophilic ability to promote self-coupling, shown by the tautomeric iminol species, compared with the stable keto-amino form.
2.3.1. IR Spectra
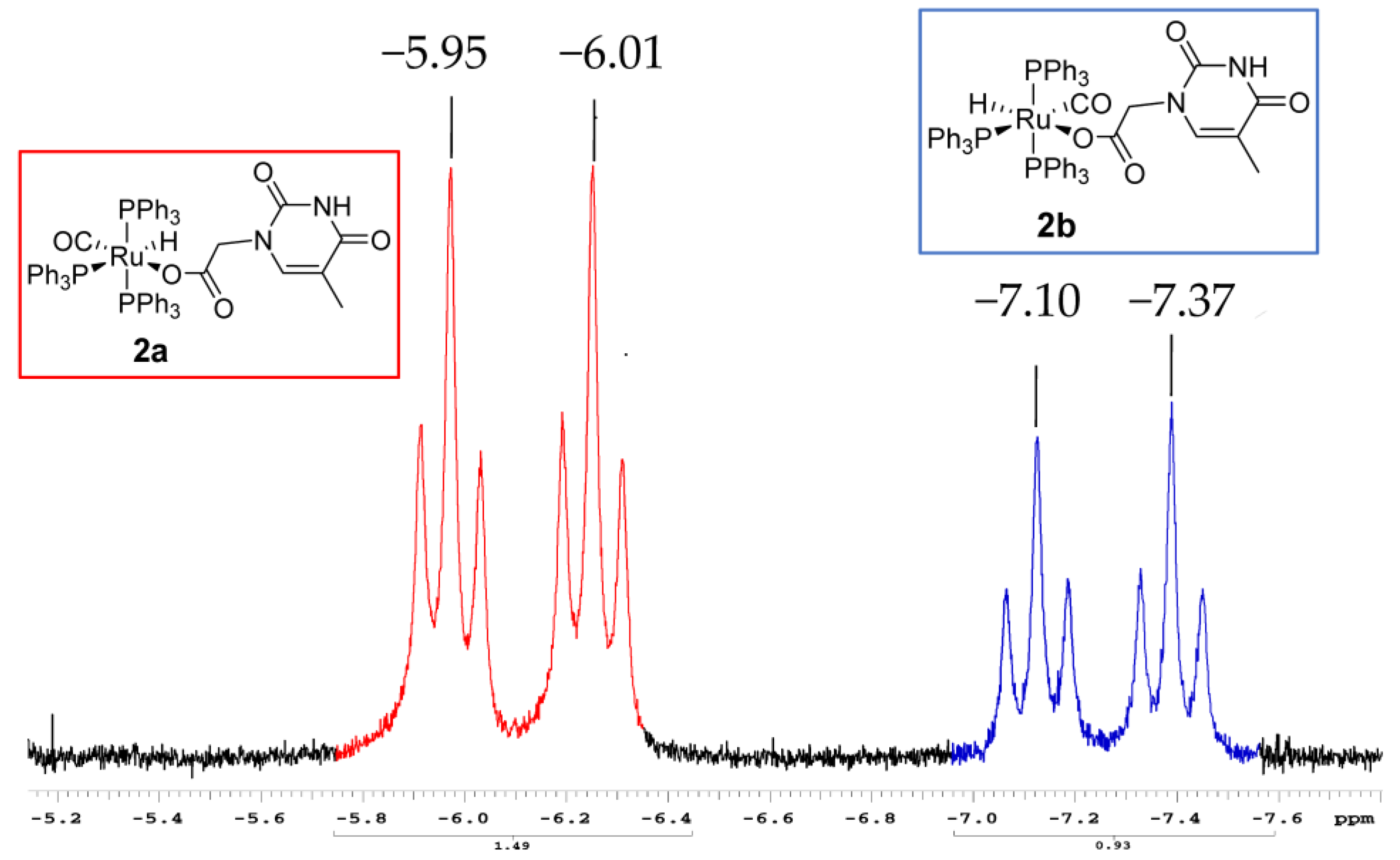
2.3.2. NMR Characterization
2.3.3. DFT Calculations
3. Materials and Methods
3.1. General
3.2. Experimental Procedure
3.2.1. Synthesis of the Complex k1(O), k2(O,O)-(Ru(CO)(PPh3)2THAc) (5)
3.2.2. Synthesis of the Complexes k1(O), k2(N,O)-(Ru(CO)(PPh3)2THAc)2 (4) + k1(O), k2(O,O)-Ru(CO)(PPh3)2(THAc)2) (5)
3.3. X-ray Crystallography
3.4. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bordoni, S.; Cerini, S.; Tarroni, R.; Monari, M.; Micheletti, G.; Boga, C. Ruthenium–Thymine Acetate Binding Modes: Experimental and Theoretical Studies. Appl. Sci. 2021, 11, 3113. [Google Scholar] [CrossRef]
- Małecki, J.G. Half-sandwich ruthenium(II) complexes with N- and N,(N,O)-donor ligands: Molecular, electronic structures, and computational study. Struct. Chem. 2012, 23, 461–472. [Google Scholar] [CrossRef]
- Małecki, J.G.; Groń, T.; Duda, H. Molecular and Spectroscopic Properties of Hydridecarbonyl Ruthenium Complexes with Pyrazine Carboxylic Acid Ligands. Polyhedron 2012, 31, 319–331. [Google Scholar] [CrossRef]
- Małecki, J.G.; Maroná, A.M.; Kusz, J. Phosphorescence of a Ruthenium(II) Hydride-Carbonyl Complex with 3-Hydroxy-2-Quinoxalinecarboxylic Acid as a Co-Ligand. Mendeleev Commun. 2015, 25, 103–105. [Google Scholar] [CrossRef]
- Małecki, J.G.; Krompiec, S.; Maroń, A.; Penkala, M. Synthesis, Molecular, Spectroscopic and Catalytic Characterization of Ruthenium(II) Complexes with Pyridine-2-Carboxylic Acid Derivatives Ligands. Polyhedron 2012, 48, 21–30. [Google Scholar] [CrossRef]
- Xiang, J.; Lo, L.T.L.; Leung, C.F.; Yiu, S.M.; Ko, C.C.; Lau, T.C. Synthesis, Structures, and Photophysical Properties of Ruthenium(II) Quinolinolato Complexes. Organometallics 2012, 31, 7101–7108. [Google Scholar] [CrossRef]
- Ruiz, J.; Villa, M.D.; Rodríguez, V.; Cutillas, N.; Vicente, C.; López, G.; Bautista, D. A Novel Metal-Binding Mode of Thymine Nucleobases: N(3) and O(4) Chelation. Inorg. Chem. 2007, 46, 5448–5449. [Google Scholar] [CrossRef] [PubMed]
- Esteruelas, M.A.; García-Raboso, J.; Oliván, M. Reactions of an Osmium-Hexahydride Complex with Cytosine, Deoxycytidine, and Cytidine: The Importance of the Minor Tautomers. Inorg. Chem. 2012, 51, 9522–9528. [Google Scholar] [CrossRef]
- Correa, R.S.; Freire, V.; Barbosa, M.I.F.; Bezerra, D.P.; Bomfim, L.M.; Moreira, D.R.M.; Soares, M.B.P.; Ellena, J.; Batista, A.A. Ru(Ii)-Thyminate Complexes: New Metallodrug Candidates against Tumor Cells. New J. Chem. 2018, 42, 6794–6802. [Google Scholar] [CrossRef]
- Silva, V.R.; Corrêa, R.S.; Santos, L.D.S.; Soares, M.B.P.; Batista, A.A.; Bezerra, D.P. A Ruthenium-Based 5-Fluorouracil Complex with Enhanced Cytotoxicity and Apoptosis Induction Action in HCT116 Cells. Sci. Rep. 2018, 8, 288. [Google Scholar] [CrossRef]
- dos Santos, E.R.; Graminha, A.E.; Schultz, M.S.; Correia, I.; Selistre-De-Araújo, H.S.; Corrêa, R.S.; Ellena, J.; Lacerda, E.D.P.S.; Pessoa, J.C.; Batista, A.A. Cytotoxic activity and structural features of Ru(II)/phosphine/amino acid complexes. J. Inorg. Biochem. 2018, 182, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, K.M.; Honorato, J.; Gonçalves, G.R.; Cominetti, M.R.; Batista, A.A.; Correa, R.S. Ru(Ii)/Diclofenac-Based Complexes: DNA, BSA Interaction and Their Anticancer Evaluation against Lung and Breast Tumor Cells. Dalton Trans. 2020, 49, 12643–12652. [Google Scholar] [CrossRef]
- Lynam, J.M.; Welby, C.E.; Whitwood, A.C. Exploitation of a Chemically Non-Innocent Acetate Ligand in the synthesis and Reactivity of Ruthenium Vinylidene Complexes. Organometallics 2009, 28, 1320–1328. [Google Scholar] [CrossRef]
- Welby, C.E.; Eschemann, T.O.; Unsworth, C.A.; Smith, E.J.; Thatcher, R.J.; Whitwood, A.C.; Lynam, J.M. Ruthenium Acetate Complexes as Versatile Probes of Metal-Ligand Interactions: Insight into the Ligand Effects of Vinylidene, Carbene, Carbonyl, Nitrosyl and Isocyanide. Eur. J. Inorg. Chem. 2012, 2012, 1493–1506. [Google Scholar] [CrossRef]
- Jeschke, J.; Gäbler, C.; Korb, M.; Rüffer, T.; Lang, H. Ruthenium Carboxylate Complexes as Efficient Catalysts for the Addition of Carboxylic Acids to Propargylic Alcohols. Eur. J. Inorg. Chem. 2015, 2015, 2939–2947. [Google Scholar] [CrossRef]
- Baldino, S.; Giboulot, S.; Lovison, D.; Nedden, H.G.; Pöthig, A.; Zanotti-Gerosa, A.; Zuccaccia, D.; Ballico, M.; Baratta, W. Preparation of Neutral Trans—Cis [Ru(O2CR)2P2(NN)], Cationic [Ru(O2CR)P2(NN)](O2CR) and Pincer [Ru(O2CR)(CNN)P2] (P = PPh3, P2= Diphosphine) Carboxylate Complexes and Their Application in the Catalytic Carbonyl Compounds Reduction. Organometallics 2021, 40, 1086–1103. [Google Scholar] [CrossRef]
- Samouei, H.; Grushin, V.V. New, Highly Efficient, Simple, Safe, and Scalable Synthesis of [(Ph3P)3Ru(CO)(H)2]. Organometallics 2013, 32, 4440–4443. [Google Scholar] [CrossRef]
- Liu, M.C.; Feng, W.; Ding, J.C. 1-(Carboxymethyl)Thymine. Acta Crystallogr. Sect. E Struct. Rep. Online 2004, 60, o1611–o1612. [Google Scholar] [CrossRef]
- Hassanein, K.; Castillo, O.; Gómez-García, C.J.; Zamora, F.; Amo-Ochoa, P. Asymmetric and Symmetric Dicopper(II) Paddle-Wheel with Modified Nucleobases. Cryst. Growth Des. 2015, 15, 5485–5494. [Google Scholar] [CrossRef]
- Lippert, B. Multiplicity of Metal Ion Binding Patterns to Nucleobases. Co-Ord. Chem. Rev. 2000, 200–202, 487–516. [Google Scholar] [CrossRef]
- Lippert, B. Rare Iminol Tautomer of 1-Methylthymine through Metal Coordination at N(3). Inorganica Chim. Acta 1981, 55, 5–14. [Google Scholar] [CrossRef]
- Renn, O.; Lippert, B. Metal-Stabilized Rare Tautomers of Nucleobases 3. (1-Methylthyminato-N3)(1-Methylthymine-N3)-Ck-Diammineplatinum(I1) Hemihexachloroplatinate(IV) Dihydrate. Inorganica Chim. Acta 1991, 190, 285–289. [Google Scholar] [CrossRef]
- Lippert, B.; Neugebauer, D. Simultaneous Binding of Two Different Transition Metals to the DNA Model Base I-Methylthymine: The X-Ray Structure of Bis[Bis(p-1-Methylthyminato-N3,04)-Cis-Diammine Platinum(II)] Silver Nitrate Pentahydrate. Inorganica Chim. Acta 1980, 46, 171–179. [Google Scholar] [CrossRef]
- Lippert, B.; Sanz Miguel, P.J. The Renaissance of Metal-Pyrimidine Nucleobase Coordination Chemistry. Acc. Chem. Res. 2016, 49, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Schöllhorn, H.; Thewalt, U.; Lippert, B. Metal-Stabilized Rare Tautomers of Nucleobases. 2. 2-Oxo-4-Hydroxo Form of Uracil: Crystal Structures and Solution Behavior of Two Platinum(II) Complexes Containing Iminol Tautomers of 1-Methyluracil. J. Am. Chem. Soc. 1989, 111, 7213–7221. [Google Scholar] [CrossRef]
- Peng, C.S.; Tokmakoff, A. Identification of Lactam-Lactim Tautomers of Aromatic Heterocycles in Aqueous Solution Using 2D IR Spectroscopy. J. Phys. Chem. Lett. 2012, 3, 3302–3306. [Google Scholar] [CrossRef]
- Peng, C.S.; Baiz, C.R.; Tokmakoff, A. Direct Observation of Ground-State Lactam-Lactim Tautomerization Using Temperature-Jump Transient 2D IR Spectroscopy. Proc. Natl. Acad. Sci. USA 2013, 110, 9243–9248. [Google Scholar] [CrossRef]
- Hammud, H.H.; El-Dakdouki, M.H.; Sonji, N.; Sonji, G.; Bouhadir, K.H. Interactions of Some Divalent Metal Ions with Thymine and Uracil Thiosemicarbazide Derivatives. Nucleosides Nucleotides Nucleic Acids 2016, 35, 259–276. [Google Scholar] [CrossRef]
- Albright, A.L.; Collins, L.; Li, J.; Harris, B.; White, J.M. Crystal Structures of the Amide and Iminol Tautomers O-(3,5-Dinitrobenzoyloxy)Benzohydroxamate. A Case of a Disappearing Solvate? Aust. J. Chem. 2016, 69, 1193–1197. [Google Scholar] [CrossRef]
- Lee, H.; Lee, J.; Lee, S.; Shin, Y.; Jung, W.; Kim, J.-H.; Park, K.; Kim, K.; Cho, S.; Ro, S.; et al. A Novel Class of Highly Potent, Selective, and Non-Peptidic Inhibitor of Ras Farnesyltransferase (FTase). Bioorganic Med. Chem. Lett. 2001, 11, 3069–3072. [Google Scholar] [CrossRef]
- Morais, T.S.; Marques, F.; Madeira, P.J.A.; Robalo, M.P.; Garcia, M.H. Design and Anticancer Properties of New Water-Soluble Ruthenium–Cyclopentadienyl Complexes. Pharmaceuticals 2022, 15, 862. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, E.; Binacchi, F.; Marotta, C.; Cirri, D.; Gabbiani, C.; Pratesi, A. Highlights of New Strategies to Increase the Efficacy of Transition Metal Complexes for Cancer Treatments. Molecules 2023, 28, 273. [Google Scholar] [CrossRef]
- Lucaciu, R.L.; Hangan, A.C.; Sevastre, B.; Oprean, L.S. Metallo-Drugs in Cancer Therapy: Past, Present and Future. Molecules 2022, 27, 6485. [Google Scholar] [CrossRef] [PubMed]
- Dandliker, P.J.; Núñez, M.E.; Barton, J.K. Oxidative Charge Transfer To Repair Thymine Dimers and Damage Guanine Bases in DNA Assemblies Containing Tethered Metallointercalators. Biochemistry 1998, 37, 6491–6502. [Google Scholar] [CrossRef]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic Anticancer Compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef]
- Coverdale, J.P.C.; Laroiya-McCarron, T.; Romero-Canelón, I. Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics 2019, 7, 31. [Google Scholar] [CrossRef]
- Yellol, J.; Pérez, S.A.; Buceta, A.; Yellol, G.; Donaire, A.; Szumlas, P.; Bednarski, P.J.; Makhloufi, G.; Janiak, C.; Espinosa, A.; et al. Novel C,N-Cyclometalated Benzimidazole Ruthenium(II) and Iridium(III) Complexes as Antitumor and Antiangiogenic Agents: A Structure-Activity Relationship Study. J. Med. Chem. 2015, 58, 7310–7327. [Google Scholar] [CrossRef] [PubMed]
- Renfrew, A.K. Transition Metal Complexes with Bioactive Ligands: Mechanisms for Selective Ligand Release and Applications for Drug Delivery. Metallomics 2014, 6, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Sáez, R.; Lorenzo, J.; Prieto, M.J.; Font-Bardia, M.; Calvet, T.; Omeñaca, N.; Vilaseca, M.; Moreno, V. Influence of PPh3 Moiety in the Anticancer Activity of New Organometallic Ruthenium Complexes. J. Inorg. Biochem. 2014, 136, 1–12. [Google Scholar] [CrossRef]
- Habtemariam, A.; Melchart, M.; Fernández, R.; Parsons, S.; Oswald, I.D.H.; Parkin, A.; Fabbiani, F.P.A.; Davidson, J.E.; Dawson, A.; Aird, R.E.; et al. Structure-Activity Relationships for Cytotoxic Ruthenium(II) Arene Complexes Containing N,N-, N,O-, and O,O-Chelating Ligands. J. Med. Chem. 2006, 49, 6858–6868. [Google Scholar] [CrossRef]
- Sivakova, S.; Rowan, S.J. Nucleobases as Supramolecular Motifs. Chem. Soc. Rev. 2005, 34, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs—A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef] [PubMed]
- Sletten, E. Metal Binding to Nucleic Acids—A Journey from the Beginning. Inorganica Chim. Acta 2016, 452, 273–278. [Google Scholar] [CrossRef]
- Van Buijtenen, J.; Meuldijk, J.; Vekemans, J.A.J.M.; Hulshof, L.A.; Kooijman, H.; Spek, A.L. Dinuclear Ruthenium Complexes Bearing Dicarboxylate and Phosphine Ligands. Acceptorless Catalytic Dehydrogenation of 1-Phenylethanol. Organometallics 2006, 25, 873–881. [Google Scholar] [CrossRef]
- Wang, W.; Hellinga, H.W.; Beese, L.S. Structural Evidence for the Rare Tautomer Hypothesis of Spontaneous Mutagenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 17644–17648. [Google Scholar] [CrossRef]
- Myron, F. Goodman Mutation Caught in the Act. Nature 1995, 378, 237–238. [Google Scholar] [CrossRef]
- Rogstad, K.N.; Jang, Y.H.; Sowers, L.C.; Goddard, W.A. First Principles Calculations of the p K a Values and Tautomers of Isoguanine and Xanthine. Chem. Res. Toxicol. 2003, 16, 1455–1462. [Google Scholar] [CrossRef]
- Ganguly, S.; Kundu, K.K. Deprotonation Energetics of Some Nucleosides in Water from EMF Measure-Ments. Can. J. Chem. 1995, 73, 70–78. [Google Scholar] [CrossRef]
- SMART & SAINT Software Reference Manuals, version 5.051, (Windows NT Version); Bruker Analytical X-ray Instruments Inc.: Madison, WI, USA, 1998.
- Sheldrick, G.M. SADABS, Program for Empirical Absorption Correction; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Cryst. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Kesharwani, M.K.; Brauer, B.; Martin, J.M.L. Frequency and Zero-Point Vibrational Energy Scale Factors for Double-Hybrid Density Functionals (and Other Selected Methods): Can Anharmonic Force Fields Be Avoided? J. Phys. Chem. A 2015, 119, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.L.; Hay, P.J.; Pratt, L.R. Hydrolysis of Ferric Ion in Water and Conformational Equilibrium. J. Phys. Chem. A 1998, 102, 3565–3573. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A New Local Density Functional for Main-Group Thermochemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thymine Acetate | 4 | THAcH |
| C6-O4 | 1.23 (1) | 1.241 (2) |
| C6-N2 | 1.35 (1) | 1.371 (2) |
| C7-N2 | 1.341 (9) | 1.379 (2) |
| C7-O3 | 1.280 (8) | 1.213 (2) |
| C7-N1 | 1.358 (9) | 1.376 (2) |
| C3-C4 | 1.33 (1) | 1.338 (2) |
| C3-N1 | 1.38 (1) | 1.375 (2) |
| Metal Center | ||
| Ru-P1 | 2.355 (2) | |
| Ru-P2 | 2.348 (2) | |
| Ru-O1 | 2.108 (5) | |
| Ru-O3 | 2.216 (5) | |
| Ru-N2 | 2.109 (7) | |
| Ru-C8 | 1.81 (1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bordoni, S.; Tarroni, R.; Monari, M.; Cerini, S.; Battaglia, F.; Micheletti, G.; Boga, C.; Drius, G. Ru-Controlled Thymine Tautomerization Frozen by a k1(O)-, k2(N,O)-Metallacycle: An Experimental and Theoretical Approach. Molecules 2023, 28, 3983. https://doi.org/10.3390/molecules28103983
Bordoni S, Tarroni R, Monari M, Cerini S, Battaglia F, Micheletti G, Boga C, Drius G. Ru-Controlled Thymine Tautomerization Frozen by a k1(O)-, k2(N,O)-Metallacycle: An Experimental and Theoretical Approach. Molecules. 2023; 28(10):3983. https://doi.org/10.3390/molecules28103983
Chicago/Turabian StyleBordoni, Silvia, Riccardo Tarroni, Magda Monari, Stefano Cerini, Fabio Battaglia, Gabriele Micheletti, Carla Boga, and Giacomo Drius. 2023. "Ru-Controlled Thymine Tautomerization Frozen by a k1(O)-, k2(N,O)-Metallacycle: An Experimental and Theoretical Approach" Molecules 28, no. 10: 3983. https://doi.org/10.3390/molecules28103983
APA StyleBordoni, S., Tarroni, R., Monari, M., Cerini, S., Battaglia, F., Micheletti, G., Boga, C., & Drius, G. (2023). Ru-Controlled Thymine Tautomerization Frozen by a k1(O)-, k2(N,O)-Metallacycle: An Experimental and Theoretical Approach. Molecules, 28(10), 3983. https://doi.org/10.3390/molecules28103983