
HPLC-PDA Method for Quantification of Bioactive Compounds in Crude Extract and Fractions of Aucklandia costus Falc. and Cytotoxicity Studies against Cancer Cells


Abstract
1. Introduction
2. Results
2.1. Structure Elucidation of Marker Compounds Isolated from A. costus
2.2. Identification of Marker Compounds in Crude Extract
2.3. Correlation Coefficient (r2), Limit of Detection (LOD) and Limit of Quantification (LOQ)
2.4. Quantification of Marker Compounds in Crude Extract and Fractions
System Suitability
2.5. Intra- and Interday Precision
2.6. Analyte Recoveries
2.7. Specificity
2.8. Stability of Analyte Solutions
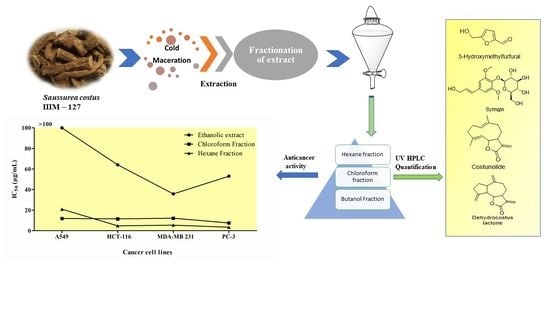
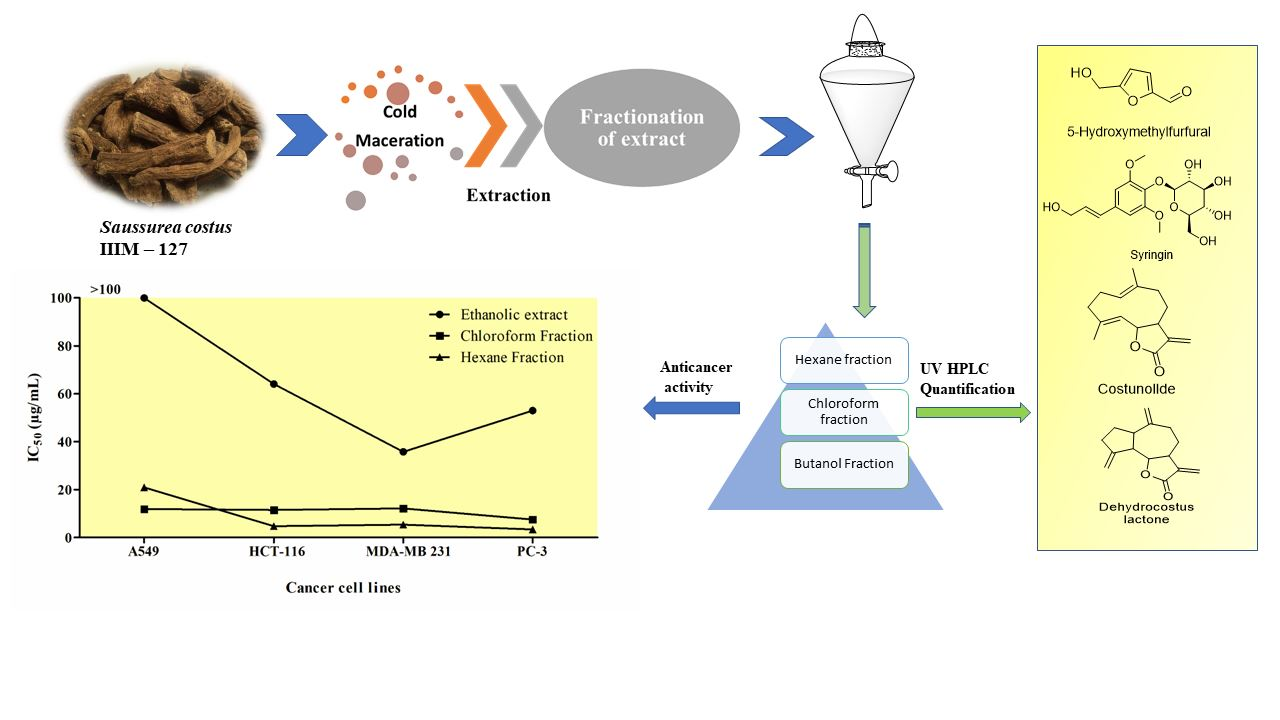
2.9. Anticancer Activity of A. costus Crude Extract and Fractions
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Chemicals, Reagents and Standard Compounds
4.3. NMR and HRMS Instrumentation
4.4. Extraction, Fractionation and Isolation of Standard Compounds
4.5. HPLC Instrumentation and Chromatographic Conditions
4.6. Preparation of Standard Solution and the Calibration Curve
4.7. Cell Culture
4.8. Cytotoxicity Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.A.P.; Thornburg, C.C.; Henrich, C.J.; Grkovic, T.; O’Keefe, B.R. Creating and screening natural product libraries. Nat. Prod. Rep. 2020, 37, 893–918. [Google Scholar] [CrossRef]
- Katiyar, C.; Gupta, A.; Kanjilal, S.; Katiyar, S. Drug discovery from plant sources: An integrated approach. Ayu 2012, 33, 10–19. [Google Scholar] [CrossRef]
- Lautié, E.; Russo, O.; Ducrot, P.; Boutin, J.A. Unraveling Plant Natural Chemical Diversity for Drug Discovery Purposes. Front. Pharmacol. 2020, 11, 397. [Google Scholar] [CrossRef]
- Altemimi, A.; Lakhssassi, N.; Baharlouei, A.; Watson, D.G.; Lightfoot, D.A. Phytochemicals: Extraction, Isolation, and Identification of Bioactive Compounds from Plant Extracts. Plants 2017, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan, S.; Chen, Y.; Saravanan, D.; Sundram, K.M.; Yoga Latha, L. Extraction, isolation and characterization of bioactive compounds from plants’ extracts. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 1–10. [Google Scholar] [CrossRef]
- Jan, R.; Asaf, S.; Numan, M.; Lubna, K.K.-M. Plant Secondary Metabolite Biosynthesis and Transcriptional Regulation in Response to Biotic and Abiotic Stress Conditions. Agronomy 2021, 11, 968. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Lahlou, M. The Success of Natural Products in Drug Discovery. Pharm. Pharmacol. 2013, 4, 15. [Google Scholar] [CrossRef]
- Shapiro, K.; Gong, W.C. Natural products used for diabetes. J. Am. Pharm. Assoc. 2002, 42, 217–226. [Google Scholar] [CrossRef]
- Rehman, M.U.; Wali, A.F.; Ahmad, A.; Shakeel, S.; Rasool, S.; Ali, R.; Rashid, S.M.; Madkhali, H.; Ganaie, M.A.; Khan, R. Neuroprotective Strategies for Neurological Disorders by Natural Products: An update. Curr. Neuropharmacol. 2019, 17, 247–267. [Google Scholar] [CrossRef]
- Thulasi, G.P.; Cherupally, K.K.N.; Devi, P.U. Natural Drugs in DNA Repair. In DNA Repair; Maddalena, M., Ed.; IntechOpen: Rijeka, Croatia, 2019; p. Ch. 10. [Google Scholar]
- Ooi, B.K.; Chan, K.G.; Goh, B.H.; Yap, W.H. The Role of Natural Products in Targeting Cardiovascular Diseases via Nrf2 Pathway: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2018, 9, 1308. [Google Scholar] [CrossRef]
- Tabatabaei-Malazy, O.; Larijani, B.; Abdollahi, M. Targeting metabolic disorders by natural products. J. Diabetes Metab. Disord. 2015, 14, 57. [Google Scholar] [CrossRef]
- Dudics, S.; Langan, D.; Meka, R.R.; Venkatesha, S.H.; Berman, B.M.; Che, C.T.; Moudgil, K.D. Natural Products for the Treatment of Autoimmune Arthritis: Their Mechanisms of Action, Targeted Delivery, and Interplay with the Host Microbiome. J. Diabetes Metab. Disord. 2018, 19, 2508. [Google Scholar] [CrossRef]
- Deng, W.; Du, H.; Liu, D.; Ma, Z. Editorial: The Role of Natural Products in Chronic Inflammation. Front. Pharmacol. 2022, 13, 901538–901541. [Google Scholar] [CrossRef]
- Demain, A.L.; Vaishnav, P. Natural products for cancer chemotherapy. Microb. Biotechnol. 2011, 4, 687–699. [Google Scholar] [CrossRef]
- Luo, L.; Yang, J.; Wang, C.; Wu, J.; Li, Y.; Zhang, X.; Li, H.; Zhang, H.; Zhou, Y.; Lu, A.; et al. Natural products for infectious microbes and diseases: An overview of sources, compounds, and chemical diversities. Sci. China Life Sci. 2022, 65, 1123–1145. [Google Scholar] [CrossRef]
- Shah, S.C.; Kayamba, V.; Peek, R.M., Jr.; Heimburger, D. Cancer Control in Low- and Middle-Income Countries: Is It Time to Consider Screening? J. Glob. Oncol. 2019, 5, 1–8. [Google Scholar] [CrossRef]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.; Silva, J.; Pinteus, S.; Gaspar, H.; Alpoim, M.C.; Botana, L.M.; Pedrosa, R. From Marine Origin to Therapeutics: The Antitumor Potential of Marine Algae-Derived Compounds. Front. Pharmacol. 2018, 9, 777. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wei, Q.; Zhou, Y.; Wang, J.; Liu, Q.; Xu, H. A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. 2017, 11, 87. [Google Scholar] [CrossRef]
- Tungmunnithum, D.; Thongboonyou, A.; Pholboon, A.; Yangsabai, A. Flavonoids and Other Phenolic Compounds from Medicinal Plants for Pharmaceutical and Medical Aspects: An Overview. Medicine 2018, 5, 93. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J. Natural products and drug discovery. Natl. Sci. Rev. 2022, 9, nwac206. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Li, S.-J.; Zhang, Z.-X.; Zhang, M.; Shi, Q.-W.; Gu, Y.-C.; Dong, M.; Kiyota, H. Chemical constituents from the genus Saussurea and their biological activities. Heterocycl. Commun. 2017, 23, 331–358. [Google Scholar] [CrossRef]
- Butola, J.; Samant, S. Saussurea Species in Indian Himalayan Region: Diversity, Distribution and Indigenous Uses. Int. J. Plant Biol. 2010, 1, e9. [Google Scholar] [CrossRef]
- Rathore, S.; Debnath, P.; Kumar, R. Kuth Saussurea costus (Falc.) Lipsch.: A critically endangered medicinal plant from Himalaya. J. Appl. Res. Med. Aromat. Plants. 2021, 20, 100277. [Google Scholar] [CrossRef]
- Kuniyal, C.; Rawat, D.; Sundriyal, R.C. Cultivation of Saussurea costus cannot be treated as ‘artificially propagated’. Curr. Sci. 2015, 108, 1587–1589. [Google Scholar]
- Su, X.; Yao, Z.; Li, S.; Sun, H. Synergism of Chinese Herbal Medicine: Illustrated by Danshen Compound. Evid. Based Complement. Altern. Med. 2016, 2016, 7279361. [Google Scholar] [CrossRef]
- Pandey, M.M.; Rastogi, S.; Rawat, A.K.S. Saussurea costus: Botanical, chemical and pharmacological review of an ayurvedic medicinal plant. J. Ethnopharmacol. 2007, 110, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Zahara, K.; Tabassum, S.; Sabir, S.; Arshad, M.; Qureshi, R.; Amjad, M.S.; Chaudhari, S.K. A review of therapeutic potential of Saussurea lappa-An endangered plant from Himalaya. Asian. Pac. J. Trop. Med. 2014, 7 (Suppl. S1), S60–S69. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Alqaseer, K.; Fatlawi, D.; Shehab, S.; Falah, M.; Hassan, M.; Shnain, W.; Radhi, O. Saussurea lappa: An Important Medicinal Plant for Treatment Different Diseases: A review. Int. J. Nurs. Sci. 2021, 11, 1–19. [Google Scholar] [CrossRef]
- Shati, A.A.; Alkahtani, M.A.; Alfaifi, M.Y.; Elbehairi, S.E.I.; Elsaid, F.G.; Prasanna, R.; Mir, M.A. Secondary Metabolites of Saussurea costus Leaf Extract Induce Apoptosis in Breast, Liver, and Colon Cancer Cells by Caspase-3-Dependent Intrinsic Pathway. Biomed. Res. Int. 2020, 2020, 1608942. [Google Scholar] [CrossRef] [PubMed]
- Mujammami, M. Clinical significance of Saussurea Costus in thyroid treatment. Saudi Med. J. 2020, 41, 1047–1053. [Google Scholar] [CrossRef]
- Moujir, L.; Callies, O.; Sousa, P.M.C.; Sharopov, F.; Seca, A.M.L. Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases. Appl. Sci. 2020, 10, 3001. [Google Scholar] [CrossRef]
- Kovács, B.; Hohmann, J.; Csupor-Löffler, B.; Kiss, T.; Csupor, D. A comprehensive phytochemical and pharmacological review on sesquiterpenes from the genus Ambrosia. Heliyon 2022, 8, e09884. [Google Scholar] [CrossRef]
- Wang, Y.F.; Ni, Z.Y.; Dong, M.; Cong, B.; Shi, Q.W.; Gu, Y.C.; Kiyota, H. Secondary metabolites of plants from the genus Saussurea: Chemistry and biological activity. Chem. Biodivers. 2010, 7, 2623–2659. [Google Scholar] [CrossRef]
- Kumar, B.R. Application of HPLC and ESI-MS techniques in the analysis of phenolic acids and flavonoids from green leafy vegetables (GLVs). J. Pharm. Anal. 2017, 7, 349–364. [Google Scholar] [CrossRef]
- Li, A.; Sun, A.; Liu, R. Preparative isolation and purification of costunolide and dehydrocostuslactone from Aucklandia lappa Decne by high-speed countercurrent chromatography. J. Chromatogr. A 2005, 1076, 193–197. [Google Scholar] [CrossRef]
- Robinson, A.; Kumar, T.V.; Sreedhar, E.; Naidu, V.G.M.; Krishna, S.R.; Babu, K.S.; Srinivas, P.V.; Rao, J.M. A new sesquiterpene lactone from the roots of Saussurea lappa: Structure-anticancer activity study. Bioorg. Med. Chem. Lett. 2008, 18, 4015–4017. [Google Scholar] [CrossRef] [PubMed]
- Sundaram Chinna Krishnan, S.; Pillai Subramanian, I.; Pillai Subramanian, S. Isolation, characterization of syringin, phenylpropanoid glycoside from Musa paradisiaca tepal extract and evaluation of its antidiabetic effect in streptozotocin-induced diabetic rats. Biomed. Prev. Nutr. 2014, 4, 105–111. [Google Scholar] [CrossRef]
- Yamada, P.; Nemoto, M.; Shigemori, H.; Yokota, S.; Isoda, H. Isolation of 5-(hydroxymethyl)furfural from Lycium chinense and its inhibitory effect on the chemical mediator release by basophilic cells. Planta. Med. 2011, 77, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Alquadeib, B.T. Development and validation of a new HPLC analytical method for the determination of diclofenac in tablets. Saudi. Pharm. J. 2019, 27, 66–70. [Google Scholar] [CrossRef]
- Khagga, B.; Kaitha, M.; Dammu, R.; Mogili, S. ICH guidelines—“Q” series (quality guidelines)—A review. GSC Biol. Pharm. Sci. 2019, 6, 89–106. [Google Scholar] [CrossRef]
- Seyed Mojtaba, M.; Sussan, G. Linearity of Calibration Curves for Analytical Methods: A Review of Criteria for Assessment of Method Reliability. In Calibration and Validation of Analytical Methods; Mark, T.S., Ed.; IntechOpen: Rijeka, Croatia, 2018; p. Ch. 6. [Google Scholar]
- Shrivastava, A. Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chron. Young Sci. 2011, 2, 21–25. [Google Scholar] [CrossRef]
- Jo, B.-G.; Park, Y.-H.; Kim, K.H.; Kim, S.-N.; Yang, M.H. Simultaneous Determination of Four Marker Compounds in Lobelia chinensis Lour. Extract by HPLC-PDA. Appl. Sci. 2021, 11, 12080. [Google Scholar] [CrossRef]
- Seo, J.H.; Kim, J.E.; Shim, J.H.; Yoon, G.; Bang, M.A.; Bae, C.S.; Lee, K.J.; Park, D.H.; Cho, S.S. HPLC Analysis, Optimization of Extraction Conditions and Biological Evaluation of Corylopsis coreana Uyeki Flos. Molecules 2016, 21, 94. [Google Scholar] [CrossRef]
- Taverniers, I.; Van Bockstaele, E.; De Loose, M. Analytical method validation and quality assurance. In Pharmaceutical Sciences Encyclopedia: Drug Discovery, Development, and Manufacturing; John and Wiley and Sons: Hoboken, NJ, USA, 2010; pp. 1–48. [Google Scholar]
- Vilkickyte, G.; Raudone, L. Optimization, Validation and Application of HPLC-PDA Methods for Quantification of Triterpenoids in Vaccinium vitis-idaea L. Molecules 2021, 26, 1645. [Google Scholar] [CrossRef]
- Yang, N.Y.; Duan, J.A.; Shang, E.X.; Tian, L.J. Analysis of sesquiterpene lactones in Eupatorium lindleyanum by HPLC-PDA-ESI-MS/MS. Phytochem. Anal. 2010, 21, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Karasová, G.; Kowalska, S.; Lehotay, J.; Buszewski, B. Mobile-phase pH influence on the retention of some benzoic acid derivatives in reversed-phase chromatography. J. Sep. Sci. 2006, 29, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Choi, B.Y. Costunolide-A Bioactive Sesquiterpene Lactone with Diverse Therapeutic Potential. Int. J. Mol. Sci. 2019, 20, 2926. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Z.; Xie, Y.; Hu, H. Antitumor activity and mechanism of costunolide and dehydrocostus lactone: Two natural sesquiterpene lactones from the Asteraceae family. Biomed. Pharmacother. 2020, 125, 109955. [Google Scholar] [CrossRef]
- Butturini, E.; Cavalieri, E.; Carcereri de Prati, A.; Darra, E.; Rigo, A.; Shoji, K.; Murayama, N.; Yamazaki, H.; Watanabe, Y.; Suzuki, H.; et al. Two Naturally Occurring Terpenes, Dehydrocostuslactone and Costunolide, Decrease Intracellular GSH Content and Inhibit STAT3 Activation. PLoS ONE 2011, 6, e20174. [Google Scholar] [CrossRef]
- Zheng, H.; Chen, Y.; Zhang, J.; Wang, L.; Jin, Z.; Huang, H.; Man, S.; Gao, W. Evaluation of protective effects of costunolide and dehydrocostuslactone on ethanol-induced gastric ulcer in mice based on multi-pathway regulation. Chem. Biol. Interact. 2016, 250, 68–77. [Google Scholar] [CrossRef]
- Hossin, A.Y.; Inafuku, M.; Takara, K.; Nugara, R.N.; Oku, H. Syringin: A Phenylpropanoid Glycoside Compound in Cirsium brevicaule A. GRAY Root Modulates Adipogenesis. Molecules 2021, 26, 1531. [Google Scholar] [CrossRef]
- Chow, P.H.; Kourghi, M.; Pei, J.V.; Nourmohammadi, S.; Yool, A.J. 5-Hydroxymethyl-Furfural and Structurally Related Compounds Block the Ion Conductance in Human Aquaporin-1 Channels and Slow Cancer Cell Migration and Invasion. Mol. Pharmacol. 2020, 98, 38. [Google Scholar] [CrossRef]
- Abdilla-Santes, R.M.; Winkelman, J.G.M.; van Zandvoort, I.; Weckhuysen, B.M.; Bruijnincx, P.C.A.; Jurak, E.; Deuss, P.J.; Heeres, H.J. 5-Hydroxy-2-Methylfurfural from Sugar Beet Thick Juice: Kinetic and Modeling Studies. ACS Sustain. Chem. Eng. 2021, 9, 2626–2638. [Google Scholar] [CrossRef]
- Shapla, U.M.; Solayman, M.; Alam, N.; Khalil, M.I.; Gan, S.H. 5-Hydroxymethylfurfural (HMF) levels in honey and other food products: Effects on bees and human health. Chem. Cent. J. 2018, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, P.K.; Doble, M. Synergistic effect of phytochemicals in combination with hypoglycemic drugs on glucose uptake in myotubes. Phytomedicine 2009, 16, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Peng, Z.; Su, C. Potential Anti-Cancer Activities and Mechanisms of Costunolide and Dehydrocostuslactone. Int. J. Mol. Sci. 2015, 16, 10888–10906. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
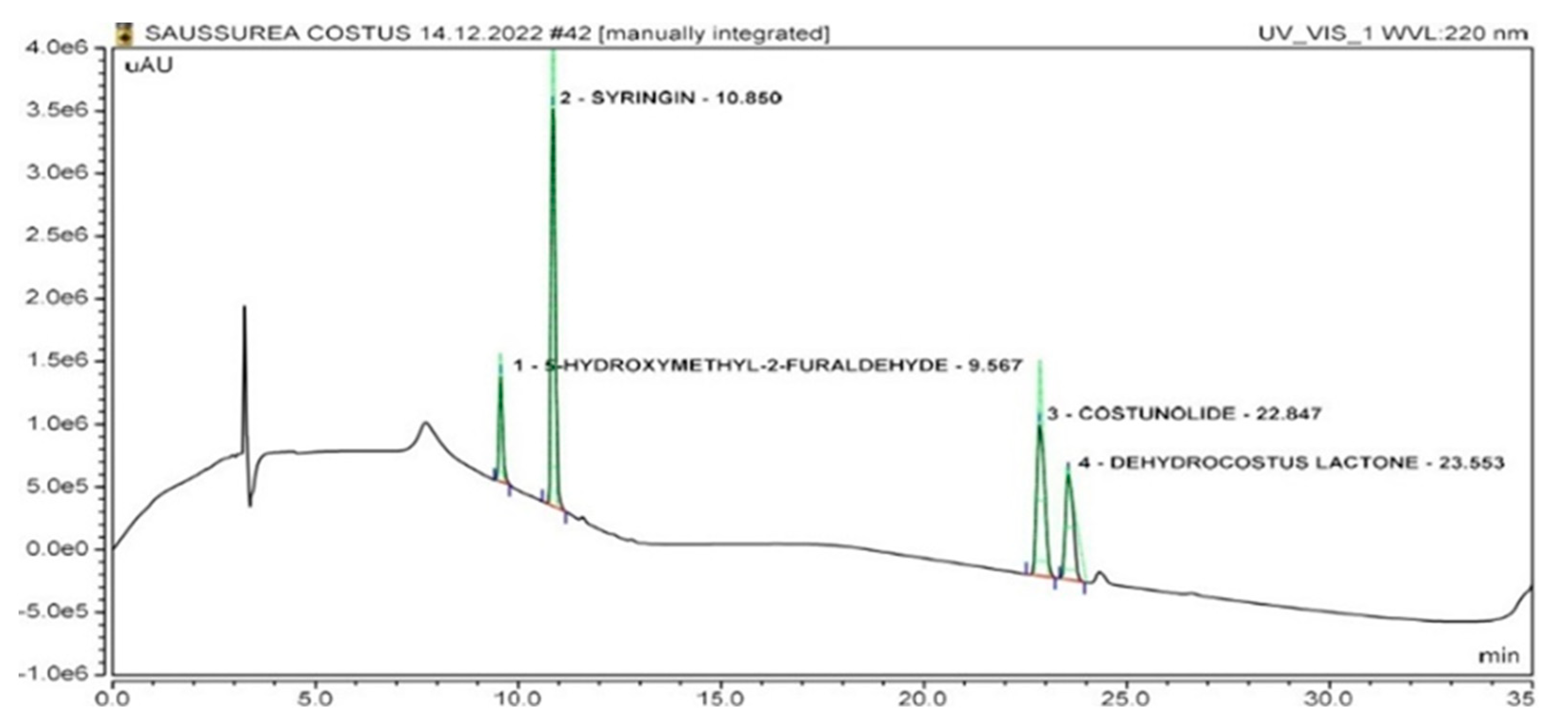
| No. | Compound | Retention Time (Minutes) | r2 | LOD (µg/mL) | LOQ (µg/mL) |
|---|---|---|---|---|---|
| 1. | Dehydrocostus lactone | 23.55 ± 0.04 | 0.9998 | 0.22 | 0.66 |
| 2. | Costunolide | 22.84 ± 0.04 | 0.9999 | 0.12 | 0.36 |
| 3. | Syringin | 10.85 ± 0.04 | 0.9938 | 0.11 | 0.33 |
| 4. | 5-Hydroxymethyl-2-furaldehyde | 9.56 ± 0.04 | 0.9998 | 0.30 | 0.90 |
| No. | Compound | Asymmetry | S/N Ratio | No. of Theoretical Plates |
|---|---|---|---|---|
| 1. | Dehydrocostus lactone | 1.30 | 52 | 56922 |
| 2. | Costunolide | 1.56 | 44.3 | 49607 |
| 3. | Syringin | 1.39 | 46 | 54850 |
| 4. | 5-Hydroxymethyl-2-furaldehyde | 1.41 | 62.2 | 60769 |
| Precision Intraday | Interday | |||||
|---|---|---|---|---|---|---|
| No | Compound | Actual Conc. of Analyte (µg/mL) | Observed Conc. of Analyte (µg/mL) | RSD (%) | Observed Conc. of Analyte (µg/mL) | RSD (%) |
| 1. | Dehydrocostus lactone | 10.00 | 9.31 | 1.07 | 9.35 | 0.05 |
| 100.00 | 100.15 | 0.05 | 100.17 | 0.02 | ||
| 250.00 | 247.16 | 0.06 | 248.36 | 0.12 | ||
| 2. | Costunolide | 5.0 | 5.15 | 0.78 | 5.09 | 0.30 |
| 50.00 | 51.15 | 0.09 | 52.10 | 0.09 | ||
| 125.00 | 125.78 | 0.12 | 124.52 | 0.08 | ||
| 3. | Syringin | 10.0 | 10.60 | 1.70 | 10.65 | 0.20 |
| 100.00 | 101.50 | 0.33 | 100.57 | 0.28 | ||
| 250.00 | 252.81 | 0.23 | 241.68 | 0.34 | ||
| 4. | 5-Hydroxymethyl-2-furaldehyde | 5.00 | 5.64 | 1.51 | 5.19 | 1.85 |
| 50.00 | 50.25 | 0.19 | 50.16 | 0.05 | ||
| 125.00 | 125.24 | 0.18 | 124.91 | 0.22 | ||
| No | Compound | Original Conc. (µg/mL) | Spike Conc. (µg/mL) | Observed Conc. (µg/mL) | %Recovery ± SD | RSD (%) |
|---|---|---|---|---|---|---|
| 1. | Dehydrocostus lactone | 5.0 | 0.50 | 5.55 | 110.0 ± 2.00 | 1.81 |
| 0.85 | 5.92 | 108.96 ± 1.78 | 1.64 | |||
| 1.13 | 6.35 | 104.22 ± 1.76 | 1.69 | |||
| 2. | Costunolide | 4.50 | 0.85 | 5.33 | 97.62 ± 1.16 | 1.94 |
| 1.50 | 6.07 | 105.02 ± 1.46 | 1.39 | |||
| 2.0 | 6.54 | 102.38 ± 1.08 | 1.06 | |||
| 3. | Syringin | 3.0 | 0.45 | 3.45 | 101.77 ± 0.66 | 0.65 |
| 0.90 | 3.92 | 103.03 ± 0.75 | 0.73 | |||
| 1.35 | 4.41 | 102.43 ± 1.07 | 1.05 | |||
| 4. | 5-Hydroxymethyl-2-furaldehyde | 6.0 | 0.50 | 6.55 | 110.20 ± 2.00 | 1.81 |
| 1.25 | 7.21 | 97.52 ± 0.36 | 0.37 | |||
| 1.75 | 7.74 | 101.14 ± 0.431 | 0.42 |
| Peak Area (AU ± SD) | ||||||||
|---|---|---|---|---|---|---|---|---|
| No | Compound | Temp | 0 h | 6 h | 12 h | 24 h | 48 h | RSD% |
| 1. | Dehydrocostus lactone | R. T | 353,737.6 ± 219.4 | 351,436.3 ± 389.7 | 349,150.6 ± 277.3 | 341,010.6 ± 245.4 | 339,356.3 ± 251.2 | 1.84 |
| 4 °C | 353,461.3 ± 186.2 | 351,467.3 ± 188.4 | 349,076.6 ± 190.0 | 340,680 ± 95 | 338,091 ± 123.1 | 1.95 | ||
| 2. | Costunolide | R. T | 518,111.3 ± 230.9 | 517,746.6 ± 205.9 | 516,795.3 ± 179.1 | 504,090 ± 278.74 | 489,646 ± 327.04 | 2.22 |
| 4 °C | 518,090.3 ± 171.7 | 517,310.6 ± 174.18 | 516,308.3 ± 175.5 | 503,310 ± 295.46 | 488,431.6 ± 267.5 | 2.52 | ||
| 3. | Syringin | R. T | 322,582 ± 182.4 | 321,731 ± 295.9 | 321,069 ± 177.6 | 320,186.6 ± 255.9 | 305,507.3 ± 335.11 | 2.24 |
| 4 °C | 322,186 ± 212.03 | 321,571.3 ± 250.63 | 320,767 ± 258.2 | 319,787 ± 338.04 | 294,418.6 ± 267.23 | 3.78 | ||
| 4. | 5-Hydroxymethyl-2-furaldehyde | R. T | 150,886.6 ± 245.8 | 149,345.6 ± 200 | 147,433.3 ± 591.6 | 143,536.6 ± 228.10 | 141,447.3 ± 389 | 2.69 |
| 4 °C | 150,116.6 ± 309.8 | 148,952 ± 245.3 | 145,400.6 ± 443.5 | 142,965 ± 258.98 | 140,599.3 ± 379.6 | 2.73 | ||
| IC50 (µg/mL) ± SD | |||||
|---|---|---|---|---|---|
| No | Sample Code | A549 (Lung Cancer Cell Line) | HCT-116 (Colon Cancer Cell Line) | MDA-MB 231 (Human Breast Cancer Cell Line) | PC-3 (Prostate Cancer Cell Line) |
| 1 | Ethanolic extract | >100 | 64.05 ± 2.26 | 35.766 ± 2.20 | 52.998 ± 2.18 |
| 2 | Chloroform fraction | 11.875 ± 0.84 | 11.53 ± 0.42 | 12.179 ± 0.32 | 7.527 ± 0.18 |
| 3 | Hexane fraction | 20.904 ± 0.75 | 4.717 ± 0.16 | 5.353 ± 0.13 | 3.37 ± 0.14 |
| 4 | Butanol fraction | >100 | >100 | >100 | >100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhushan, A.; Rani, D.; Tabassum, M.; Kumar, S.; Gupta, P.N.; Gairola, S.; Gupta, A.P.; Gupta, P. HPLC-PDA Method for Quantification of Bioactive Compounds in Crude Extract and Fractions of Aucklandia costus Falc. and Cytotoxicity Studies against Cancer Cells. Molecules 2023, 28, 4815. https://doi.org/10.3390/molecules28124815
Bhushan A, Rani D, Tabassum M, Kumar S, Gupta PN, Gairola S, Gupta AP, Gupta P. HPLC-PDA Method for Quantification of Bioactive Compounds in Crude Extract and Fractions of Aucklandia costus Falc. and Cytotoxicity Studies against Cancer Cells. Molecules. 2023; 28(12):4815. https://doi.org/10.3390/molecules28124815
Chicago/Turabian StyleBhushan, Anil, Dixhya Rani, Misbah Tabassum, Saajan Kumar, Prem N. Gupta, Sumeet Gairola, Ajai P. Gupta, and Prasoon Gupta. 2023. "HPLC-PDA Method for Quantification of Bioactive Compounds in Crude Extract and Fractions of Aucklandia costus Falc. and Cytotoxicity Studies against Cancer Cells" Molecules 28, no. 12: 4815. https://doi.org/10.3390/molecules28124815
APA StyleBhushan, A., Rani, D., Tabassum, M., Kumar, S., Gupta, P. N., Gairola, S., Gupta, A. P., & Gupta, P. (2023). HPLC-PDA Method for Quantification of Bioactive Compounds in Crude Extract and Fractions of Aucklandia costus Falc. and Cytotoxicity Studies against Cancer Cells. Molecules, 28(12), 4815. https://doi.org/10.3390/molecules28124815