

Systematic Modification of the Substitution Pattern of the 7-Hydroxy-5-oxopyrazolo[4,3-b]pyridine-6-carboxamide Scaffold Enabled the Discovery of New Ligands with High Affinity and Selectivity for the Cannabinoid Type 2 Receptor

 , , , , ,
, , , , , 
 , and
, and 
Abstract
:1. Introduction
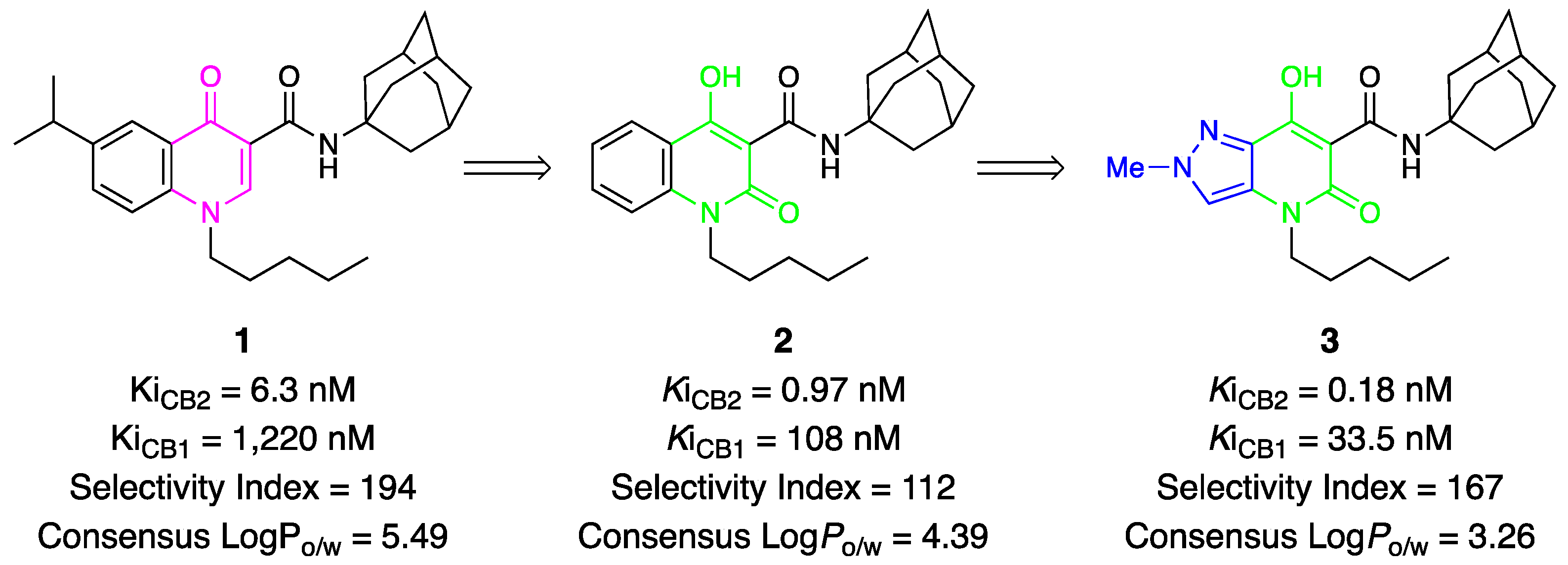
2. Results and Discussion
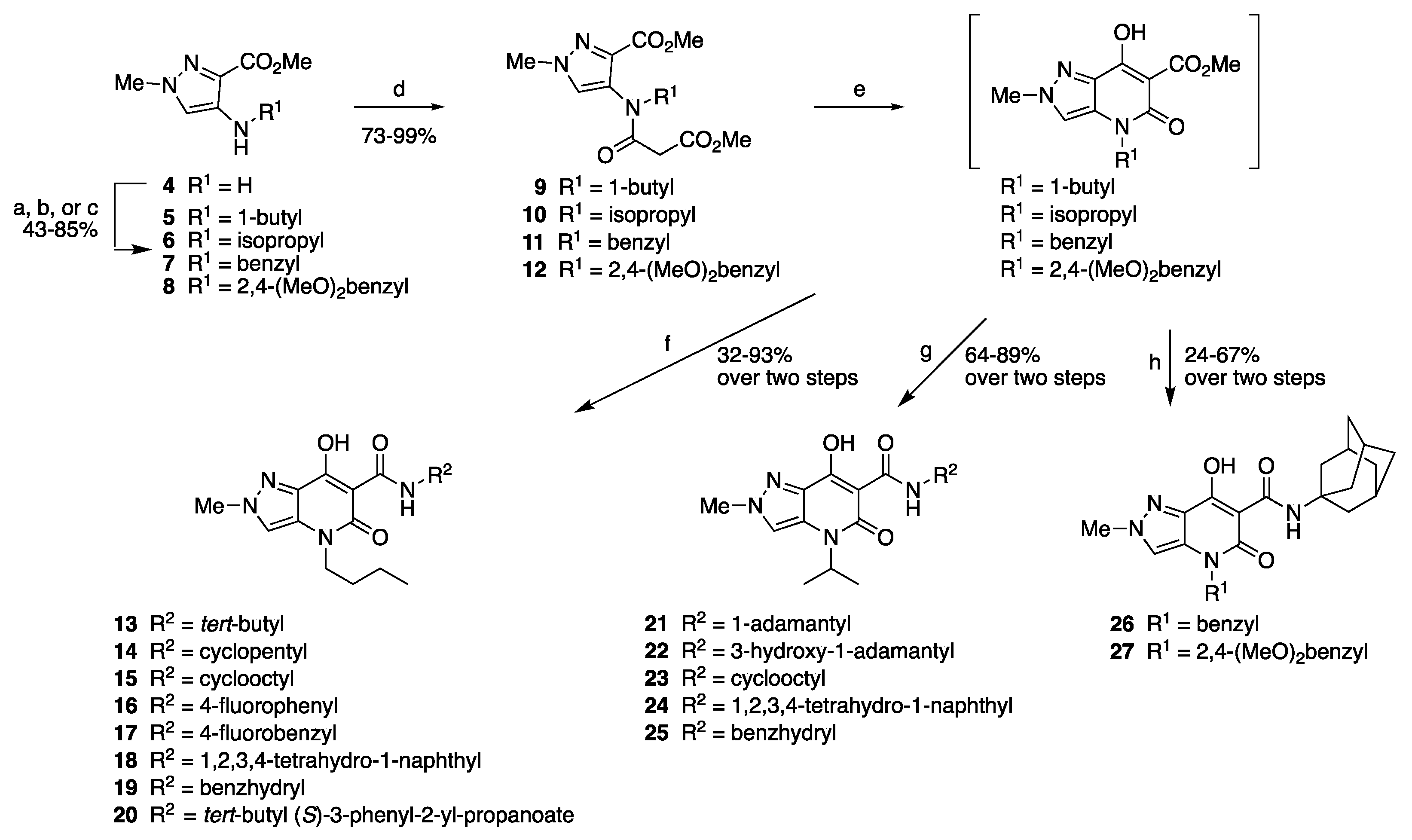
2.1. Synthesis
2.2. Estimated Drug-like Properties
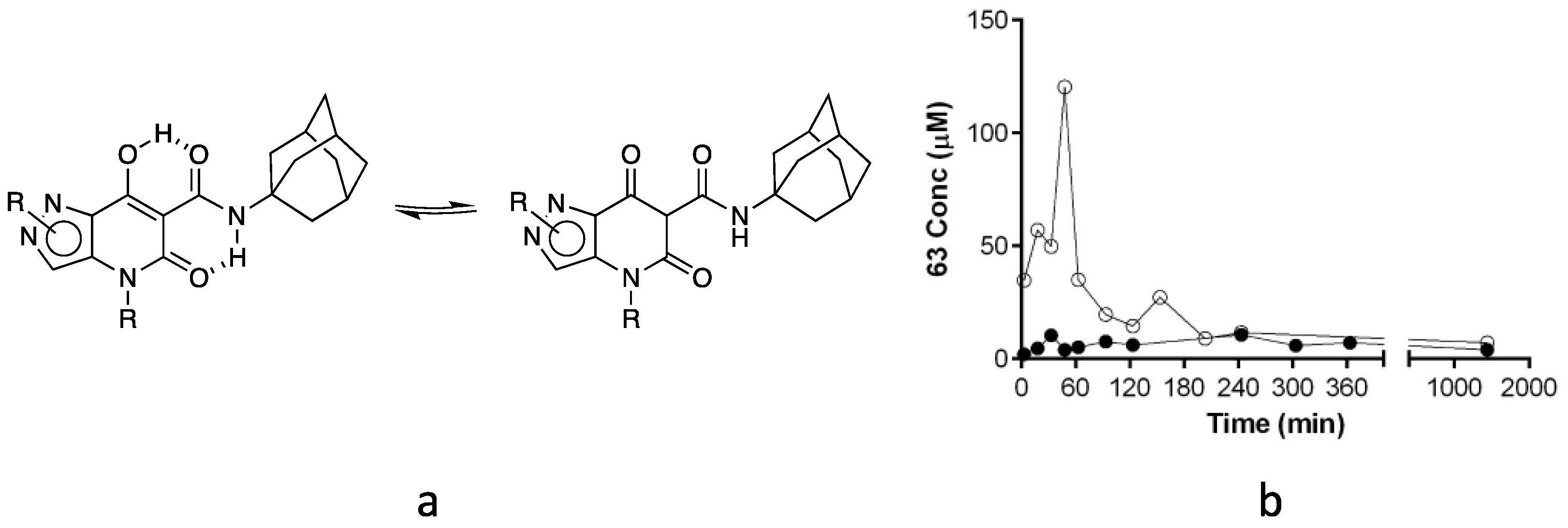
2.3. Water Solubility Evaluation
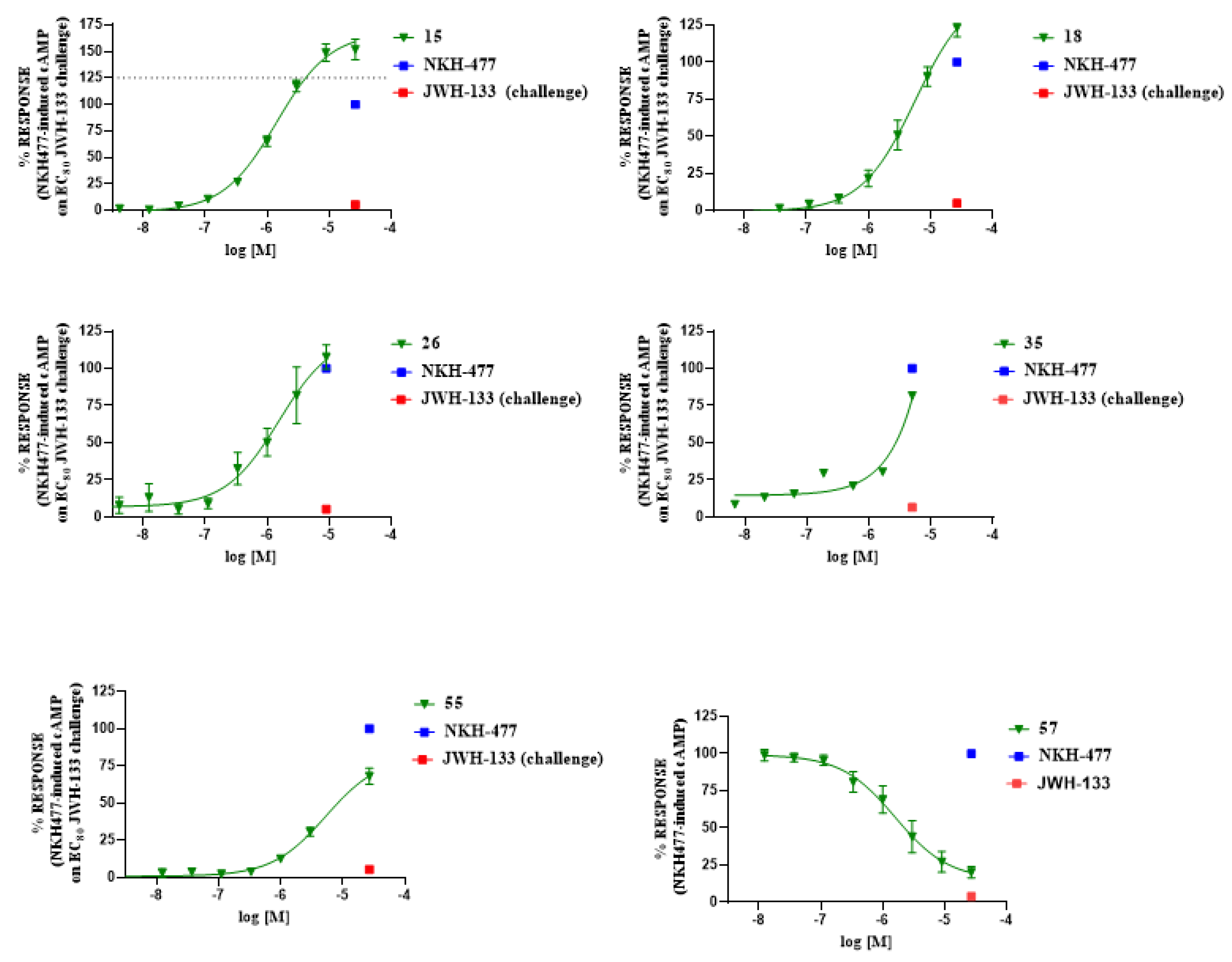
2.4. In Vitro Pharmacology
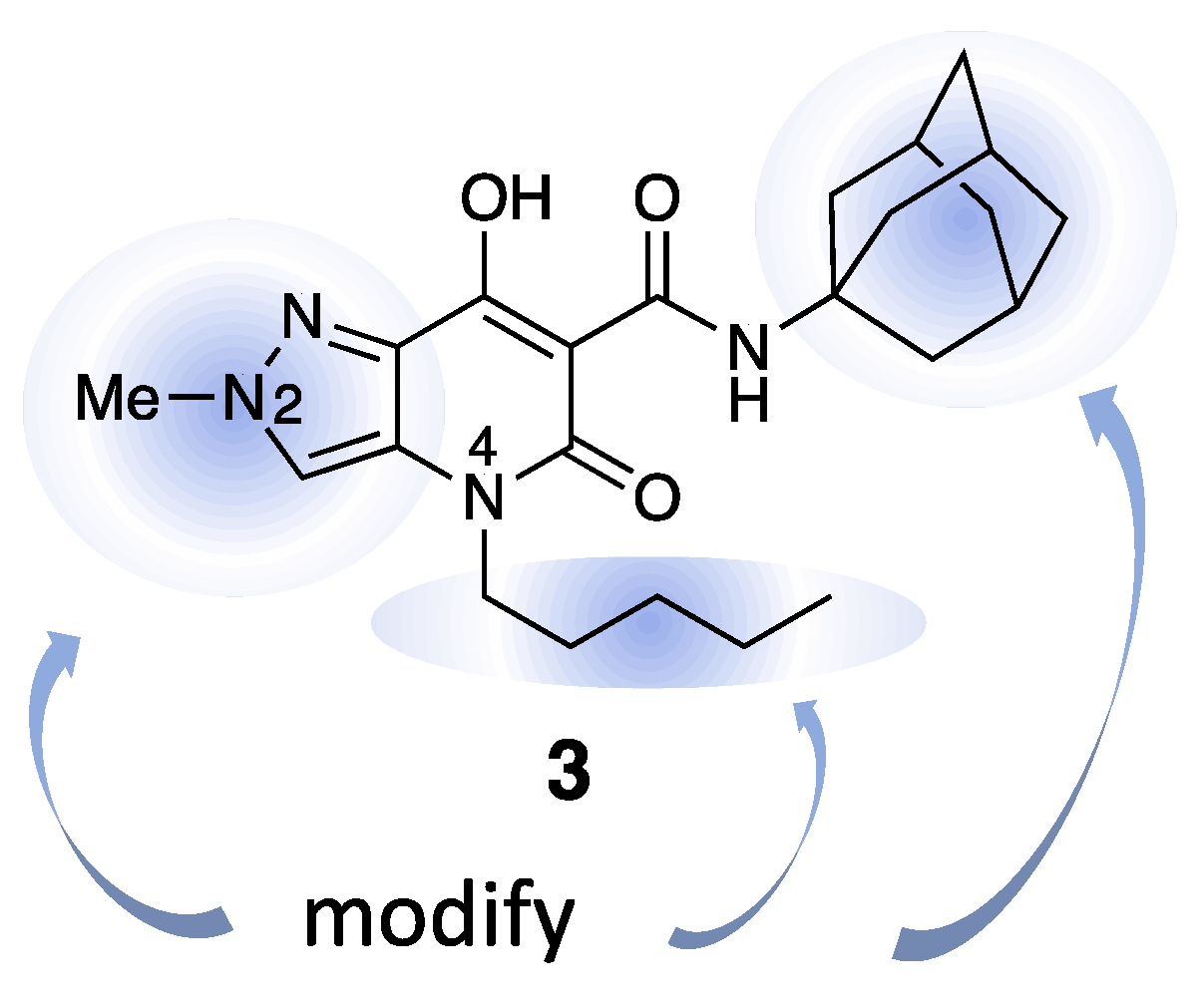
2.5. Structure–Activity Relationship (SAR)
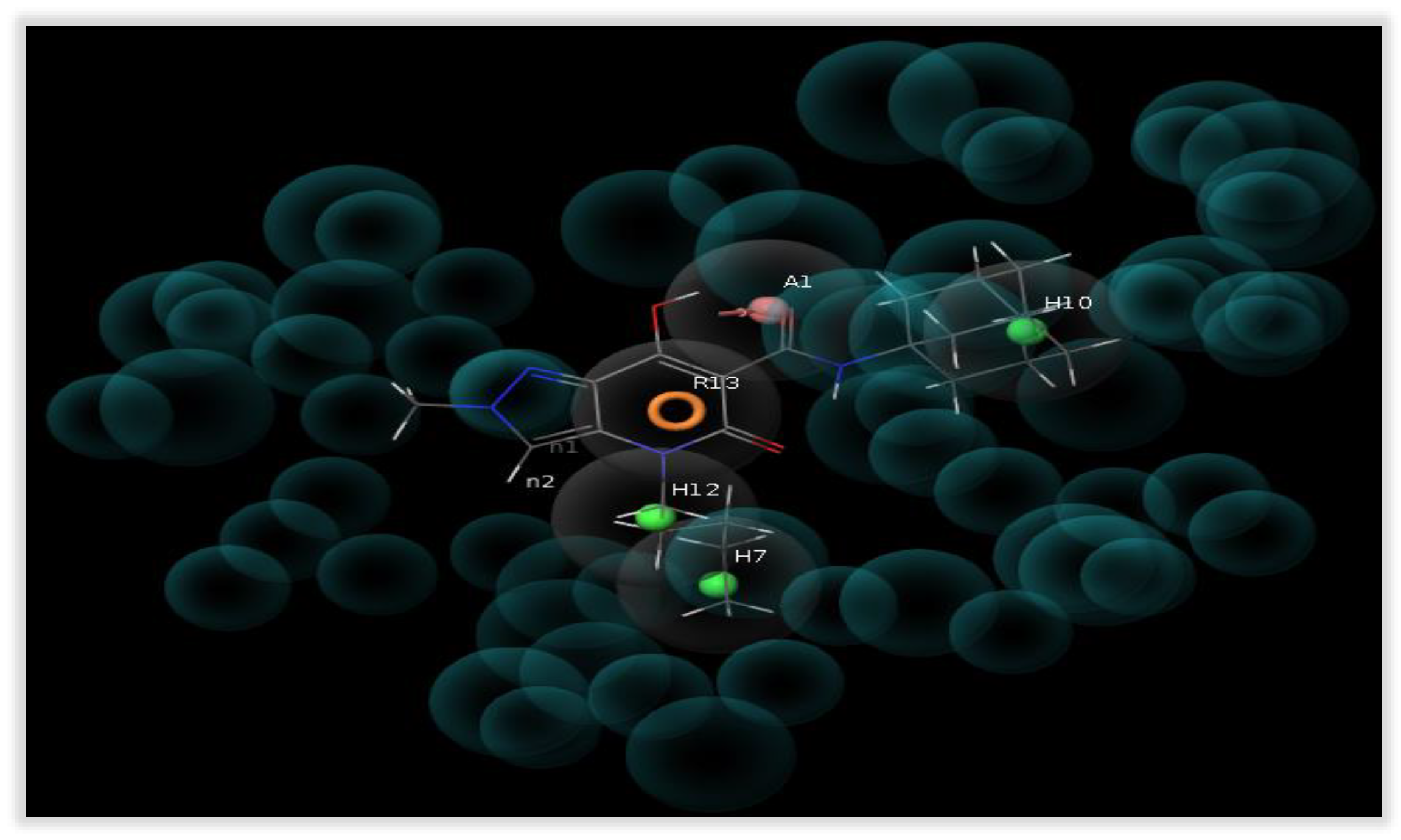
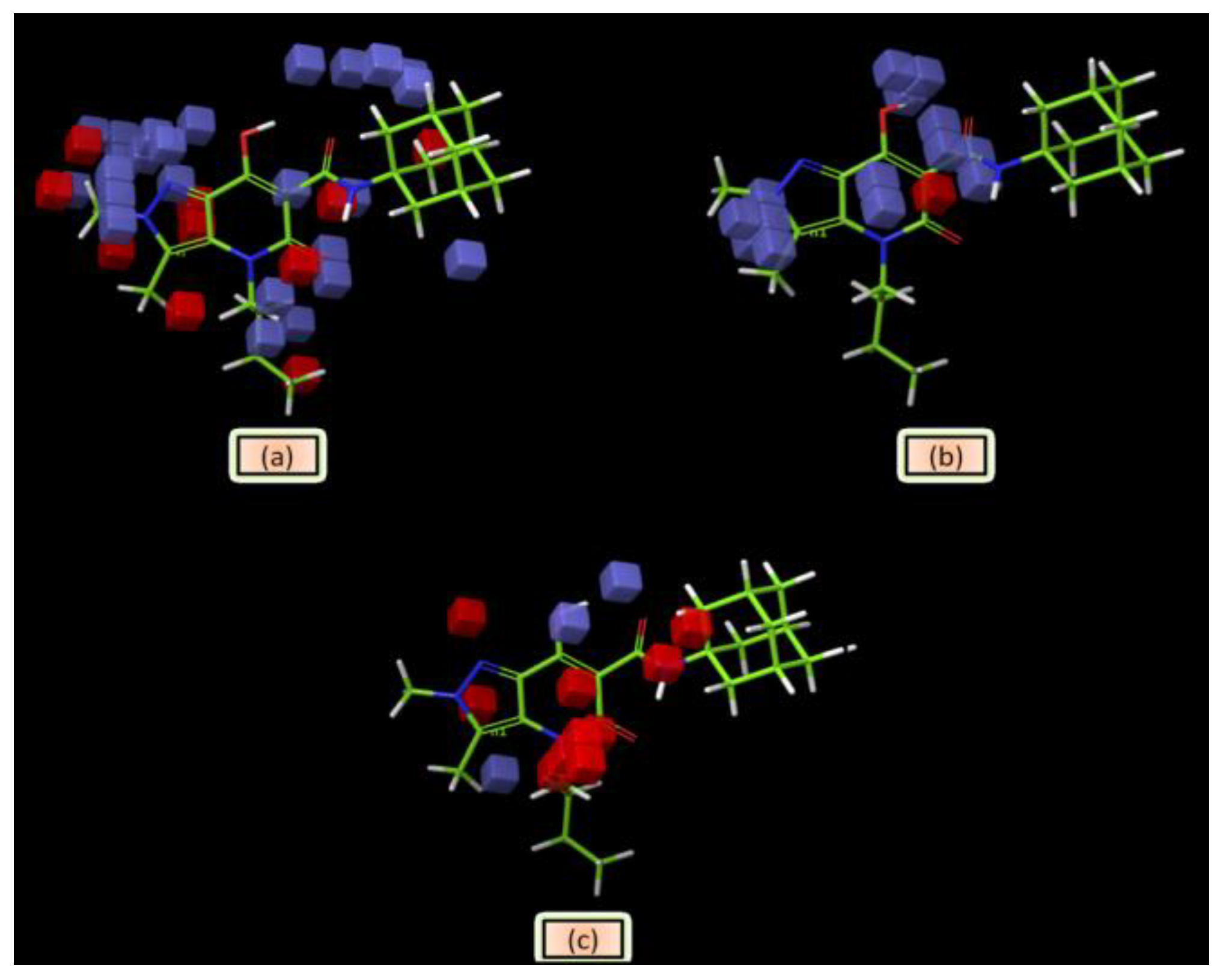
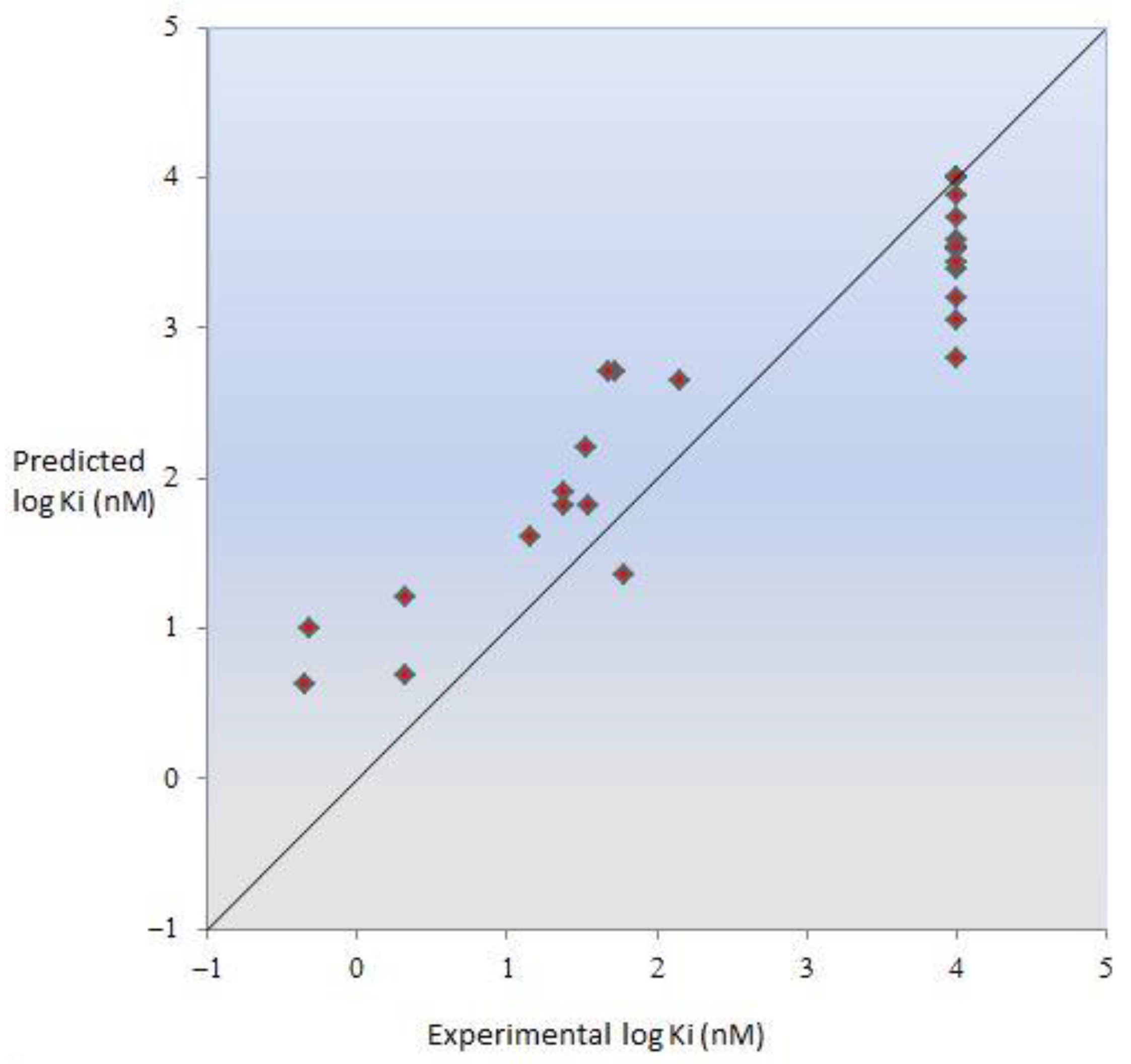
2.6. Molecular Modeling
3. Materials and Methods
3.1. Synthesis
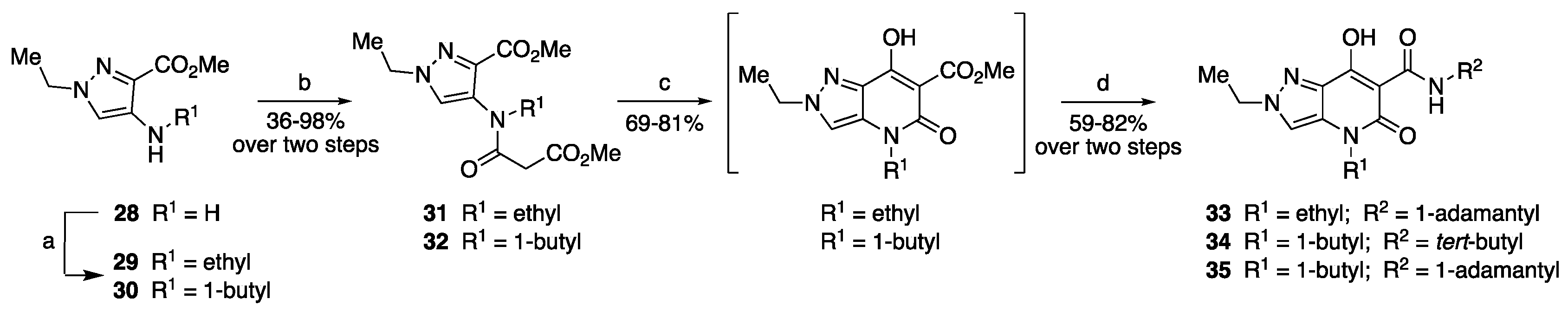
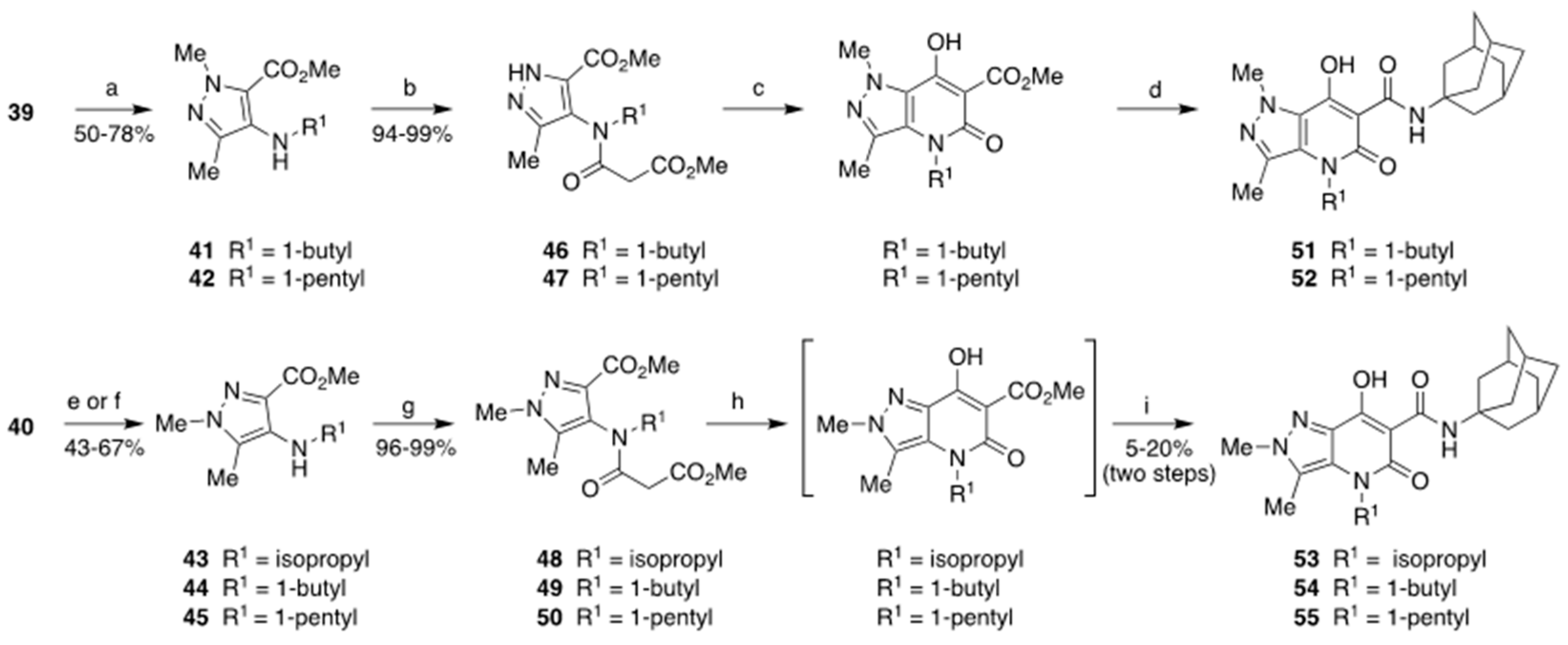
3.1.1. General Procedure for the Synthesis of Compounds 5, 29, 30, 41, 42, 44, 45
3.1.2. General Procedure for the Synthesis of Compounds 6, 43
3.1.3. General Procedure for the Synthesis of Compounds 7, 8
3.1.4. General Procedure for the Synthesis of Compounds 9–12, 31, 32, 46–50
3.1.5. General Procedure for the Synthesis of the Final Compounds 13–27, 33–35, 51–55
- 4-(1-Butyl)-N-(tert-butyl)-7-hydroxy-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (13)
- 4-(1-Butyl)-N-cyclopentyl-7-hydroxy-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (14)
- 4-(1-Butyl)-N-cyclooctyl-7-hydroxy-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (15)
- 4-(1-Butyl)-N-(4-fluorophenyl)-7-hydroxy-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (16)
- 4-(1-Butyl)-N-(4-fluorobenzyl)-7-hydroxy-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (17)
- 4-(1-Butyl)-7-hydroxy-2-methyl-5-oxo-N-(1,2,3,4-tetrahydronaphthalen-1-yl)-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (18)
- N-Benzhydryl-4-(1-butyl)-7-hydroxy-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (19)
- tert-Butyl (4-Butyl-7-hydroxy-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carbonyl)-L-phenylalaninate (20)
- N-(Adamantan-1-yl)-7-hydroxy-4-isopropyl-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (21)
- 7-Hydroxy-N-(1-hydroxyadamantan-3-yl)-4-isopropyl-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (22)
- N-Cyclooctyl-7-hydroxy-4-isopropyl-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (23)
- 7-Hydroxy-4-isopropyl-2-methyl-5-oxo-N-(1,2,3,4-tetrahydronaphthalen-1-yl)-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (24)
- N-Benzhydryl-7-hydroxy-4-isopropyl-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (25)
- N-(Adamantan-1-yl)-4-benzyl-7-hydroxy-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (26)
- N-(Adamantan-1-yl)-4-(2,4-dimethoxybenzyl)-7-hydroxy-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (27)
- N-(Adamantan-1-yl)-2,4-diethyl-7-hydroxy-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (33)
- 4-(1-Butyl)-N-(tert-butyl)-2-ethyl-7-hydroxy-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (34)
- N-(Adamantan-1-yl)-4-(1-butyl)-2-ethyl-7-hydroxy-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (35)
- N-(Adamantan-1-yl)-4-(1-butyl)-7-hydroxy-1,3-dimethyl-5-oxo-4,5-dihydro-1H-pyrazolo[4,3-b]pyridine-6-carboxamide (51)
- N-(Adamantan-1-yl)-7-hydroxy-1,3-dimethyl-5-oxo-4-(1-pentyl)-4,5-dihydro-1H-pyrazolo[4,3-b]pyridine-6-carboxamide (52)
- N-(Adamantan-1-yl)-7-hydroxy-4-isopropyl-2,3-dimethyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (53)
- N-(Adamantan-1-yl)-4-(1-butyl)-7-hydroxy-2,3-dimethyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (54)
- N-(Adamantan-1-yl)-7-hydroxy-2,3-dimethyl-5-oxo-4-(1-pentyl)-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide (55)
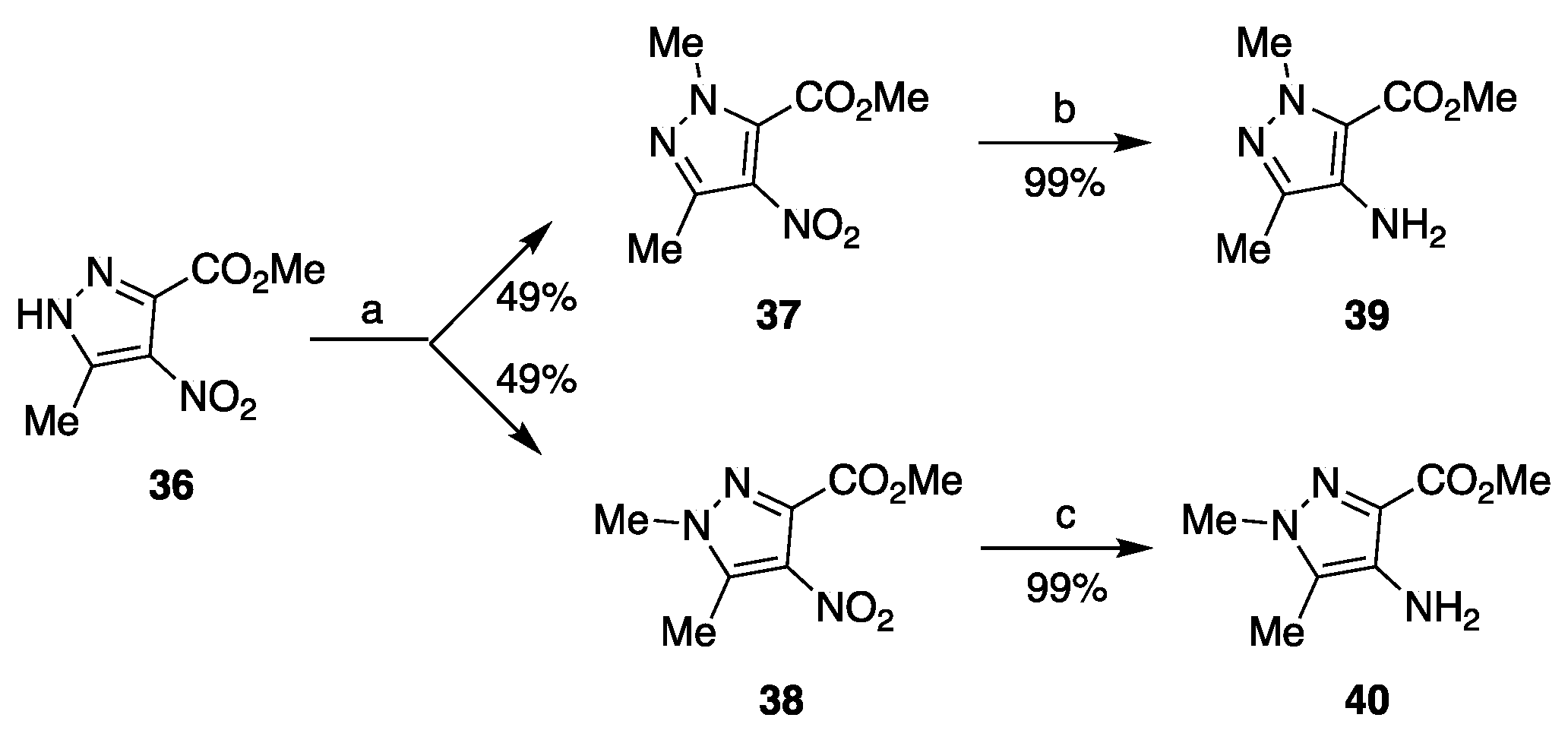
3.1.6. General Procedure for the Synthesis of Compounds 37 and 38
3.1.7. General Procedure for the Synthesis of Compounds 39 and 40
3.2. Preparation of Salts of Compounds 63
3.2.1. N-(Adamantan-1-yl)-4-(1-butyl)-7-hydroxy-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide Sodium Salt
3.2.2. N-(Adamantan-1-yl)-4-(1-butyl)-7-hydroxy-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide Potassium Salt
3.2.3. N-(Adamantan-1-yl)-4-(1-butyl)-7-hydroxy-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide Cesium Salt
3.2.4. N-(Adamantan-1-yl)-4-(1-butyl)-7-hydroxy-2-methyl-5-oxo-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-6-carboxamide Tetramethylammonium Salt
3.3. Dissolution of 63-Salts in 0.9% NaCl Solution
3.4. In Vitro Pharmacology
3.4.1. Competition Binding Assay
3.4.2. Functional Activity at CB2 Receptor In Vitro
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug. Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tudorancea, I.M.; Ciorpac, M.; Stanciu, G.D.; Caratasu, C.; Sacarescu, A.; Ignat, B.; Burlui, A.; Rezus, E.; Creanga, I.; Alexa-Stratulat, T.; et al. The Therapeutic Potential of the Endocannabinoid System in Age-Related Diseases. Biomedicines 2022, 10, 2492. [Google Scholar] [CrossRef]
- Davis, M.P. Future Therapeutic Potential of Synthetic Cannabinoids and Endocannabinoid System Modulators. In Cannabis and Cannabinoid-Based Medicines in Cancer Care; Cyr, C., Davis, M.P., Schecter, D., Daeninck, P., Eds.; Springer: Cham, Switzerland, 2022; pp. 91–115. [Google Scholar] [CrossRef]
- Kendall, D.A.; Yudowsi, G.A. Cannabinoid Receptors in the Central Nervous System: Their Signaling and Roles in Disease. Front. Cell. Neurosci. 2017, 10, 294. [Google Scholar] [CrossRef] [Green Version]
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. An overview of the cannabinoid type 2 (CB2) receptor and its therapeutic potential. Curr. Opin. Anaesthesiol. 2018, 31, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-J.; Gao, M.; Gao, F.-f.; Su, Q.-X.; Wu, J. Brain cannabinoid receptor 2: Expression, function and modulation. Acta Pharmacol. Sin. 2017, 38, 312–316. [Google Scholar] [CrossRef] [Green Version]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal structure of the human cannabinoid receptor CB1. Cell 2016, 167, 750–762.e14. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Hua, T.; Vemuri, K.; Ho, J.-H.; Wu, Y.; Wu, L.; Popov, P.; Benchama, O.; Zvonok, N.; Locke, K.; et al. Crystal structure of the human cannabinoid receptor CB2. Cell 2019, 176, 459–467.e13. [Google Scholar] [CrossRef] [Green Version]
- Xing, C.; Zhuang, Y.; Xu, T.-H.; Feng, Z.; Zhou, X.E.; Chen, M.; Wang, L.; Meng, X.; Xue, Y.; Wang, J.; et al. Cryo-EM structure of the human cannabinoid receptor CB2-Gi signaling complex. Cell 2020, 180, 645–654.e13. [Google Scholar] [CrossRef]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Brust, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef]
- Aghazadeh Tabrizi, M.; Baraldi, P.G.; Borea, P.A.; Varani, K. Medicinal chemistry, pharmacology, and potential therapeutic benefits of cannabinoid CB2 receptor agonists. Chem. Rev. 2016, 116, 519–560. [Google Scholar] [CrossRef]
- Whiting, Z.M.; Yin, J.; de la Harpe, S.M.; Vernall, A.J.; Grimsey, N.L. Developing the cannabinoid receptor 2 (CB2) pharmacopoeia: Past, present, and future. Trends Pharmacol. Sci. 2022, 43, 754–771. [Google Scholar] [CrossRef] [PubMed]
- Contartese, A.; Valoti, M.; Corelli, F.; Pasquini, S.; Mugnaini, C.; Pessina, F.; Aldinucci, C.; Sgaragli, G.P.; Frosini, M. A novel CB2 agonist, COR167, potently protects rat brain cortical slices against OGD and reperfusion injury. Pharm. Res. 2012, 66, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, C.; Blanchet, M.R.; Laviolette, M.; Flamand, N. The CB2 receptor and its role as a regulator of inflammation. Cell Mol. Life Sci. 2016, 73, 4449–4470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassano, T.; Calcagnini, S.; Pace, L.; De Marco, F.; Romano, A.; Gaetani, S. Cannabinoid receptor 2 signaling in neurodegenerative disorders: From pathogenesis to a promising therapeutic target. Front. Neurosci. 2017, 11, 30. [Google Scholar] [CrossRef] [Green Version]
- Annunziata, P.; Cioni, C.; Mugnaini, C.; Corelli, F. Potent immunomodulatory activity of a highly selective cannabinoid CB2 agonist on immune cells from healthy subjects and patients with multiple sclerosis. J. Neuroimmunol. 2017, 303, 66–74. [Google Scholar] [CrossRef]
- Fulmer, M.L.; Thewke, D.P. The endocannabinoid system and heart disease: The role of cannabinoid receptor type 2. Cardiovasc. Hematol. Disord. Drug Targets 2018, 18, 34–51. [Google Scholar] [CrossRef]
- Kolb, B.; Saber, H.; Fadel, H.; Rajah, G. The endocannabinoid system and stroke: A focused review. Brain Circ. 2019, 5, 1–7. [Google Scholar] [CrossRef]
- Shang, Y.; Tang, Y. The central cannabinoid receptor type-2 (CB2) and chronic pain. Int. J. Neurosci. 2017, 127, 812–823. [Google Scholar] [CrossRef]
- Laezza, C.; Pagano, C.; Navarra, G.; Pastorino, O.; Proto, M.C.; Fiore, D.; Piscopo, C.; Gazzerro, P.; Bifulco, M. The endocannabinoid system: A target for cancer treatment. Int. J. Mol. Sci. 2020, 21, 747. [Google Scholar] [CrossRef] [Green Version]
- Cioni, C.; Tassi, M.; Marotta, G.; Mugnaini, C.; Corelli, F.; Annunziata, P. A novel highly selective cannabinoid CB2 agonist reduces in vitro growth of human glial cell tumors with a mechanism involving TGF-beta. Cent. Nerv. Syst. Agents Med. Chem. 2019, 19, 206–214. [Google Scholar] [CrossRef]
- Vivek, S.K.; Ginpreet, K. Therapeutic potential of cannabinoid receptor 2 in the treatment of diabetes mellitus and its complications. Eur. J. Pharmacol. 2019, 862, 172628–172632. [Google Scholar] [CrossRef]
- Lunn, C.A.; Reich, E.-P.; Fine, J.S.; Lavey, B.; Kozlowski, J.A.; Hipkin, R.W.; Lundell, D.J.; Bober, L. Biology and therapeutic potential of cannabinoid CB2 receptor inverse agonists. Br. J. Pharmacol. 2008, 153, 226–239. [Google Scholar] [CrossRef] [Green Version]
- Pasquini, S.; Botta, L.; Semeraro, T.; Mugnaini, C.; Ligresti, A.; Palazzo, E.; Maione, S.; Di Marzo, V.; Corelli, F. Investigations on the 4-Quinolone-3-carboxylic Acid Motif. 2. Synthesis and Structure-Activity Relationship of Potent and Selective Cannabinoid-2 Receptor Agonists Endowed with Analgesic Activity in Vivo. J. Med. Chem. 2008, 51, 5075–5084. [Google Scholar] [CrossRef] [PubMed]
- Cascio, M.G.; Bolognini, D.; Pertwee, R.G.; Palazzo, E.; Corelli, F.; Pasquini, S.; Di Marzo, V.; Maione, S. In vitro and in vivo pharmacological characterization of two novel selective cannabinoid CB2 receptor inverse agonists. Pharm. Res. 2010, 61, 349–354. [Google Scholar] [CrossRef]
- Pasquini, S.; Ligresti, A.; Mugnaini, C.; Semeraro, T.; Cicione, L.; De Rosa, M.; Guida, F.; Luongo, L.; De Chiaro, M.; Cascio, M.G.; et al. Investigations on the 4-Quinolone-3-carboxylic Acid Motif. 3. Synthesis, Structure-Affinity Relationships, and Pharmacological Characterization of 6-Substituted 4-Quinolone-3-carboxamides as Highly Selective Cannabinoid-2 Receptor Ligands. J. Med. Chem. 2010, 53, 5915–5928. [Google Scholar] [CrossRef] [PubMed]
- Brogi, S.; Corelli, F.; Di Marzo, V.; Ligresti, A.; Mugnaini, C.; Pasquini, S.; Tafi, A. Three-Dimensional Quantitative Structure-Selectivity Relationships (3D-QSSR) Analysis Guided Rational Design of a Highly Selective Ligand for the Cannabinoid Receptor 2. Eur. J. Med. Chem. 2011, 46, 547–555. [Google Scholar] [CrossRef]
- Pasquini, S.; De Rosa, M.; Pedani, V.; Mugnaini, C.; Guida, F.; Luongo, L.; De Chiaro, M.; Maione, S.; Dragoni, S.; Frosini, M.; et al. Investigations on the 4-Quinolone-3-carboxylic Acid Motif. 4. Identification of New Potent and Selective Ligands for the Cannabinoid Type 2 Receptor with Diverse Substitution Patterns and Anti-Hyperalgesic Effects in Mice. J. Med. Chem. 2011, 54, 5444–5453. [Google Scholar] [CrossRef]
- Mugnaini, C.; Nocerino, S.; Pedani, V.; Pasquini, S.; Tafi, A.; De Chiaro, M.; Bellucci, L.; Valoti, M.; Guida, F.; Luongo, L.; et al. Investigations on the 4-quinolone-3-carboxylic acid motif. 5. Modulation of the physicochemical profile of a set of potent and selective CB2 ligands through a bioisosteric approach. ChemMedChem 2012, 7, 920–934. [Google Scholar] [CrossRef] [Green Version]
- Pasquini, S.; De Rosa, M.; Ligresti, A.; Mugnaini, C.; Brizzi, A.; Caradonna, N.P.; Cascio, M.G.; Bolognini, D.; Pertwee, R.G.; Di Marzo, V.; et al. Investigations on the 4-Quinolone-3-carboxylic Acid Motif. 6. Synthesis and Pharmacological Evaluation of 7-Substituted 4-Quinolone-3-carboxamide Derivatives as High Affinity Ligands for Cannabinoid Receptors. Eur. J. Med. Chem. 2012, 58, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Mugnaini, C.; Brizzi, A.; Ligresti, A.; Allarà, M.; Lamponi, S.; Vacondio, F.; Silva, C.; Mor, M.; Di Marzo, V.; Corelli, F. Investigations on the 4-Quinolone-3-carboxylic Acid Motif. 7. Synthesis and Pharmacological Evaluation of 4-Quinolone-3-carboxamides and 4-Hydroxy-2-quinolone-3-carboxamides as High Affinity Cannabinoid Receptor 2 (CB2R) Ligands with Improved Aqueous Solubility. J. Med. Chem. 2016, 59, 1052–1067. [Google Scholar] [CrossRef]
- Mugnaini, C.; Kostrzewa, M.; Bryk, M.; Mahmoud, A.M.; Brizzi, A.; Lamponi, S.; Giorgi, G.; Ferlenghi, F.; Vacondio, F.; Maccioni, P.; et al. Overcoming the Physicochemical Limitations of Cannabinoid Type-2 Receptor Ligands: Design, Synthesis, Physicochemical and Pharmacological Profiling of 7-Hydroxy-5-oxopyrazolo[4,3-b]pyridine-6-carboxamide Derivatives with Anti-Osteoarthritis Activity in Vivo. J. Med. Chem. 2020, 63, 7369–7391. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, S.; Benicchi, T.; Bettinetti, L.; Ceccarelli, I.; Diodato, E.; Federico, C.; Fiengo, P.; Franceschini, D.; Gokce, O.; Heitz, F.; et al. Fused 3-hydroxy-3-trifluoromethylpyrazoles inhibit mutant Huntingtin toxicity. ACS Med. Chem. Lett. 2013, 4, 979–984. [Google Scholar] [CrossRef] [Green Version]
- O’Dowd, C.; Harrison, T.; Hewitt, P.; Rountree, S.; Hugues, M.; Burkamp, F.; Jordan, L.; Helm, M.; Broccatelli, F.; Crawford, J.J.; et al. Piperidine Derivative as Inhibitors of Ubiquitin Specific Protease 7. WO 2018/073602 A1, 26 April 2018. [Google Scholar]
- Kadam, S.S.; Maier, L.; Kostakis, I.; Pouli, N.; Toušek, J.; Necčas, M.; Marakos, P.; Marek, R. Synthesis and tautomerism of substituted pyrazolo[4,3-c]pyrazoles. Eur. J. Org. Chem. 2013, 6811–6822. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS-97, Rel. 97-2, A Program for Automatic Solution of Crystal Structures; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Mugnaini, C.; Brizzi, A.; Vinciarelli, G.; Paolino, M.; Corelli, F. New synthesis of pyrazolo[4,3-b]pyridine derivatives as CB2 receptor ligands. New J. Chem. 2020, 44, 16218–16226. [Google Scholar] [CrossRef]
- Gleeson, M.P.; Hersey, A.; Montanari, D.; Overington, J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat. Rev. Drug Discov. 2011, 10, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Leeson, P.D.; St-Gallay, S.A. The influence of the ‘organizational factor’ on compound quality in drug discovery. Nat. Rev. Drug Discov. 2011, 10, 749–765. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- SwissADME. Available online: http://www.swissadme.ch/ (accessed on 25 May 2023).
- Murray, C.W.; Erlanson, D.A.; Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H.; Richmond, N.J. Validity of ligand efficiency metrics. ACS Med. Chem. Lett. 2014, 5, 616–618. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.W.; Gallego, R.A.; Edwards, M.P. Lipophilic efficiency as an important metric in drug design. J. Med. Chem. 2018, 61, 6401–6420. [Google Scholar] [CrossRef]
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability enhancement techniques for poorly aqueous soluble drugs and therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef] [PubMed]
- Jansson, K.; Fristedt, T.; Olsson, A.; Svensson, B.; Jönsson, S. Synthesis and reactivity of laquinimod, a quinoline-3-carboxamide: Intramolecular transfer of the enol proton to a nitrogen atom as a plausible mechanism for ketene formation. J. Org. Chem. 2006, 71, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Ghonim, A.E.; Ligresti, A.; Rabbito, A.; Mahmoud, A.M.; Di Marzo, V.; Osman, N.A.; Abadi, A.H. Structure-activity relationships of thiazole and benzothiazole derivatives as selective cannabinoid CB2 agonists with in vivo anti-inflammatory properties. Eur. J. Med. Chem. 2019, 180, 154–170. [Google Scholar] [CrossRef]
- Mugnaini, C.; Rabbito, A.; Brizzi, A.; Palombi, N.; Petrosino, S.; Verde, R.; Di Marzo, V.; Ligresti, A.; Corelli, F. Synthesis of novel 2-(1-adamantanylcarboxamido)thiophene derivatives. Selective cannabinoid type 2 (CB2) receptor agonists as potential agents for the treatment of skin inflammatory disease. Eur. J. Med. Chem. 2019, 161, 239–251. [Google Scholar] [CrossRef]
- Silvestri, R.; Ligresti, A.; La Regina, G.; Piscitelli, F.; Gatti, V.; Brizzi, A.; Pasquini, S.; Lavecchia, A.; Allarà, M.; Fantini, N.; et al. Synthesis, cannabinoid receptor affinity, molecular modeling studies and in vivo pharmacological evaluation of new substituted 1-aryl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamides. 2. Effect of the 3- 92 carboxamide substituent on the affinity and selectivity profile. Bioorg. Med. Chem. 2009, 17, 5549–5564. [Google Scholar] [CrossRef]
- Kallinen, A.; Boyd, R.; Lane, S.; Bhalla, R.; Mardon, K.; Stimson, D.H.R.; Werry, E.L.; Fulton, R.; Connor, M.; Kassiou, M. Synthesis and in vitro evaluation of fluorine-18 benzimidazole sulfones as CB2 PET-radioligands. Org. Biomol. Chem. 2019, 17, 5086–5098. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.H.M.; van der Neut, M.A.W.; Wals, H.C.; Kuil, G.D.; Borst, A.J.M.; Mulder, A.; den Hartog, A.P.; Zilaout, H.; Goutier, W.; van Stuivenberg, H.H.; et al. Synthesis and SAR of novel imidazoles as potent and selective cannabinoid CB2 receptor antagonists with high binding efficiencies. Bioorg. Med. Chem. Lett. 2010, 20, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Fulp, A.; Bortoff, K.; Zhang, Y.; Seltzman, H.; Mathews, J.; Snyder, R.; Fennell, T.; Maitra, R. Diphenyl purine derivatives as peripherally selective cannabinoid receptor 1 antagonists. J. Med. Chem. 2012, 55, 10022–10032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadipuram, A.K.; Krishnamurthy, M.; Ferreira, A.M.; Li, W.; Moore, B.M. Synthesis and testing of novel classical cannabinoids: Exploring the side chain ligand binding pocket of the CB1 and CB2 receptors. Bioorg. Med. Chem. 2003, 11, 3121–3132. [Google Scholar] [CrossRef]
- Durdagi, S.; Kapou, A.; Kourouli, T.; Andreou, T.; Nikas, S.P.; Nahmias, V.R.; Papahatjis, D.P.; Papadopoulos, M.G.; Mavromoustakos, T. The application of 3D-QSAR studies for novel cannabinoid ligands substituted at the C1’ position of the alkyl side chain on the structural requirements for binding to cannabinoid receptors CB1 and CB2. J. Med. Chem. 2007, 50, 2875–2885. [Google Scholar] [CrossRef] [PubMed]
- Järbe, T.U.; DiPatrizio, N.V.; Lu, D.; Makriyannis, A. (–)-Adamantyl-delta8-tetrahydrocannabinol (AM-411), a selective cannabinoid CB1 receptor agonist: Effects on open-field behaviors and antagonism by SR-141716 in rats. Behav. Pharmacol. 2004, 15, 517–521. [Google Scholar] [CrossRef]
- Ross, R.A.; Brockie, H.C.; Stevenson, L.A.; Murphy, V.L.; Templeton, F.; Makriyannis, A.; Pertwee, R.G. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630. Br. J. Pharmacol. 1999, 126, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, J.M.; Dart, M.J.; Tietje, K.R.; Garrison, T.R.; Grayson, G.K.; Daza, A.V.; El-Kouhen, O.F.; Yao, B.B.; Hsieh, G.C.; Pai, M.; et al. Indol-3-ylcycloalkyl ketones: Effects of N1 substituted indole side chain variations on CB(2) cannabinoid receptor activity. J. Med. Chem. 2010, 53, 295–315. [Google Scholar] [CrossRef]
- Stern, E.; Muccioli, G.G.; Millet, R.; Goossens, J.-F.; Farce, A.; Chavatte, P.; Poupaert, J.H.; Lambert, D.M.; Depreux, P.; Hénichart, J.P. Novel 4-oxo-1,4-dihydroquinoline-3-carboxamide derivatives as new CB2 cannabinoid receptors agonists: Synthesis, pharmacological properties and molecular modeling. J. Med. Chem. 2006, 49, 70–79. [Google Scholar] [CrossRef]
- Yao, B.B.; Mukherjee, S.; Fan, Y.; Garrison, T.R.; Daza, A.V.; Grayson, G.K.; Hooker, B.A.; Dart, M.J.; Sullivan, J.P.; Meyer, M.D. In vitro pharmacological characterization of AM1241: A protean agonist at the cannabinoid CB2 receptor? Br. J. Pharmacol. 2006, 149, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.E.; Tang, Y.; Alt, A.; Burford, N.T.; Gerritz, S.W.; Ogawa, L.M.; Zhang, L.; Kendall, D.A. Identification and biochemical analyses of selective CB2 agonists. Eur. J. Pharmacol. 2019, 854, 1–8. [Google Scholar] [CrossRef]
- Jordan, C.J.; Feng, Z.W.; Galaj, E.; Bi, G.H.; Xue, Y.; Liang, Y.; McGuire, T.; Xie, X.Q.; Xi, Z.X. Xie2-64, a novel CB2 receptor inverse agonist, reduces cocaine abuse-related behaviors in rodents. Neuropharmacology 2020, 176, 108241. [Google Scholar] [CrossRef]
- Khanolkar, A.D.; Lu, D.; Fan, P.; Tian, X.; Makriyannis, A. Novel conformationally restricted tetracyclic analogs of delta8-tetrahydrocannabinol. Bioorg. Med. Chem. Lett. 1999, 9, 2119–2124. [Google Scholar] [CrossRef] [PubMed]
- Osman, N.A.; Ligresti, A.; Klein, C.D.; Allarà, M.; Rabbito, A.; Di Marzo, V.; Abouzid, K.A.; Abadi, A.H. Discovery of novel Tetrahydrobenzo[b]thiophene and pyrrole-based scaffolds as potent and selective CB2 receptor ligands: The structural elements controlling binding affinity, selectivity and functionality. Eur. J. Med. Chem. 2016, 122, 619–634. [Google Scholar] [CrossRef]
- Angerer, V.; Mogler, L.; Steitz, J.P.; Bisel, P.; Hess, C.; Schoeder, C.T.; Müller, C.E.; Huppertz, L.M.; Westphal, F.; Schäper, J.; et al. Structural characterization and pharmacological evaluation of the new synthetic cannabinoid CUMYL-PEGACLONE. Drug Test. Anal. 2018, 10, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Gianquinto, E.; Sodano, F.; Rolando, B.; Kostrzewa, M.; Allarà, M.; Mahmoud, A.M.; Kumar, P.; Spyrakis, F.; Ligresti, A.; Chegaev, K. N-[1,3-Dialkyl(aryl)-2-oxoimidazolidin-4-ylidene]aryl(alkyl)sulphonamides as Novel Selective Human Cannabinoid Type 2 Receptor (hCB2R) Ligands; Insights into the Mechanism of Receptor Activation/Deactivation. Molecules 2022, 27, 8152. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | Structure | CB1R c Ki f (nM) | CB2R d Ki f (nM) | SI e | logKiCB2 (nM) Exp. | logKiCB2 (nM) Pred. |
|---|---|---|---|---|---|---|
| 13 |  | >10,000 | >10,000 | – | 4 | 3.19 |
| 14 |  | >10,000 | >10,000 | – | 4 | 3.53 |
| 15 |  | >10,000 | 9.03 ± 4.84 | >1107 | 1.16 | 1.6 |
| 16 |  | >10,000 | >10,000 | – | 4 | 4 |
| 17 |  | >10,000 | >10,000 | – | 4 | 3.73 |
| 18 |  | >10,000 | 17.31 ± 8.31 | >578 | 1.38 | 1.9 |
| 19 |  | >10,000 | 142.25 ± 109.73 | >70 | 2.15 | 2.65 |
| 20 |  | >10,000 | >10,000 | – | 4 | 4 |
| 21 |  | >10,000 | >10,000 | – | 4 | 3.59 |
| 22 |  | >10,000 | >10,000 | – | 4 | 3.39 |
| 23 |  | >10,000 | >10,000 | – | 4 | 3.05 |
| 24 |  | >10,000 | >10,000 | – | 4 | 3.43 |
| 25 |  | >10,000 | >10,000 | – | 4 | 4 |
| 26 |  | 115.63 ± 23.66 | 0.48 ± 0.12 | 241 | −0.32 | 1 |
| 27 |  | >10,000 | >10,000 | – | 4 | 3.52 |
| 33 |  | >10,000 | 24.04 ± 15.34 | >416 | 1.38 | 1.81 |
| 34 |  | >10,000 | 105.85 ± 18.12 | >95 | 2.02 | 1.61 |
| 35 |  | 102.30 ± 15.23 | 0.45 ± 0.01 | 227 | −0.35 | 0.63 |
| 51 |  | >10,000 | 35.61 ± 19.46 | >280 | 1.55 | 1.81 |
| 52 |  | >10,000 | 34.10 ± 4.88 | >293 | 1.53 | 2.2 |
| 53 |  | >10,000 | >10,000 | – | 4 | 3.88 |
| 54 |  | >10,000 | 2.16 ± 1.7 | >4630 | 0.33 | 1.2 |
| 55 |  | >10,000 | 2.11 ± 0.74 | >4739 | 0.32 | 0.69 |
| 56 |  | >10,000 | >10,000 | – | 4 | 3.54 |
| 57 |  | >10,000 | 54.31 ± 12.28 | >184 | 1.73 | 2.7 |
| 58 |  | >10,000 | 61.76 ± 20.78 | >162 | 1.79 | 1.36 |
| 59 |  | >10,000 | >10,000 | – | 4 | 2.8 |
| 60 |  | >10,000 | 48.46 ± 10.94 | >206 | 1.68 | 2.7 |
| 61 |  | >10,000 | >10,000 | – | 4 | 4 |
| 62 |  | >10,000 | >10,000 | – | 4 | 4 |
| 63 [34] |  | 231 ± 76 | 2.5 ± 0.2 | 92 | – | – |
| 3 [34] |  | 33.5 ± 0.9 | 0.18 ± 0.01 | 167 | – | – |
| SR144528 g,h |  | 116 ± 22 | 1.8 ± 0.5 | 64 | – | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mugnaini, C.; Kostrzewa, M.; Casini, M.; Kumar, P.; Catallo, V.; Allarà, M.; Guastaferro, L.; Brizzi, A.; Paolino, M.; Tafi, A.; et al. Systematic Modification of the Substitution Pattern of the 7-Hydroxy-5-oxopyrazolo[4,3-b]pyridine-6-carboxamide Scaffold Enabled the Discovery of New Ligands with High Affinity and Selectivity for the Cannabinoid Type 2 Receptor. Molecules 2023, 28, 4958. https://doi.org/10.3390/molecules28134958
Mugnaini C, Kostrzewa M, Casini M, Kumar P, Catallo V, Allarà M, Guastaferro L, Brizzi A, Paolino M, Tafi A, et al. Systematic Modification of the Substitution Pattern of the 7-Hydroxy-5-oxopyrazolo[4,3-b]pyridine-6-carboxamide Scaffold Enabled the Discovery of New Ligands with High Affinity and Selectivity for the Cannabinoid Type 2 Receptor. Molecules. 2023; 28(13):4958. https://doi.org/10.3390/molecules28134958
Chicago/Turabian StyleMugnaini, Claudia, Magdalena Kostrzewa, Marta Casini, Poulami Kumar, Valeria Catallo, Marco Allarà, Laura Guastaferro, Antonella Brizzi, Marco Paolino, Andrea Tafi, and et al. 2023. "Systematic Modification of the Substitution Pattern of the 7-Hydroxy-5-oxopyrazolo[4,3-b]pyridine-6-carboxamide Scaffold Enabled the Discovery of New Ligands with High Affinity and Selectivity for the Cannabinoid Type 2 Receptor" Molecules 28, no. 13: 4958. https://doi.org/10.3390/molecules28134958
APA StyleMugnaini, C., Kostrzewa, M., Casini, M., Kumar, P., Catallo, V., Allarà, M., Guastaferro, L., Brizzi, A., Paolino, M., Tafi, A., Kapatais, C., Giorgi, G., Vacondio, F., Mor, M., Corelli, F., & Ligresti, A. (2023). Systematic Modification of the Substitution Pattern of the 7-Hydroxy-5-oxopyrazolo[4,3-b]pyridine-6-carboxamide Scaffold Enabled the Discovery of New Ligands with High Affinity and Selectivity for the Cannabinoid Type 2 Receptor. Molecules, 28(13), 4958. https://doi.org/10.3390/molecules28134958