3.3. Procedure for the Synthesis of Compounds 3
In a reaction tube, pyrazol-5-ones 1 (0.24 mmol), pyrazole-maleimide 2 (0.20 mmol) and catalyst C10 (0.02 mmol) were added into toluene (4 mL). The reaction solution was stirred at 25 °C. After the reaction was complete (monitored by TLC), the crude product was purified by column chromatography (ethyl acetate/petroleum ether = 1/10 to 1/3) on silica gel to produce the product 3.
(S)-3-((R)-4-benzyl-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3aa)
This was prepared according to the procedure within 1 h as a white solid (121.8 mg, 98% yield, dr = 1:1). mp 127.1–127.9 °C; = −33.206 (c 0.52, CH2Cl2); 1H NMR (400 MHz, Chloroform-d) δ: 8.00 (dq, J = 6.7, 2.6, 1.6 Hz, 4H), 7.69–7.48 (m, 15H), 7.40–7.28 (m, 9H), 7.23–7.16 (m, 2H), 7.12–7.01 (m, 10H), 6.29 (s, 1H), 5.50 (s, 1H), 4.22 (dd, J = 17.6, 7.4 Hz, 2H), 3.99 (dd, J = 9.3, 7.4 Hz, 1H), 3.76 (dd, J = 9.4, 5.5 Hz, 1H), 3.61 (dd, J = 19.5, 13.5 Hz, 2H), 3.50 (d, J = 13.2 Hz, 1H), 3.18 (dd, J = 17.8, 9.4 Hz, 1H), 3.01 (dd, J = 18.6, 9.7 Hz, 1H), 2.78 (s, 1H), 1.53 (s, 9H), 1.38 (s, 9H). 13C NMR (151 MHz, CDCl3) δ: 174.89, 173.87, 173.72, 173.43, 173.07, 158.07, 157.37, 148.93, 148.66, 137.00, 136.82, 133.31, 133.24, 132.50, 131.10, 131.02, 130.99, 130.71, 129.42, 129.36, 129.35, 129.17, 128.99, 128.79, 128.71, 128.49, 128.47, 128.44, 128.39, 127.95, 127.79, 127.69, 127.61, 127.00, 126.29, 126.05, 125.40, 125.34, 120.04, 119.89, 103.68, 61.50, 57.13, 44.93, 43.58, 41.24, 40.02, 31.06, 30.04, 29.81, 29.69. HRMS (ESI) m/z Calcd for C39H36N5O3, [M + H]+, 622.2813, found: 622.2806. Enantiomeric excess was determined to be 99% (determined by HPLC using chiral IB-H column, hexane/2-propanol = 7/3, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 21.9 min, tminor = 16.6 min).
(S)-3-((R)-4-benzyl-5-oxo-1-phenyl-3-(o-tolyl)-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3ba)
This was prepared according to the procedure within 1.2 h as a white solid (125.8 mg, 99% yield, dr = 1:1), mp 108.1–108.9 °C; = −35.030 (c 0.33, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.83 (d, J = 11.5 Hz, 2H), 7.77 (q, J = 3.5 Hz, 2H), 7.69–7.61 (m, 6H), 7.58 (d, J = 8.0 Hz, 2H), 7.46–7.41 (m, 2H), 7.41–7.26 (m, 12H), 7.22–7.15 (m, 2H), 7.08 (d, J = 6.9 Hz, 8H), 7.03 (dd, J = 7.8, 1.8 Hz, 2H), 6.29 (s, 1H), 5.51 (s, 1H), 4.31–4.13 (m, 2H), 3.98 (dd, J = 9.3, 7.4 Hz, 1H), 3.75 (dd, J = 9.4, 5.4 Hz, 1H), 3.60 (dd, J = 26.7, 13.5 Hz, 2H), 3.48 (d, J = 13.2 Hz, 1H), 3.18 (dd, J = 17.8, 9.4 Hz, 1H), 2.99 (dd, J = 18.6, 9.7 Hz, 1H), 2.66 (d, J = 58.4 Hz, 1H), 2.43 (s, 6H), 1.38 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 173.89, 173.75, 173.46, 173.08, 158.23, 157.50, 148.90, 148.64, 139.37, 139.09, 137.00, 136.83, 133.39, 133.33, 133.27, 132.55, 131.90, 131.63, 130.98, 130.89, 129.39, 129.26, 128.97, 128.88, 128.81, 128.70, 128.50, 128.46, 128.41, 128.37, 128.17, 127.93, 127.75, 127.68, 127.60, 126.27, 126.03, 125.39, 125.26, 124.66, 123.88, 120.09, 119.94, 103.70, 103.63, 61.50, 57.14, 44.95, 43.55, 41.28, 40.04, 31.10, 30.07, 29.81, 29.70, 21.68, 21.65. HRMS (ESI) m/z Calcd for C40H38N5O3, [M + H]+, 636.2969, found: 636.2972. Enantiomeric excess was determined to be 98% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 7/3, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 25.1 min, tminor = 12.1 min).
(S)-3-((R)-4-benzyl-5-oxo-1-phenyl-3-(m-tolyl)-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3ca)
This was prepared according to the procedure within 1.2 h as a white solid (125.8 mg, 99% yield, dr = 1:1), mp 109.1–109.9 °C; = −33.491 (c 0.42, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.85 (s, 1H), 7.82 (s, 1H), 7.78 (d, J = 7.2 Hz, 2H), 7.68 (d, J = 1.5 Hz, 1H), 7.67–7.61 (m, 5H), 7.60–7.55 (m, 2H), 7.43 (dd, J = 12.2, 6.9 Hz, 3H), 7.40–7.27 (m, 11H), 7.23–7.16 (m, 2H), 7.09 (d, J = 5.6 Hz, 8H), 7.04 (dd, J = 7.7, 1.9 Hz, 2H), 6.29 (d, J = 1.0 Hz, 1H), 5.50 (s, 1H), 4.31–4.15 (m, 2H), 3.99 (dd, J = 9.3, 7.5 Hz, 1H), 3.75 (dd, J = 9.5, 5.4 Hz, 1H), 3.65 (d, J = 13.2 Hz, 1H), 3.57 (d, J = 13.8 Hz, 1H), 3.49 (d, J = 13.2 Hz, 1H), 3.19 (dd, J = 17.8, 9.4 Hz, 1H), 3.00 (dd, J = 18.6, 9.7 Hz, 1H), 2.66 (d, J = 49.9 Hz, 1H), 2.44 (s, 6H), 1.54 (s, 9H), 1.38 (d, J = 1.0 Hz, 9H). 13C NMR (101 MHz, CDCl3) δ: 174.94, 173.90, 173.76, 173.46, 173.11, 173.08, 158.23, 157.51, 148.90, 148.65, 139.37, 139.09, 137.00, 136.82, 133.39, 133.33, 133.26, 132.55, 131.90, 131.63, 130.98, 130.89, 129.39, 129.26, 128.97, 128.88, 128.81, 128.70, 128.50, 128.47, 128.41, 128.37, 128.17, 127.93, 127.75, 127.69, 127.61, 126.28, 126.04, 125.40, 125.27, 124.66, 123.88, 120.09, 119.94, 103.70, 103.63, 61.50, 57.14, 44.95, 43.55, 41.28, 40.04, 31.10, 30.07, 29.81, 29.70, 21.65. HRMS (ESI) m/z Calcd for C40H38N5O3, [M + H]+, 636.2969, found: 636.2974. Enantiomeric excess was determined to be 99% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 7/3, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 22.5 min, tminor = 12.1 min).
(S)-3-((R)-4-benzyl-5-oxo-1-phenyl-3-(p-tolyl)-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3da)
This was prepared according to the procedure within 1.2 h as a white solid (125.8 mg, 99% yield, dr = 1:1), mp 127.1–127.9 °C; = −61.572 (c 0.23, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.89 (dd, J = 8.0 Hz, 4H), 7.68–7.56 (m, 9H), 7.39–7.28 (m, 14H), 7.19 (dd, J = 16.7, 8.2 Hz, 2H), 7.09 (d, J = 6.7 Hz, 8H), 7.06–7.01 (m, 2H), 6.28 (s, 1H), 5.48 (s, 1H), 4.20 (d, J = 26.5 Hz, 2H), 3.97 (dd, J = 9.3, 7.4 Hz, 1H), 3.74 (dd, J = 9.7, 5.2 Hz, 1H), 3.61 (dd, J = 27.2, 13.5 Hz, 2H), 3.49 (d, J = 13.2 Hz, 1H), 3.17 (dd, J = 17.8, 9.4 Hz, 1H), 2.99 (dd, J = 18.6, 9.7 Hz, 1H), 2.63 (d, J = 27.3 Hz, 1H), 2.50 (s, 3H), 2.44 (s, 3H), 1.38 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 174.98, 173.84, 173.76, 173.38, 173.08, 173.01, 158.07, 157.37, 148.92, 148.63, 141.58, 141.11, 137.03, 136.86, 133.46, 133.33, 133.26, 132.59, 130.09, 129.89, 129.37, 128.95, 128.80, 128.67, 128.48, 128.45, 128.40, 128.35, 128.32, 128.20, 127.89, 127.71, 127.58, 126.90, 126.21, 125.96, 125.38, 125.36, 120.04, 119.87, 103.68, 103.66, 61.51, 61.48, 57.15, 44.97, 43.50, 41.21, 39.88, 31.12, 30.07, 29.80, 29.67, 21.62, 21.55. HRMS (ESI) m/z Calcd for C40H38N5O3, [M + H]+, 636.2969, found: 636.2979. Enantiomeric excess was determined to be 98% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 7/3, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 25.3 min, tminor = 12.1 min).
(S)-3-((R)-4-benzyl-3-(naphthalen-2-yl)-5-oxo-1-phenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3ea)
This was prepared according to the procedure within 2.5 h as a white solid (106.1 mg, 79% yield, dr = 1:1), mp 110.1–110.9 °C; = −46.491 (c 0.79, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 8.42 (d, J = 2.3 Hz, 1H), 8.39 (s, 1H), 8.20–8.12 (m, 3H), 8.00–7.92 (m, 7H), 7.91–7.85 (m, 1H), 7.72–7.61 (m, 9H), 7.61–7.51 (m, 4H), 7.37 (dt, J = 15.5, 7.6 Hz, 5H), 7.32–7.17 (m, 15H), 7.15–7.01 (m, 13H), 6.26 (d, J = 2.0 Hz, 1H), 4.95 (s, 1H), 4.47 (s, 1H), 4.20 (dd, J = 10.1, 5.1 Hz, 1H), 4.11 (t, J = 8.2 Hz, 1H), 3.81 (q, J = 6.2, 5.6 Hz, 2H), 3.66 (dd, J = 13.9, 1.6 Hz, 1H), 3.56 (d, J = 13.2 Hz, 1H), 3.22 (dd, J = 17.6, 9.1 Hz, 1H), 3.00 (dd, J = 18.7, 9.8 Hz, 1H), 2.47 (s, 1H), 1.52 (d, J = 1.2 Hz, 9H), 1.35 (d, J = 2.1 Hz, 9H). 13C NMR (151 MHz, CDCl3) δ: 175.41, 173.92, 173.74, 173.51, 173.14, 173.03, 157.91, 157.12, 148.68, 137.00, 136.82, 134.37, 134.28, 133.50, 133.29, 132.95, 132.92, 132.85, 132.56, 129.43, 129.39, 129.29, 129.23, 129.04, 128.77, 128.71, 128.46, 128.43, 128.40, 128.23, 128.15, 128.04, 128.00, 127.97, 127.76, 127.67, 127.60, 127.48, 127.05, 126.56, 126.40, 126.16, 125.38, 125.28, 124.13, 124.04, 120.13, 119.97, 103.66, 103.58, 61.49, 57.31, 45.19, 43.54, 41.45, 40.11, 31.21, 30.12, 29.82, 29.65. HRMS (ESI) m/z Calcd for C43H38N5O3, [M + H]+, 672.2969, found: 672.2976. Enantiomeric excess was determined to be 98% (determined by HPLC using chiral OD-H-AD-H column, hexane/2-propanol = 7/3, λ = 254 nm, 25 °C, 0.6 mL/min, tmajor = 61.0 min, tminor = 34.0 min).
(S)-3-((R)-4-benzyl-5-oxo-1-phenyl-3-(thiophen-2-yl)-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3fa)
This was prepared according to the procedure within 1.1 h as a white solid (124.2 mg, 99% yield, dr = 1:1), mp 120.1–120.9 °C; = −35.474 (c 0.65, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.64 (ddd, J = 19.8, 10.9, 7.0 Hz, 9H), 7.53 (dd, J = 10.3, 6.5 Hz, 3H), 7.39–7.28 (m, 9H), 7.25–7.17 (m, 4H), 7.16–7.00 (m, 11H), 6.33 (s, 1H), 5.80 (s, 1H), 4.09 (d, J = 28.1 Hz, 2H), 4.00–3.94 (m, 1H), 3.77 (dd, J = 9.4, 5.6 Hz, 1H), 3.59–3.51 (m, 2H), 3.47 (d, J = 13.1 Hz, 1H), 3.19 (dd, J = 17.8, 9.3 Hz, 1H), 3.03 (dd, J = 18.5, 9.6 Hz, 1H), 2.79 (s, 1H), 1.57 (s, 9H), 1.38 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 173.69, 173.56, 172.98, 172.69, 172.39, 154.24, 149.01, 148.68, 136.79, 136.61, 134.42, 134.31, 133.30, 133.21, 132.36, 129.63, 129.46, 129.40, 128.96, 128.92, 128.70, 128.50, 128.46, 128.44, 128.24, 127.97, 127.91, 127.78, 127.72, 127.61, 127.39, 126.33, 126.10, 125.39, 125.35, 120.08, 119.96, 103.67, 61.59, 61.51, 45.35, 41.07, 39.72, 31.05, 30.11, 29.83, 29.65. HRMS (ESI) m/z Calcd for C37H34N5O3S, [M + H]+, 628.2377, found: 628.2387. Enantiomeric excess was determined to be 99% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 8/2, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 32.4 min, tminor = 21.3 min).
(S)-3-((R)-4-benzyl-3-methyl-5-oxo-1-phenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3ga)
This was prepared according to the procedure within 1.1 h as a white solid (108.5mg, 97% yield, dr = 1:1), mp 124.1–124.9 °C; = 80.357 (c 0.45, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.75–7.70 (m, 2H), 7.66–7.61 (m, 2H), 7.61–7.56 (m, 2H), 7.53–7.48 (m, 2H), 7.38–7.26 (m, 10H), 7.21–7.08 (m, 12H), 6.38 (s, 1H), 6.18 (s, 1H), 4.17 (dd, J = 18.1, 6.8 Hz, 1H), 3.56–3.35 (m, 4H), 3.28 (t, J = 14.2 Hz, 2H), 3.13–3.03 (m, 2H), 3.02–2.96 (m, 1H), 2.28 (s, 3H), 2.26 (s, 3H), 1.58 (s, 7H), 1.38 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 174.51, 174.00, 173.76, 173.29, 172.63, 172.49, 160.48, 159.29, 149.12, 148.67, 137.04, 136.92, 133.30, 133.14, 133.09, 132.60, 129.20, 129.13, 128.93, 128.91, 128.85, 128.74, 128.70, 128.66, 128.61, 128.53, 128.47, 128.02, 127.92, 127.75, 127.69, 127.25, 125.91, 125.70, 125.43, 125.41, 125.12, 119.63, 119.07, 103.61, 103.51, 61.59, 61.52, 59.86, 57.34, 53.71, 44.04, 43.24, 40.46, 39.44, 33.77, 30.78, 29.87, 29.68, 29.61, 15.32, 14.71. HRMS (ESI) m/z Calcd for C34H34N5O3, [M + H]+, 560.2656, found: 560.2659. Enantiomeric excess was determined to be 99% (determined by HPLC using chiral IA-H-OD-H column, hexane/2-propanol = 8/2, λ = 254 nm, 25 °C, 0.6 mL/min, tmajor = 113.3 min, tminor = 71.1 min).
(S)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)-3-((R)-4-(2-fluorobenzyl)-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)pyrrolidine-2,5-dione (3ha)
This was prepared according to the procedure within 1.5 h as a white solid (126.6 mg, 99% yield, dr = 1:1), mp 113.1–113.9 °C; = −42.005 (c 0.89, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.99–7.93 (m, 4H), 7.76–7.71 (m, 2H), 7.67 (ddd, J = 8.1, 3.3, 1.2 Hz, 4H), 7.64–7.55 (m, 4H), 7.55–7.44 (m, 5H), 7.42–7.28 (m, 9H), 7.23–7.03 (m, 6H), 6.92–6.81 (m, 4H), 6.28 (s, 1H), 5.54 (s, 1H), 4.36–4.14 (m, 2H), 4.02 (dd, J = 9.3, 7.4 Hz, 1H), 3.81–3.71 (m, 2H), 3.66 (d, J = 14.2 Hz, 1H), 3.52 (d, J = 13.7 Hz, 1H), 3.18 (dd, J = 17.8, 9.4 Hz, 1H), 2.99 (dd, J = 18.5, 9.7 Hz, 1H), 2.82 (s, 1H), 1.52 (s, 9H), 1.38 (s, 9H). 13C NMR (151 MHz, CDCl3) δ: 174.85, 173.83, 173.72, 173.42, 173.01, 172.89, 161.74 (d, J = 8.08 Hz), 160.14, 160.06, 158.39, 157.77, 148.94, 148.63, 137.06, 136.88, 133.29, 133.23, 131.29, 131.26, 131.20, 131.18, 131.02, 130.71, 130.64, 129.88, 129.83, 129.73, 129.67, 129.54, 129.28, 129.22, 129.08, 129.05, 128.93, 128.79, 128.50, 128.47, 128.40, 127.74, 127.69, 127.62, 127.09, 126.89, 126.27, 126.04, 125.39, 125.35, 124.15, 124.12, 124.08, 120.70, 120.60, 120.03, 119.93, 119.84, 119.78, 119.70, 115.69, 115.67, 115.53, 103.71, 61.50, 56.25, 49.45, 45.07, 44.90, 43.50, 33.77, 32.63, 31.04, 30.09, 29.81, 29.75, 29.69, 29.67, 17.67. 19F NMR (376 MHz, CDCl3) δ: −113.91, −114.43. HRMS (ESI) m/z Calcd for C39H35FN5O3, [M + H]+, 640.2718, found: 640.2726. Enantiomeric excess was determined to be 94% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 9/1, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 86.7 min, tminor = 34.5 min).
(S)-3-((R)-4-(2-bromobenzyl)-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3ia)
This was prepared according to the procedure within 2 h as a white solid (137.1 mg, 98% yield, dr = 1:1), mp 125.1–125.9 °C; = −7.206 (c 0.68, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.99–7.95 (m, 2H), 7.93–7.89 (m, 2H), 7.87–7.82 (m, 2H), 7.78–7.73 (m, 2H), 7.69–7.65 (m, 2H), 7.63–7.58 (m, 3H), 7.55–7.41 (m, 10H), 7.40–7.35 (m, 3H), 7.34–7.30 (m, 2H), 7.29–7.26 (m, 2H), 7.23–7.19 (m, 2H), 7.07 (ddtd, J = 28.0, 14.7, 7.3, 1.8 Hz, 6H), 6.29 (d, J = 2.9 Hz, 1H), 5.37 (s, 1H), 4.26 (d, J = 7.9 Hz, 1H), 4.21–4.03 (m, 3H), 3.89 (d, J = 14.4 Hz, 1H), 3.77 (q, J = 7.1 Hz, 2H), 3.17 (dd, J = 17.5, 9.0 Hz, 1H), 2.97 (dd, J = 18.6, 9.6 Hz, 1H), 2.72 (s, 1H), 1.53 (s, 9H), 1.33 (s, 9H). 13C NMR (151 MHz, CDCl3) δ: 173.73, 173.70, 172.98, 172.93, 172.27, 158.50, 157.85, 148.93, 148.65, 137.16, 137.05, 133.55, 133.37, 133.29, 133.23, 132.93, 131.10, 130.80, 130.74, 130.71, 130.33, 129.95, 129.49, 129.37, 129.25, 129.13, 129.07, 128.92, 128.84, 128.76, 128.69, 128.46, 128.30, 127.76, 127.67, 127.60, 127.56, 127.24, 126.23, 126.04, 125.77, 125.40, 125.33, 119.77, 119.54, 103.65, 103.63, 61.49, 61.47, 56.05, 45.22, 39.18, 37.65, 30.88, 30.09, 29.82, 29.65. HRMS (ESI) m/z Calcd for C39H35BrN5O3, [M + H]+, 700.1918, found: 700.1926. Enantiomeric excess was determined to be 96% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 9/1, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 51.7 min, tminor = 37.6 min).
(S)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)-3-((R)-4-(2-nitrobenzyl)-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)pyrrolidine-2,5-dione (3ja)
This was prepared according to the procedure within 1.2 h as a white solid (129.3 mg, 97% yield, dr = 1:1), mp 120.1–120.9 °C; = −42.404 (c 0.44, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.97–7.89 (m, 5H), 7.78–7.72 (m, 3H), 7.68–7.64 (m, 4H), 7.63–7.59 (m, 3H), 7.55 (t, J = 7.6 Hz, 3H), 7.50 (dd, J = 5.2, 2.0 Hz, 4H), 7.47–7.41 (m, 2H), 7.40 (s, 1H), 7.39–7.33 (m, 6H), 7.32 (s, 1H), 7.31–7.26 (m, 4H), 7.22 (dd, J = 5.6, 3.2 Hz, 3H), 6.27 (s, 1H), 5.37 (s, 1H), 4.43 (q, J = 14.2 Hz, 2H), 4.18 (d, J = 14.1 Hz, 1H), 4.09 (q, J = 4.4, 3.8 Hz, 2H), 4.06 (s, 1H), 3.73 (s, 1H), 3.24–3.12 (m, 1H), 2.93 (dd, J = 18.6, 9.6 Hz, 1H), 2.61 (s, 1H), 1.52 (s, 9H), 1.31 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 174.87, 173.65, 173.42, 172.82, 172.12, 157.74, 157.28, 149.97, 148.93, 148.64, 136.88, 136.81, 133.27, 133.20, 133.03, 132.65, 132.32, 132.01, 131.27, 131.08, 130.42, 130.20, 129.54, 129.27, 129.09, 128.99, 128.91, 128.81, 128.72, 128.48, 128.44, 128.26, 127.78, 127.69, 127.60, 127.43, 126.69, 126.32, 126.13, 125.46, 125.37, 125.32, 119.61, 119.55, 103.61, 61.52, 61.47, 56.51, 45.05, 35.22, 34.06, 30.89, 30.03, 29.80, 29.62. HRMS (ESI) m/z Calcd for C39H35N6O5, [M + H]+, 667.2663, found: 667.2673. Enantiomeric excess was determined to be 86% (determined by HPLC using chiral IG-H column, hexane/2-propanol = 7/3, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 55.8 min, tminor = 22.1 min).
(S)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)-3-((R)-4-(3-methylbenzyl)-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)pyrrolidine-2,5-dione (3ka)
This was prepared according to the procedure within 1 h as a white solid (120.7 mg, 95% yield, dr = 1:1), mp 109.1–109.9 °C; = −40.141 (c 0.14, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 8.04–7.97 (m, 4H), 7.71 (d, J = 7.8 Hz, 4H), 7.66–7.52 (m, 10H), 7.38 (ddt, J = 16.5, 12.1, 7.9 Hz, 9H), 7.26–7.20 (m, 2H), 7.03–6.80 (m, 9H), 6.33 (s, 1H), 5.55 (s, 1H), 4.32–4.12 (m, 2H), 4.02 (dd, J = 9.4, 7.3 Hz, 1H), 3.85–3.75 (m, 1H), 3.60 (dd, J = 13.5, 4.0 Hz, 2H), 3.51 (d, J = 13.2 Hz, 1H), 3.22 (dd, J = 17.8, 9.4 Hz, 1H), 3.05 (dd, J = 18.4, 9.6 Hz, 1H), 2.10 (d, J = 8.6 Hz, 6H), 1.56 (s, 9H), 1.42 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 173.90, 173.75, 173.62, 173.24, 158.24, 148.92, 148.64, 138.00, 137.01, 136.87, 133.17, 132.35, 131.15, 130.94, 130.65, 130.10, 129.32, 129.08, 128.95, 128.78, 128.69, 128.62, 128.46, 128.25, 128.20, 127.66, 127.59, 127.02, 126.27, 126.24, 126.02, 120.05, 119.87, 103.67, 61.48, 57.11, 44.90, 41.26, 31.07, 30.04, 29.80, 29.67, 21.16, 21.10. HRMS (ESI) m/z Calcd for C40H38N5O3, [M + H]+, 636.2969, found: 636.2976. Enantiomeric excess was determined to be 96% (determined by HPLC using chiral IB-H column, hexane/2-propanol = 4/1, λ = 254 nm, 25 °C, 0.6 mL/min, tmajor = 51.1 min, tminor = 41.1 min).
(S)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)-3-((R)-4-(4-methylbenzyl)-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)pyrrolidine-2,5-dione (3la)
This was prepared according to the procedure within 1.5 h as a white solid (124.5 mg, 98% yield, dr = 1:1), mp 112.1–112.9 °C; [ = −30.363 (c 0.85, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 8.04–7.95 (m, 4H), 7.67 (ddd, J = 8.1, 5.5, 1.2 Hz, 4H), 7.65–7.58 (m, 5H), 7.58– 7.47 (m, 5H), 7.41–7.25 (m, 10H), 7.24–7.16 (m, 2H), 6.97 (d, J = 7.8 Hz, 2H), 6.93–6.84 (m, 6H), 6.29 (s, 1H), 5.50 (s, 1H), 4.20 (dd, J = 18.6, 6.7 Hz, 2H), 3.96 (dd, J = 9.3, 7.3 Hz, 1H), 3.74 (dd, J = 9.3, 5.5 Hz, 1H), 3.57 (dd, J = 13.6, 10.3 Hz, 2H), 3.46 (d, J = 13.3 Hz, 1H), 3.16 (dd, J = 17.9, 9.4 Hz, 1H), 2.99 (dd, J = 18.5, 9.7 Hz, 1H), 2.83 (s, 1H), 2.16 (d, J = 2.5 Hz, 6H), 1.37 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 174.84, 173.87, 173.72, 173.55, 173.19, 173.09, 158.20, 157.51, 148.93, 148.65, 137.58, 137.39, 137.09, 136.93, 133.34, 133.27, 131.15, 131.04, 130.94, 130.65, 130.19, 129.40, 129.35, 129.19, 129.12, 129.07, 128.96, 128.82, 128.67, 128.46, 127.66, 127.60, 127.02, 126.23, 125.99, 125.39, 125.34, 120.07, 119.91, 103.67, 61.48, 57.20, 44.95, 43.63, 40.90, 39.63, 31.04, 30.02, 29.80, 29.67, 20.99. HRMS (ESI) m/z Calcd for C40H38N5O3, [M + H]+, 636.2969, found: 636.2976. Enantiomeric excess was determined to be 99% (determined by HPLC using chiral ID-H-OD-H column, hexane/2-propanol = 7/3, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 48.6 min, tminor = 31.9 min).
(S)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)-3-((R)-4-(4-methoxybenzyl)-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)pyrrolidine-2,5-dione (3ma)
This was prepared according to the procedure within 1.5 h as a white solid (129.0 mg, 99% yield, dr = 1:1), mp 112.1–112.9 °C; = −29.344 (c 0.78, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 8.04–7.96 (m, 4H), 7.71–7.65 (m, 4H), 7.64–7.58 (m, 5H), 7.58–7.48 (m, 5H), 7.41–7.27 (m, 9H), 7.24–7.17 (m, 2H), 7.04–6.98 (m, 2H), 6.96–6.91 (m, 2H), 6.64–6.57 (m, 4H), 6.29 (s, 1H), 5.51 (s, 1H), 4.20 (dd, J = 18.1, 7.4 Hz, 2H), 3.96 (dd, J = 9.3, 7.3 Hz, 1H), 3.74 (dd, J = 9.0, 5.8 Hz, 1H), 3.63 (d, J = 2.0 Hz, 6H), 3.56 (dd, J = 18.2, 13.7 Hz, 2H), 3.44 (d, J = 13.3 Hz, 1H), 3.16 (dd, J = 17.8, 9.4 Hz, 1H), 3.00 (dd, J = 18.5, 9.6 Hz, 1H), 2.83 (s, 1H), 1.52 (s, 9H), 1.38 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 174.85, 173.91, 173.75, 173.57, 173.22, 173.11, 159.15, 159.02, 158.20, 157.51, 148.92, 148.65, 137.08, 136.91, 133.33, 133.25, 131.15, 131.04, 130.97, 130.67, 130.45, 129.39, 129.15, 128.98, 128.83, 128.70, 128.46, 127.67, 127.60, 127.56, 126.99, 126.24, 126.00, 125.39, 125.34, 125.23, 124.45, 120.02, 119.88, 113.81, 113.76, 103.67, 61.49, 57.30, 55.11, 44.88, 43.59, 40.52, 39.26, 31.04, 30.00, 29.80, 29.68. HRMS (ESI) m/z Calcd for C40H38N5O4, [M + H]+, 652.2918, found: 652.2922. Enantiomeric excess was determined to be 99% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 9/1, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 74.5 min, tminor = 51.3 min).
(S)-3-((R)-4-(3,5-bis(trifluoromethyl)benzyl)-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3na)
This was prepared according to the procedure within 2.5 h as a white solid (146.9 mg, 97% yield, dr = 1:1), mp 112.1–112.9 °C; = 1.1617 (c 0.80, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.94 (d, J = 7.4 Hz, 4H), 7.72–7.47 (m, 20H), 7.41–7.30 (m, 10H), 7.23 (d, J = 8.7 Hz, 2H), 6.32 (s, 1H), 5.58 (s, 1H), 4.23 (s, 2H), 4.11 (t, J = 8.2 Hz, 1H), 3.81 (t, J = 7.3 Hz, 1H), 3.70 (dd, J = 22.0, 13.6 Hz, 2H), 3.60 (d, J = 13.1 Hz, 1H), 3.21 (dd, J = 17.6, 9.1 Hz, 1H), 3.03 (dd, J = 18.6, 9.7 Hz, 1H), 2.78 (s, 1H), 1.56 (s, 9H), 1.35 (s, 9H). 13C NMR (151 MHz, CDCl3) δ: 173.53, 173.32, 172.71, 172.68, 172.32, 157.34, 156.70, 148.97, 148.70, 136.49, 136.38, 135.93, 135.13, 133.21, 133.09, 131.96, 131.79, 131.74, 131.57, 131.52, 131.35, 131.29, 130.36 (d, J=14.14 Hz), 129.75, 129.62, 129.42, 129.10, 128.84, 128.63, 128.53, 128.50, 128.29, 127.78, 127.70, 127.13, 126.63, 126.60, 126.44, 125.51, 125.37, 125.30, 123.74, 123.69, 121.93, 121.89, 121.83, 119.66, 119.48, 103.64, 103.59, 61.58, 61.53, 56.82, 44.86, 43.37, 40.35, 39.14, 30.90, 29.81, 29.77, 29.62. 19F NMR (376 MHz, CDCl3) δ: −63.14, −63.17. HRMS (ESI) m/z Calcd for C41H34F6N5O3, [M + H]+, 758.2560, found: 758.2570. Enantiomeric excess was determined to be 95% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 9/1, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 41.5 min, tminor = 24.1 min).
(S)-1-(1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)-3-((R)-4-(naphthalen-2-ylmethyl)-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)pyrrolidine-2,5-dione (3oa)
This was prepared according to the procedure within 4 h as a white solid (99.4 mg, 74% yield, dr = 1:1), mp 116.1–116.9 °C; = −66.632 (c 0.96, CH2Cl2). 1H NMR (600 MHz, Chloroform-d) δ: 8.19 (d, J = 8.3 Hz, 1H), 8.08–8.03 (m, 1H), 7.96–7.93 (m, 2H), 7.92–7.89 (m, 2H), 7.76–7.71 (m, 2H), 7.69–7.56 (m, 8H), 7.52–7.38 (m, 13H), 7.37–7.26 (m, 11H), 7.24–7.14 (m, 9H), 7.13–7.09 (m, 1H), 6.32 (s, 1H), 5.49 (s, 1H), 4.60 (s, 1H), 4.40 (s, 1H), 4.29 (d, J = 15.0 Hz, 1H), 4.17 (dd, J = 15.4, 6.8 Hz, 2H), 4.00–3.86 (m, 2H), 3.29 (dd, J = 17.9, 9.4 Hz, 1H), 3.04 (dd, J = 18.9, 9.9 Hz, 1H), 2.88 (s, 1H), 1.56 (d, J = 2.0 Hz, 9H), 1.34 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ: 173.91, 173.71, 173.46, 173.02, 158.36, 148.97, 148.62, 136.74, 136.68, 133.82, 133.75, 133.33, 133.25, 131.82, 131.68, 131.31, 131.06, 130.97, 130.60, 129.91, 129.35, 129.06, 128.98, 128.88, 128.79, 128.73, 128.69, 128.54, 128.51, 128.45, 127.90, 127.66, 127.58, 127.04, 126.74, 126.17, 126.04, 125.97, 125.75, 125.70, 125.39, 125.34, 124.92, 124.76, 123.67, 123.50, 120.06, 119.71, 103.67, 103.63, 61.51, 61.47, 56.60, 45.40, 43.90, 36.34, 34.65, 31.09, 30.26, 29.84, 29.65. HRMS (ESI) m/z Calcd for C43H38N5O3, [M + H]+, 672.2969, found: 672.2981. Enantiomeric excess was determined to be 94% (determined by HPLC using chiral IB-H column, hexane/2-propanol = 4/1, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 56.4 min, tminor = 47.9 min).
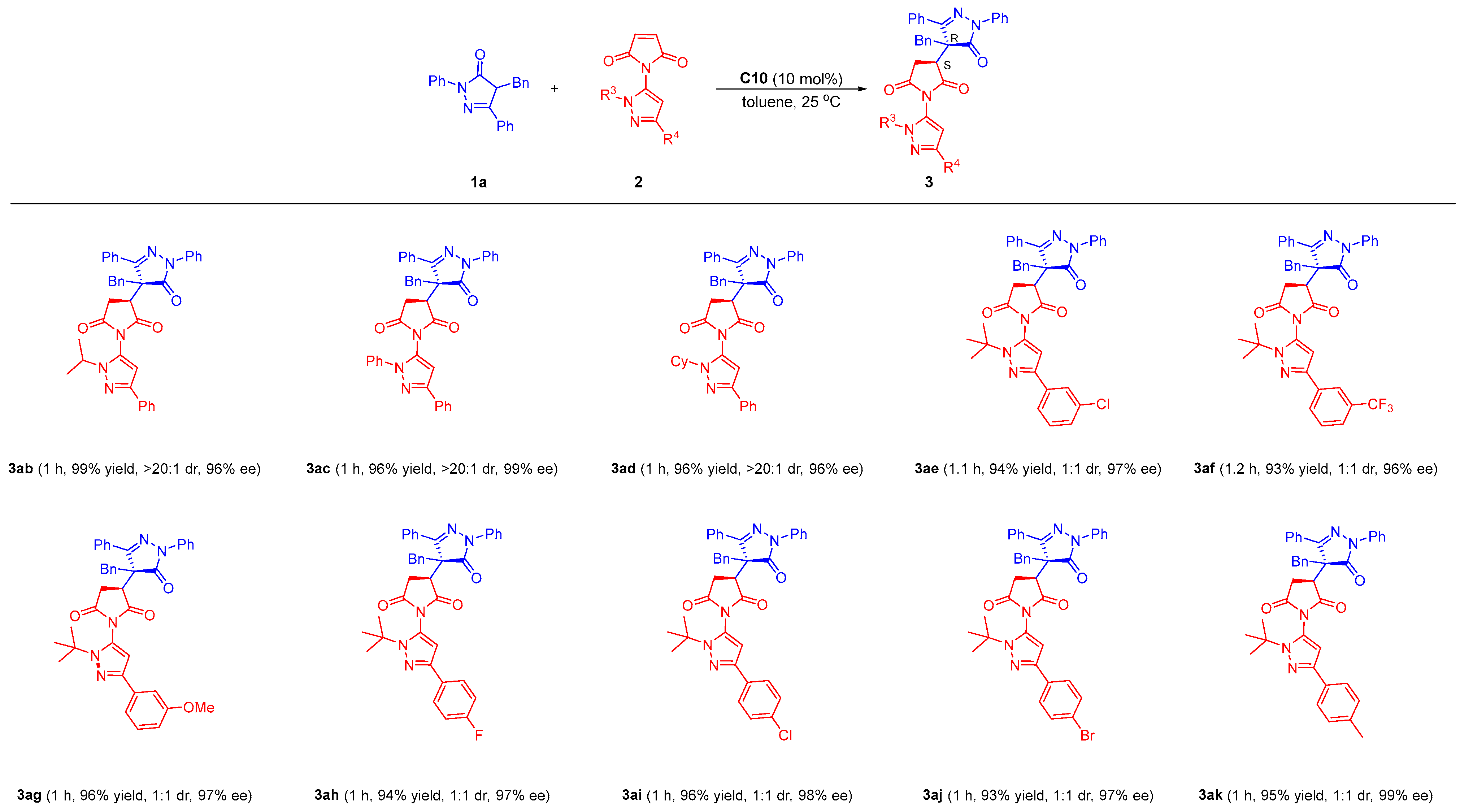
(S)-3-((R)-4-benzyl-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-isopropyl-3-phenyl-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3ab)
This was prepared according to the procedure within 1 h as a white solid (120.2 mg, 99% yield, dr > 20:1), mp 105.1–105.9 °C; = 9.067 (c 1.15, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 8.04–7.95 (m, 2H), 7.76–7.62 (m, 4H), 7.53 (dt, J = 5.3, 2.7 Hz, 3H), 7.34 (q, J = 7.4 Hz, 4H), 7.27 (d, J = 1.4 Hz, 1H), 7.20 (dd, J = 7.4 Hz, 1H), 7.15–7.01 (m, 5H), 6.17 (s, 1H), 3.94 (dd, J = 9.4, 5.7 Hz, 1H), 3.82 (s, 2H), 3.56 (d, J = 13.4 Hz, 1H), 3.16 (dd, J = 18.3, 9.7 Hz, 1H), 1.58 (s, 1H), 1.35 (d, J = 6.6 Hz, 3H), 1.10 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 173.72, 173.51, 172.82, 150.49, 136.88, 133.39, 132.76, 130.98, 130.85, 129.31, 129.21, 128.83, 128.49, 128.40, 128.12, 127.91, 127.68, 127.17, 126.14, 125.51, 119.77, 100.97, 50.25, 44.09, 40.68, 30.51, 22.49, 22.11. HRMS (ESI) m/z Calcd for C38H34N5O3, [M + H]+, 608.2656, found: 608.2666. Enantiomeric excess was determined to be 96% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 7/3, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 25.3 min, tminor = 13.6 min).
(S)-3-((R)-4-benzyl-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1,3-diphenyl-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3ac)
This was prepared according to the procedure within 1 h as a white solid (123.1 mg, 96% yield, dr > 20:1), mp 108.1–108.9 °C; = 11.915 (c 1.13, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.98–7.91 (m, 2H), 7.78–7.72 (m, 2H), 7.59–7.48 (m, 5H), 7.43–7.26 (m, 9H), 7.22 (d, J = 11.9 Hz, 2H), 7.12–6.99 (m, 5H), 6.28 (s, 1H), 4.02–3.09 (m, 4H), 2.95 (dd, J = 18.1, 9.4 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 173.51, 173.01, 172.25, 157.43, 151.85, 137.82, 136.86, 132.51, 131.02, 130.82, 129.78, 129.42, 129.32, 129.25, 128.82, 128.64, 128.35, 128.32, 127.84, 127.19, 126.19, 125.68, 124.19, 120.15, 103.42, 44.16, 40.63, 30.50. HRMS (ESI) m/z Calcd for C41H32N5O3, [M + H]+, 642.2500, found: 642.2507. Enantiomeric excess was determined to be 99% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 7/3, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 33.7 min, tminor = 22.2 min).
(S)-3-((R)-4-benzyl-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-cyclohexyl-3-phenyl-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3ad)
This was prepared according to the procedure within 1 h as a white solid (124.3 mg, 96% yield, dr > 20:1), mp 112.1–112.9 °C; = 10.511 (c 1.05, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.99 (dd, J = 6.6, 3.0 Hz, 2H), 7.71–7.65 (m, 2H), 7.63–7.57 (m, 2H), 7.56–7.49 (m, 3H), 7.33 (t, J = 7.4 Hz, 4H), 7.27 (d, J = 1.4 Hz, 1H), 7.18 (dd, J = 7.4 Hz, 1H), 7.09 (q, J = 5.0, 3.3 Hz, 5H), 6.16 (s, 1H), 3.96 (t, J = 7.9 Hz, 1H), 3.78 (s, 1H), 3.55 (d, J = 13.3 Hz, 1H), 3.47 (dt, J = 11.3, 6.7 Hz, 1H), 3.17 (dd, J = 18.2, 9.6 Hz, 1H), 1.87 (q, J = 6.6 Hz, 4H), 1.56–0.76 (m, 6H). 13C NMR (151 MHz, CDCl3) δ: 173.86, 172.95, 157.77, 150.33, 136.82, 133.41, 130.97, 130.82, 129.36, 129.25, 128.81, 128.51, 128.39, 128.25, 127.95, 127.67, 127.10, 126.24, 125.52, 120.25, 100.94, 57.88, 44.11, 40.89, 32.82, 32.72, 30.52, 25.39, 25.07. HRMS (ESI) m/z Calcd for C41H38N5O3, [M + H]+, 648.2969, found: 648.2978. Enantiomeric excess was determined to be 96% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 7/3, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 20.6 min, tminor = 11.3 min).
(S)-3-((R)-4-benzyl-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-(3-chlorophenyl)-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3ae)
This was prepared according to the procedure within 1.1 h as a white solid (123.2 mg, 94% yield, dr = 1:1), mp 120.1–120.9 °C; = −17.123 (c 0.22, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 8.03–7.97 (m, 4H), 7.71–7.45 (m, 15H), 7.42–7.27 (m, 5H), 7.24–7.16 (m, 4H), 7.13–7.05 (m, 8H), 7.04–7.00 (m, 2H), 6.28 (s, 1H), 5.45 (s, 1H), 4.23 (d, J = 26.3 Hz, 2H), 4.00 (dd, J = 9.4, 7.4 Hz, 1H), 3.80–3.74 (m, 1H), 3.61 (dd, J = 18.0, 13.5 Hz, 2H), 3.50 (d, J = 13.2 Hz, 1H), 3.19 (dd, J = 17.8, 9.4 Hz, 1H), 3.02 (dd, J = 18.4, 9.7 Hz, 1H), 2.83 (s, 1H), 1.52 (s, 9H), 1.38 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 174.73, 173.82, 173.63, 173.42, 173.01, 158.05, 157.35, 147.57, 147.29, 136.97, 136.79, 135.09, 134.99, 134.44, 133.23, 132.45, 131.07, 131.03, 131.00, 130.73, 129.75, 129.42, 129.34, 129.17, 129.04, 128.99, 128.72, 128.45, 128.39, 127.96, 127.81, 127.62, 127.58, 126.98, 126.39, 126.06, 125.39, 123.47, 123.36, 120.01, 119.87, 103.89, 103.86, 61.78, 61.75, 57.11, 44.94, 43.72, 41.24, 40.01, 31.03, 30.04, 29.78, 29.65. HRMS (ESI) m/z Calcd for C39H35ClN5O3, [M + H]+, 656.2423, found: 656.2434. Enantiomeric excess was determined to be 97% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 8/2, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 36.5 min, tminor = 19.4 min).
(S)-3-((R)-4-benzyl-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-(3-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3af)
This was prepared according to the procedure within 1 h as a white solid (128.2 mg, 93% yield, dr = 1:1), mp 118.1–118.9 °C; = −24.115 (c 0.68, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 8.03–7.97 (m, 4H), 7.92 (s, 1H), 7.86–7.79 (m, 3H), 7.66 (d, J = 8.1 Hz, 2H), 7.62 (d, J = 7.0 Hz, 1H), 7.58 (dd, J = 8.3, 3.0 Hz, 4H), 7.51 (dtd, J = 11.6, 8.9, 7.8, 3.9 Hz, 6H), 7.44 (d, J = 8.1 Hz, 1H), 7.36 (dt, J = 16.5, 7.8 Hz, 4H), 7.24–7.17 (m, 2H), 7.13–7.06 (m, 8H), 7.03 (d, J = 7.0 Hz, 2H), 6.35 (s, 1H), 5.46 (s, 1H), 4.31–4.15 (m, 2H), 4.00 (dd, J = 9.4, 7.4 Hz, 1H), 3.78 (dd, J = 9.3, 5.4 Hz, 1H), 3.62 (dd, J = 18.9, 13.5 Hz, 2H), 3.51 (d, J = 13.2 Hz, 1H), 3.20 (dd, J = 17.9, 9.4 Hz, 1H), 3.03 (dd, J = 18.5, 9.7 Hz, 1H), 2.83 (s, 1H), 1.54 (s, 10H), 1.39 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 174.72, 173.81, 173.62, 173.43, 173.01, 158.05, 157.32, 147.51, 147.25, 136.97, 136.78, 134.05, 133.97, 133.24, 132.43, 131.06, 131.00, 130.95, 130.74, 130.67, 129.44, 129.33, 129.20, 129.17, 128.98, 128.93, 128.87, 128.72, 128.46, 128.39, 127.97, 127.81, 127.64, 126.97, 126.37, 126.07, 124.19, 122.14, 120.02, 119.85, 103.92, 103.89, 61.88, 61.85, 57.10, 44.96, 43.68, 41.25, 40.00, 31.07, 30.05, 29.78, 29.65. 19F NMR (376 MHz, CDCl3) δ: −62.62, −62.70. HRMS (ESI) m/z Calcd for C40H35F3N5O3, [M + H]+, 690.2687, found: 690.2694. Enantiomeric excess was determined to be 96% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 8/2, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 34.9 min, tminor = 16.9 min).
(S)-3-((R)-4-benzyl-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-(3-methoxyphenyl)-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3ag)
This was prepared according to the procedure within 1 h as a white solid (125.0 mg, 96% yield, dr = 1:1), mp 114.1–114.9 °C; = −37.751 (c 0.25, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.98 (d, J = 7.6 Hz, 4H), 7.67–7.46 (m, 10H), 7.34 (dt, J = 15.4, 7.7 Hz, 4H), 7.26–7.15 (m, 8H), 7.07 (d, J = 9.5 Hz, 8H), 7.00 (d, J = 7.5 Hz, 2H), 6.85–6.75 (m, 2H), 6.29 (s, 1H), 5.50 (s, 1H), 4.21 (q, J = 7.8, 7.3 Hz, 2H), 3.97 (t, J = 8.3 Hz, 1H), 3.80 (d, J = 21.3 Hz, 7H), 3.58 (dd, J = 13.6, 6.5 Hz, 2H), 3.47 (d, J = 13.2 Hz, 1H), 3.15 (dd, J = 17.8, 9.4 Hz, 1H), 3.00 (dd, J = 18.5, 9.6 Hz, 1H), 1.52 (s, 9H), 1.35 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 174.88, 173.92, 173.79, 173.47, 173.10, 159.83, 159.78, 158.11, 157.40, 148.78, 148.50, 137.01, 136.82, 134.73, 134.63, 133.31, 132.53, 131.09, 131.04, 131.00, 130.73, 129.54, 129.44, 129.37, 129.19, 129.00, 128.85, 128.73, 128.45, 128.38, 127.94, 127.81, 127.60, 126.99, 126.34, 126.07, 120.04, 119.91, 118.07, 118.01, 113.42, 110.77, 103.90, 103.88, 61.55, 57.12, 55.29, 55.25, 44.94, 43.60, 41.23, 40.04, 31.05, 30.03, 29.82, 29.70. HRMS (ESI) m/z Calcd for C40H38N5O4, [M + H]+, 652.2918, found: 562.2926. Enantiomeric excess was determined to be 97% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 8/2, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 43.0 min, tminor = 23.3 min).
(S)-3-((R)-4-benzyl-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-(4-fluorophenyl)-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3ah)
This was prepared according to the procedure within 1 h as a white solid (120.2 mg, 94% yield, dr = 1:1), mp 104.1–104.9 °C; = −24.525 (c 0.58, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.99 (ddt, J = 6.7, 4.2, 1.9 Hz, 4H), 7.67–7.48 (m, 14H), 7.41–7.30 (m, 4H), 7.23– 7.16 (m, 2H), 7.13–6.96 (m, 14H), 6.24 (s, 1H), 5.47 (s, 1H), 4.22 (dd, J = 17.9, 7.2 Hz, 2H), 3.99 (dd, J = 9.3, 7.4 Hz, 1H), 3.81–3.72 (m, 1H), 3.61 (dd, J = 18.5, 13.5 Hz, 2H), 3.49 (d, J = 13.2 Hz, 1H), 3.18 (dd, J = 17.8, 9.4 Hz, 1H), 3.02 (dd, J = 18.4, 9.6 Hz, 1H), 1.52 (s, 9H), 1.37 (s, 9H). 13C NMR (151 MHz, CDCl3) δ: 173.86, 173.71, 173.42, 173.09, 173.04, 163.36, 163.32, 161.72, 161.69, 158.05, 157.35, 148.07, 147.79, 136.97, 136.82, 133.22, 132.45, 131.08, 131.01, 130.95, 130.72, 129.55, 129.48, 129.39, 129.34, 129.17, 128.98, 128.89, 128.71, 128.57, 128.44, 128.39, 127.96, 127.81, 127.56, 127.07, 127.01, 126.99, 126.96, 126.28, 126.06, 120.02, 119.88, 115.37 (d, J = 15.15Hz), 103.47, 103.43, 61.55, 57.11, 44.93, 41.24, 40.04, 31.03, 30.04, 29.79, 29.66. 19F NMR (376 MHz, CDCl3) δ: −114.70, −114.83. HRMS (ESI) m/z Calcd for C39H35FN5O3, [M + H]+, 640.2718, found: 640.2731. Enantiomeric excess was determined to be 97% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 8/2, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 34.2 min, tminor = 18.0 min).
(S)-3-((R)-4-benzyl-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-(4-chlorophenyl)-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3ai)
This was prepared according to the procedure within 1 h as a white solid (125.8 mg, 96% yield, dr = 1:1), mp 101.1–101.9 °C; = −29.557 (c 0.77, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 8.07–8.00 (m, 4H), 7.71–7.66 (m, 2H), 7.65–7.52 (m, 12H), 7.43–7.29 (m, 8H), 7.27–7.18 (m, 2H), 7.11 (q, J = 6.8, 5.1 Hz, 8H), 7.09–7.02 (m, 2H), 6.30 (s, 1H), 5.51 (s, 1H), 4.26 (d, J = 25.9 Hz, 2H), 4.02 (dd, J = 9.3, 7.4 Hz, 1H), 3.83–3.75 (m, 1H), 3.65 (dd, J = 21.6, 13.5 Hz, 2H), 3.53 (d, J = 13.2 Hz, 1H), 3.22 (dd, J = 17.8, 9.4 Hz, 1H), 3.05 (dd, J = 18.4, 9.6 Hz, 1H), 2.87 (s, 1H), 1.55 (s, 9H), 1.40 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 174.77, 173.84, 173.66, 173.41, 173.06, 173.03, 158.04, 157.34, 147.84, 147.55, 136.97, 136.82, 133.36, 133.30, 133.21, 132.44, 131.83, 131.76, 131.08, 131.02, 130.94, 130.72, 129.38, 129.33, 129.17, 128.98, 128.71, 128.64, 128.44, 128.39, 127.96, 127.81, 127.55, 126.98, 126.62, 126.57, 126.28, 126.06, 103.66, 103.63, 61.68, 57.10, 44.93, 43.70, 41.24, 40.04, 31.02, 30.04, 29.78, 29.65. HRMS (ESI) m/z Calcd for C39H35ClN5O3, [M + H]+, 656.2423, found: 656.2433. Enantiomeric excess was determined to be 98% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 8/2, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 34.4 min, tminor = 18.5 min).
(S)-3-((R)-4-benzyl-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(3-(4-bromophenyl)-1-(tert-butyl)-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3aj)
This was prepared according to the procedure within 1 h as a white solid (130.0 mg, 93% yield, dr = 1:1), mp 126.1–126.9 °C; = −27.602 (c 0.22, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 7.99 (ddt, J = 6.9, 5.4, 2.5 Hz, 4H), 7.68–7.62 (m, 2H), 7.61–7.49 (m, 10H), 7.48–7.30 (m, 10H), 7.24–7.16 (m, 2H), 7.14–6.99 (m, 10H), 6.27 (s, 1H), 5.49 (s, 1H), 4.23 (dd, J = 18.1, 7.4 Hz, 2H), 3.99 (dd, J = 9.4, 7.4 Hz, 1H), 3.82–3.75 (m, 1H), 3.61 (dd, J = 20.1, 13.5 Hz, 2H), 3.50 (d, J = 13.2 Hz, 1H), 3.19 (dd, J = 17.9, 9.4 Hz, 1H), 3.02 (dd, J = 18.3, 9.6 Hz, 1H), 2.84 (s, 1H), 1.52 (s, 9H), 1.37 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 173.83, 173.65, 173.41, 173.05, 173.02, 158.03, 157.33, 147.85, 147.55, 136.96, 136.81, 133.20, 132.43, 132.27, 132.20, 131.58, 131.07, 131.02, 130.94, 130.72, 129.38, 129.33, 129.17, 128.98, 128.71, 128.68, 128.44, 128.39, 127.96, 127.81, 127.54, 126.97, 126.93, 126.89, 126.28, 126.06, 121.55, 121.49, 120.02, 119.87, 103.66, 103.63, 61.70, 57.10, 44.93, 43.69, 41.24, 40.04, 31.02, 30.04, 29.78, 29.65. HRMS (ESI) m/z Calcd for C39H35BrN5O3, [M + H]+, 700.1918, found: 700.1924. Enantiomeric excess was determined to be 97% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 8/2, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 36.0 min, tminor = 19.1 min).
(S)-3-((R)-4-benzyl-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)-1-(1-(tert-butyl)-3-(p-tolyl)-1H-pyrazol-5-yl)pyrrolidine-2,5-dione (3ak)
This was prepared according to the procedure within 1 h as a white solid (120.7 mg, 95% yield, dr = 1:1), mp 125.1–125.9 °C; = −39.223 (c 0.28, CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ: 8.01–7.92 (m, 4H), 7.67–7.62 (m, 2H), 7.62–7.45 (m, 12H), 7.33 (dt, J = 16.2, 7.9 Hz, 4H), 7.23–7.17 (m, 2H), 7.17–7.09 (m, 5H), 7.06 (d, J = 11.2 Hz, 7H), 6.99 (dd, J = 7.9, 1.8 Hz, 2H), 6.26 (s, 1H), 5.49 (s, 1H), 4.24–4.12 (m, 2H), 3.94 (dd, J = 9.3, 7.3 Hz, 1H), 3.75 (dd, J = 8.9, 5.8 Hz, 1H), 3.57 (d, J = 14.2 Hz, 2H), 3.45 (d, J = 13.2 Hz, 1H), 3.12 (dd, J = 17.8, 9.4 Hz, 1H), 2.98 (dd, J = 18.4, 9.6 Hz, 1H), 2.33 (s, 3H), 2.29 (s, 3H), 1.51 (s, 9H), 1.37 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 174.89, 173.95, 173.82, 173.48, 173.17, 173.12, 158.13, 157.45, 149.05, 148.77, 137.39, 137.32, 137.05, 136.88, 133.35, 132.58, 131.14, 131.03, 130.71, 130.61, 130.53, 129.42, 129.39, 129.21, 129.18, 129.01, 128.78, 128.73, 128.45, 128.42, 128.38, 127.93, 127.81, 127.59, 127.02, 126.31, 126.06, 125.34, 125.29, 120.06, 119.92, 103.47, 61.41, 57.15, 44.95, 43.67, 41.23, 40.06, 31.04, 30.03, 29.83, 29.73, 21.34, 21.30. HRMS (ESI) m/z Calcd for C40H38N5O3, [M + H]+, 636.2969, found: 636.2976. Enantiomeric excess was determined to be 99% (determined by HPLC using chiral OD-H column, hexane/2-propanol = 8/2, λ = 254 nm, 25 °C, 0.8 mL/min, tmajor = 31.7 min, tminor = 16.6 min).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

