
Determination of Response Factors for Analytes Detected during Migration Studies, Strategy and Internal Standard Selection for Risk Minimization



Abstract
:1. Introduction
2. Materials and Methods
2.1. Representative Analytes
2.2. Reagents
2.3. Optimization of the MS Signal
2.4. LC-MS Method
2.5. GC-MS
2.6. Preparation of Standard Solutions for LC-MS Analysis
2.7. Preparation of Standard Solutions for GC-MS Analysis
3. Results and Discussion
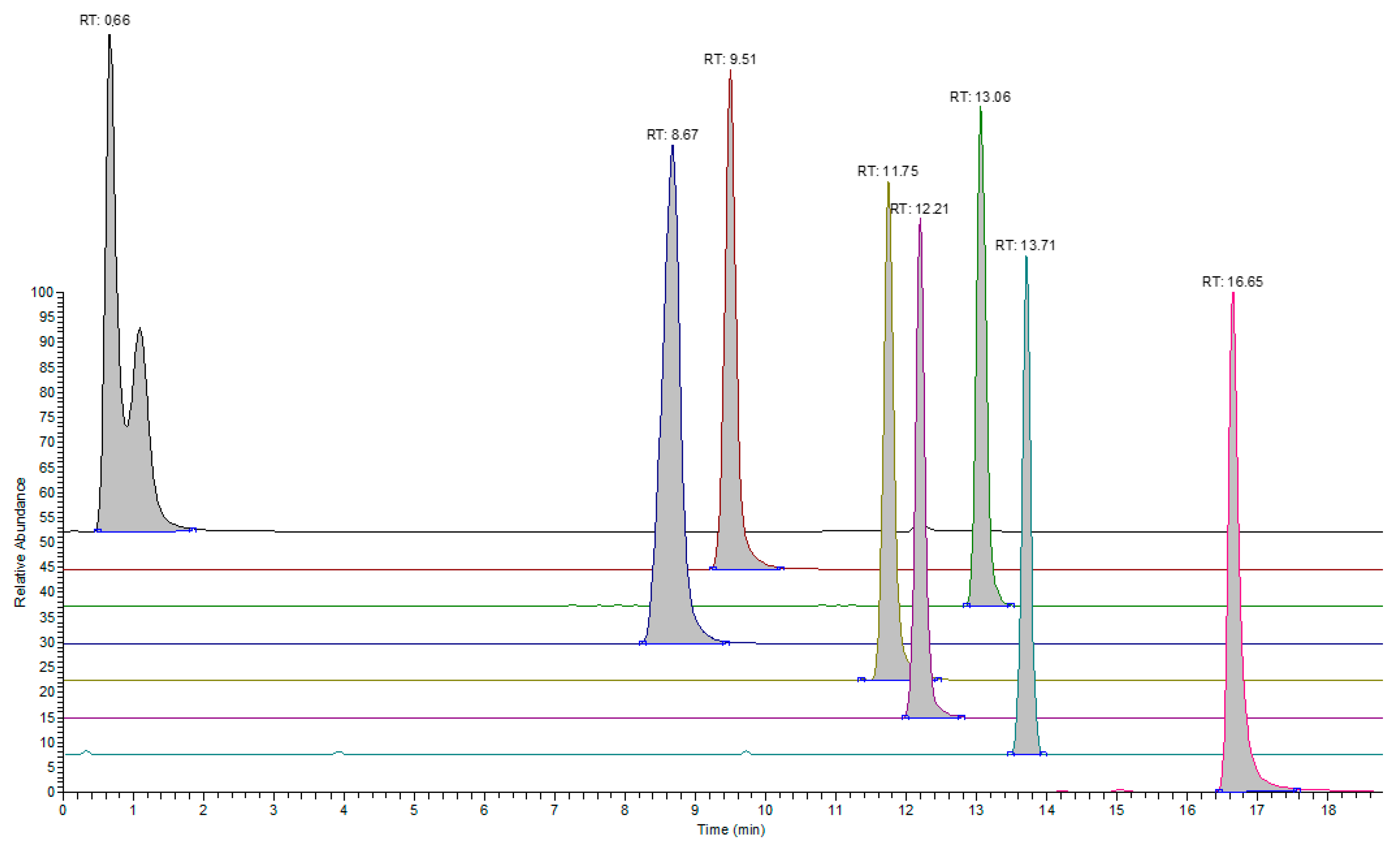
3.1. LC-MS Analysis
3.2. GC-MS Analysis
3.3. Descriptive Statistics for LC-MS Data
- The substances that are based on polarizable carbon–oxygen or carbon–nitrogen bonds (i.e., carbamides, esters, ketones) are highly relevant in terms of response. Both have small variability and their medians are approximately equal, implying that the polarizability of the bond is similar for both pairs: carbon–oxygen and carbon–nitrogen.
- The substances corresponding to a polarizable phosphorus–oxygen bond appear to have one of the highest variabilities. This is due to the unstable bis(2-ethylhexoxy)-oxophosphanium species included. The median of the population is over 3-fold higher than for all other groups.
- Amines, corresponding to ionizable nitrogen-based substances, have a median that is higher than that of carbonyl and carbamide species. They, however, exhibit a high variability that is not due solely to outlying values but rather a natural diversity (the box is wider, not just the extended lines).
- 4.
- All sulfur-based polarizable bonds (sulfate esters, sulfur ethers, sulfonamides, etc.) have low but similar responses compared with other chemical categories, meaning that sulfur–oxygen and/or sulfur–nitrogen bonds do not differ much in propensity.
3.4. Strategy for LC-MS Analysis
- -
- The substance should present a response approximately equal to the Q1 (1st quartile) of the critical compound population to be covered. The positive ionization includes amines and species with polarizable carbon–oxygen and carbon–nitrogen bonds (amides, esters, ketones, etc.), while the negative ionization includes both acids and phenolics.
- -
- The substance should, ideally, not belong to a typically expected analyte.
3.5. Evaluation for LC-MS Analysis
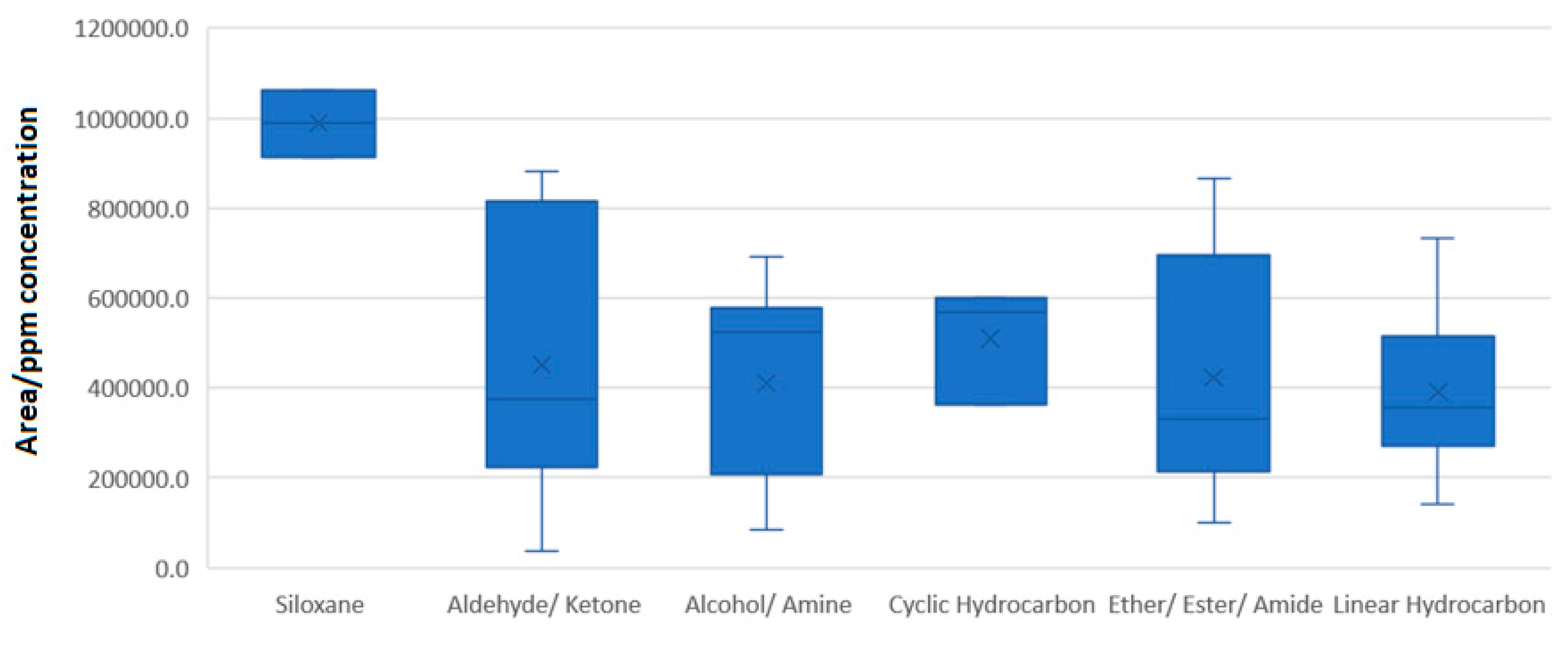
3.6. Descriptive Statistics of GC-MS Data
3.7. Strategy for GC-MS Analysis
3.8. Evaluation for GC-MS Analysis
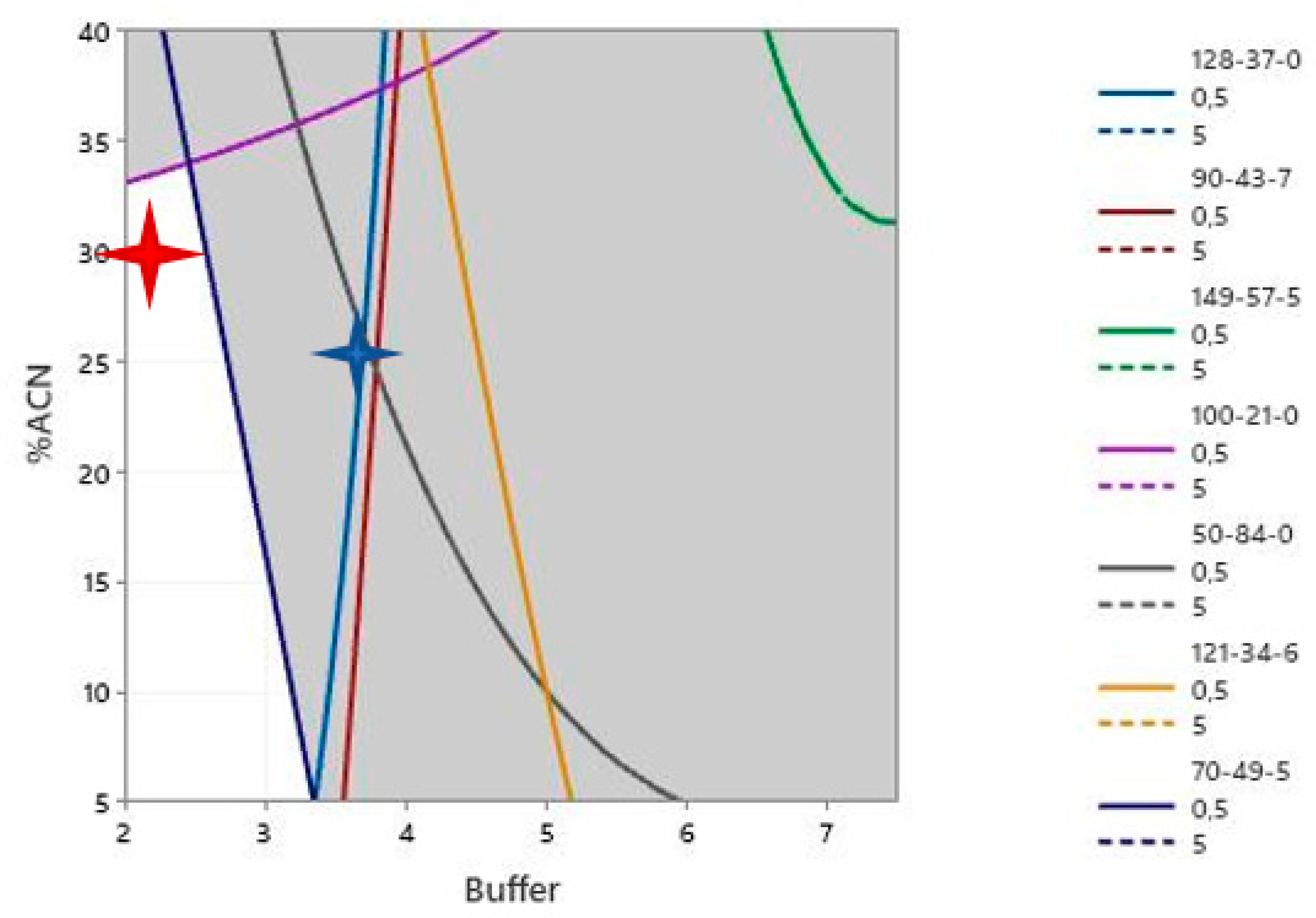
3.9. Robustness Testing of the LC-MS Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- United States Pharmacopeia. General Chapter, 〈1663〉 Assessment of Extractables Associated with Pharmaceutical Packaging/Delivery Systems; USP-NF: Rockville, MD, USA, 2022. [Google Scholar]
- United States Pharmacopeia. General Chapter, 〈1664〉 Assessment of Drug Product Leachables Associated with Pharmaceutical Packaging/Delivery Systems; USP-NF: Rockville, MD, USA, 2022. [Google Scholar]
- Paskiet, D.; Jenke, D.; Ball, D.; Houston, C.; Norwood, D.L.; Markovic, I. The Product Quality Research Institute (PQRI) Leachables and Extractables Working Group Initiatives for Parenteral and Ophthalmic Drug Product (PODP). PDA J. Pharm. Sci. Technol. 2013, 67, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Wadie, M.; Abdel-Moety, E.M.; Rezk, M.R.; Marzouk, H.M. Sustainable and smart HPTLC determination of Silodosin and Solifenacin using a constructed two illumination source chamber with a smartphone camera as a detector: Comparative study with conventional densitometric scanner. Sustain. Chem. Pharm. 2023, 33, 101095. [Google Scholar] [CrossRef]
- Wadie, M.; Abdel-Moety, E.M.; Rezk, M.R.; Marzouk, H.M. A novel smartphone HPTLC assaying platform versus traditional densitometric method for simultaneous quantification of alfuzosin and solifenacin in their dosage forms as well as monitoring content uniformity and drug residues on the manufacturing equipment. RSC Adv. 2023, 13, 11642–11651. [Google Scholar] [CrossRef] [PubMed]
- International Council for Harmonisation Guideline M7(R1). Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk; ICH: Geneva, Switzerlad, 2017. [Google Scholar]
- Jordi, M.A. Reducing relative response factor variation using a multidetector approach for extractables and leachables (E&L) analysis to mitigate the need for uncertainty factors. J. Pharm. Biomed. Anal. 2020, 186, 113334. [Google Scholar] [PubMed]
- Jenke, D.; Odufu, A. Utilization of internal standard response factors to estimate the concentration of organic compounds leached from pharmaceutical packaging systems and application of such estimated concentrations to safety assessment. J. Chromatogr. Sci. 2012, 50, 206–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, G.; Lu, D.; Liu, C.; Hower, D. Analytical challenges and recent advances in the identification and quantitation of extractables and leachables in pharmaceutical and medical products. TrAC Trends Anal. Chem. 2021, 141, 116286. [Google Scholar] [CrossRef]
- Jenke, D. Correcting the analytical evaluation threshold (AET) and reported extractable’s concentrations for analytical response factor uncertainty associated with chromatographic screening for extractables/leachables. PDA J. Pharm. Sci. Technol. 2020, 74, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Liigand, P.; Liigand, J.; Kaupmees, K.; Kruve, A. 30 Years of research on ESI/MS response: Trends, contradictions and applications. Anal. Chim. Acta 2021, 1152, 238117. [Google Scholar] [CrossRef] [PubMed]
- Jordi, M.A.; Khera, S.; Roland, K.; Jiang, L.; Solomon, P.; Nelson, J.; Lateef, S.S.; Woods, J.; Martin, L.; Martin, S.; et al. Qualitative assessment of extractables from single-use components and the impact of reference standard selection. J. Pharm. Biomed. Anal. 2018, 150, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Kruve, A.; Kaupmees, K. Predicting ESI/MS signal change for anions in different solvents. Anal. Chem. 2017, 89, 5079–5086. [Google Scholar] [CrossRef] [PubMed]
- Leardi, R. Experimental design in chemistry: A tutorial. Anal. Chim. Acta 2009, 652, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.J.; Hunter, W.G. A Review of Response Surface Methodology: A Literature Survey. Technometrics 1966, 8, 571–590. [Google Scholar]
- Billen, J.; Desmet, G. Understanding and design of existing and future chromatographic support formats. J. Chromatogr. A 2007, 1168, 73–99. [Google Scholar] [CrossRef] [PubMed]
- Crotti, S.; Seraglia, R.; Traldi, P. Some thoughts on electrospray ionization mechanisms. Eur. J. Mass Spectrom. 2011, 17, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Konermann, L.; Ahadi, E.; Rodriguez, A.D.; Vahidi, S. Unraveling the mechanism of electrospray ionization. Anal. Chem. 2013, 85, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Eliuk, S.; Makarov, A. Evolution of Orbitrap Mass Spectrometry Instrumentation. Annu. Rev. Anal. Chem. 2015, 8, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Herrera, A.V.; Asensio-Ramos, M.; Hernández-Borges, J.; Rodríguez-Delgado, M.A. Dispersive liquid-liquid microextraction for determination of organic analytes. TrAC Trends Anal. Chem. 2010, 29, 728–751. [Google Scholar] [CrossRef]
- Jenke, D.; Liu, N.; Hua, Y.; Swanson, S.; Bogseth, R. A Means of Establishing and Justifying Binary Ethanol/Water Mixtures as Simulating Solvents in Extractables Studies. PDA J. Pharm. Sci. Technol. 2015, 69, 366–382. [Google Scholar] [CrossRef] [PubMed]
- Jenke, D.; Carlson, T. A Compilation of Safety Impact Information for Extractables Associated with Materials Used in Pharmaceutical Packaging, Administration and Manufacturing systems. PDA J. Pharm. Sci. Technol. 2014, 68, 407–455. [Google Scholar] [CrossRef] [PubMed]
- Kritikos, N.; Iliou, A.; Kalampaliki, A.D.; Gikas, E.; Kostakis, I.K.; Michel, B.Y.; Dotsikas, Y. Chemometrically Assisted Optimization of Pregabalin Fluorescent Derivatization Reaction with a Novel Xanthone Analogue and Validation of the Method for the Determination of Pregabalin in Bulk via a Plate Reader. Molecules 2022, 27, 1954. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Lower Level (−1) | Central Level (0) | Upper Level (+1) |
|---|---|---|---|
| Ammonium Formate (mM) | 2.0 | 5.0 | 7.5 |
| %Acetonitrile in Mobile Phase B | 5% | 10% | 40% |
| Time | %B | Flow |
|---|---|---|
| 0.0 | 2 | 0.25 |
| 2.0 | 2 | 0.25 |
| 16.3 | 95 | 0.25 |
| 19.0 | 100 | 0.40 |
| 23.0 | 100 | 0.40 |
| Rate (°C∙min−1) | Temperature (°C) | Hold Time (min) |
|---|---|---|
| - | 40 | 1.5 |
| 10.0 | 130 | 0.0 |
| 15.0 | 260 | 1.5 |
| 15.0 | 310 | 7.5 |
| CAS | Name | Retention Time (min) | Response Slope (Peak Area/ppm Concentration) |
|---|---|---|---|
| 122-20-3 | 1-[bis(2-hydroxypropyl) amino] propan-2-ol | 1.71 | 26,366,509.7 |
| 111-92-2 | N-butylbutan-1-amine | 6.19 | 406,919.2 |
| 88-19-7 | 2-methylbenzenesulfonamide | 7.13 | 24,803.8 |
| 738-70-5 | 5-[(3,4,5-trimethoxyphenyl) methyl] pyrimidine-2,4-diamine | 7.36 | 32,593,173.1 |
| 97-39-2 | 1,2-bis(2-methylphenyl) guanidine | 8.52 | 59,877,027.0 |
| 127-63-9 | Benzenesulfonylbenzene | 10.96 | 5,598,634.0 |
| 3622-84-2 | N-butylbenzenesulfonamide | 11.38 | 1,760,919.5 |
| 109-43-3 | dibutyl decanedioate | 16.67 | 31,284,068.5 |
| 301-02-0 | (Z)-octadec-9-enamide | 17.1 | 7,128,308.5 |
| 103-23-1 | bis(2-ethylhexyl) hexanedioate | 17.71 | 27,337,269.8 |
| 78-33-1 | tris(4-tert-butylphenyl) phosphate | 17.81 | 129,976,466.5 |
| 540-10-3 | Hexadecyl hexadecanoate | 17.78 | 468,840.4 |
| 2403-88-5 | 2,2,6,6-tetramethylpiperidin-4-ol | 1.08 | 19,585,172.7 |
| 80-09-1 | 4-(4-hydroxyphenyl) sulfanylphenol | 8.65 | 3,766,994.7 |
| 78-40-0 | Triethyl phosphate | 9.49 | 23,399,462.7 |
| 778-28-9 | butyl 4-methylbenzenesulfonate | 10.45 | 24,345.5 |
| 134-62-3 | N,N-diethyl-3-methylbenzamide | 11.74 | 36,836,082.3 |
| 4986-89-4 | [2-(hydroxymethyl)-3-prop-2-enoyloxy-2-(prop-2-enoyloxymethyl)propyl] prop-2-enoate | 12.21 | 6,454,678.4 |
| 80-18-2 | methyl benzenesulfonate | 13.07 | 64,883.4 |
| 71360-06-0 | bis(3,5-dimethylphenyl) phosphane | 13.7 | 127,148,710.4 |
| 124-30-1 | octadecan-1-amine | 16.64 | 63,886,631.2 |
| 629-54-9 | Hexadecanamide | 16.84 | 24,642,537.3 |
| 121-44-8 | N,N-diethylethanamine | 1.16 | 6,841,673.3 |
| 502-44-3 | oxepan-2-one | 4.96 | 1,851,724.8 |
| 149-30-4 | 3H-1,3-benzothiazole-2-thione | 9.68 | 1,344,481.4 |
| 629-01-6 | Octanamide | 11.43 | 5,178,127.2 |
| 2358-84-1 | 2-[2-(2-methylprop-2-enoyloxy) ethoxy] ethyl 2-methylprop-2-enoate | 11.94 | 6,987,279.9 |
| 1541-67-9 | 2-[dodecyl(2-hydroxyethyl) amino] ethanol | 13.89 | 132,161,614.0 |
| 115-86-6 | Triphenyl Phosphate | 14.33 | 126,329,399.2 |
| 1620-98-0 | 3,5-di-tert-butyl-4-hydroxybenzaldehyde | 14.43 | 10,122,291.2 |
| 3658-48-8 | bis(2-ethylhexoxy)-oxophosphanium | 15.04 | 8551.6 |
| 78-51-3 | Tris(2-butoxyethyl) phosphate | 15.42 | 98,345,028.7 |
| 78-30-8 | Tris(2-methylphenyl) phosphate | 15.70 | 143,057,783.9 |
| 32509-66-3 | 2-[3,3-bis(3-tert-butyl-4-hydroxyphenyl) butanoyloxy] ethyl 3,3-bis(3-tert-butyl-4-hydroxyphenyl) butanoate | 16.88 | 17,810,936.6 |
| 112-84-5 | (Z)-docos-13-enamide | 17.96 | 6,052,480.5 |
| CAS | Name | Retention Time (min) | Response Slope (Peak Area/ppm Concentration) |
|---|---|---|---|
| 70-49-5 | 2-sulfanylbutanedioic acid | 0.6 | 65,531.6 |
| 121-34-6 | 4-Hydroxy-3-methoxybenzoic acid, | 1.14 | 265,404.7 |
| 50-84-0 | 2,4-Dichlorobenzoic acid | 6.81 | 287,978.1 |
| 20434-05-3 | Bis(4-methoxyphenyl) phosphinic acid | 9.61 | 5,837,765.8 |
| 115-39-9 | 2,6-dibromo-4-[3-(3,5-dibromo-4-hydroxyphenyl)-1,1-dioxo-2,1lambda6-benzoxathiol-3-yl] phenol | 11.59 | 585,533.2 |
| 88-26-6 | 2,6-ditert-butyl-4-(hydroxymethyl) phenol | 14.15 | 2,062,965.5 |
| 506-13-8 | 16-Hydroxy-hexadecanoic acid | 15.44 | 3,415,425.7 |
| 514-10-3 | (1R,4aR,4bR,10aR)-1,4a-dimethyl-7-propan-2-yl-2,3,4,4b,5,6,10,10a-octahydrophenanthrene-1-carboxylic acid | 17.1 | 2,330,407.5 |
| 57-10-3 | hexadecanoic acid | 17.46 | 1,177,771.3 |
| 506-30-9 | icosanoic acid | 18.45 | 4,163,768.5 |
| 6683-19-8 | [3-[3-(3,5-ditert-butyl-4-hydroxyphenyl) propanoyloxy]-2,2-bis [3-(3,5-ditert-butyl-4-hydroxyphenyl) propanoyloxymethyl] propyl] 3-(3,5-ditert-butyl-4-hydroxyphenyl) propanoate | 19.31 | 2,763,898.8 |
| 100-21-0 | Terephthalic Acid | 0.6 | 465,975.7 |
| 90-64-2 | 2-hydroxy-2-phenylacetic acid | 1.36 | 1,015,852.8 |
| 149-57-5 | 2-Ethylhexanoic acid | 11.41 | 845,491.4 |
| 90-43-7 | 2-Phenylphenol | 12.7 | 165,700.2 |
| 4376-20-9 | 2-(2-ethylhexoxycarbonyl) benzoic acid | 13.82 | 3,050,868.3 |
| 20170-32-5 | 3-(3,5-Di-tert-butyl-4-hydroxyphenyl) propionic acid | 13.94 | 3,376,097.3 |
| 128-37-0 | 2,6-ditert-butyl-4-methylphenol | 16.41 | 232,745.4 |
| 36443-68-2 | 2-[2-[2-[3-(3-tert-butyl-4-hydroxy-5-methylphenyl) propanoyloxy] ethoxy] ethoxy] ethyl 3-(3-tert-butyl-4-hydroxy-5-methylphenyl) propanoate | 16.25 | 5,584,911.8 |
| 88-24-4 | 2-tert-butyl-6-[(3-tert-butyl-5-ethyl-2-hydroxyphenyl) methyl]-4-ethylphenol | 17.61 | 18,395,081.3 |
| 57-11-4 | octadecanoic acid | 17.98 | 2,294,766.8 |
| 1709-70-2 | 4-[[3,5-bis[(3,5-ditert-butyl-4-hydroxyphenyl) methyl]-2,4,6-trimethylphenyl] methyl]-2,6-ditert-butylphenol | 19.56 | 3,527,892.8 |
| CAS | IUPAC Name | Retention Time (min) | Response Slope (Peak Area/ppm Concentration) |
|---|---|---|---|
| 541-05-9 | 2,2,4,4,6,6-hexamethyl-1,3,5,2,4,6-trioxatrisilinane | 4.3 | 914,666.8 |
| 123-05-7 | 2-ethylhexanal | 6.4 | 842,858.9 |
| 100-52-7 | benzaldehyde | 6.5 | 341,982.6 |
| 111-13-7 | octan-2-one | 7.0 | 816,866.4 |
| 556-67-2 | 2,2,4,4,6,6,8,8-octamethyl-1,3,5,7,2,4,6,8-tetraoxatetrasilocane | 7.1 | 1,062,178.4 |
| 104-76-7 | 2-ethylhexan-1-ol | 7.6 | 550,482.7 |
| 6294-40-2 | 1-bromo-4-methylcyclohexane | 7.7 | 567,268.8 |
| 1678-93-9 | butylcyclohexane | 7.7 | 362,041.2 |
| 823-76-7 | 1-cyclohexylethanone | 7.7 | 37,097.9 |
| 98-86-2 | 1-phenylethanone | 8.3 | 881,780.5 |
| 617-94-7 | 2-phenylpropan-2-ol | 8.6 | 691,246.1 |
| 122-00-9 | 1-(4-methylphenyl) ethanone | 10.1 | 403,324.6 |
| 1126-79-0 | butoxybenzene | 10.1 | 821,422.5 |
| 112-41-4 | dodec-1-ene | 10.2 | 313,423.4 |
| 7169-34-8 | 1-benzofuran-3-one | 10.5 | 54,448.2 |
| 1731-84-6 | methyl nonanoate | 10.7 | 522,552.7 |
| 103-11-7 | 2-ethylhexyl prop-2-enoate | 10.7 | 380,414.3 |
| 7473-98-5 | 2-hydroxy-2-methyl-1-phenylpropan-1-one | 11.5 | 360,269.4 |
| 148-53-8 | 2-hydroxy-3-methoxybenzaldehyde | 11.8 | 83,272.7 |
| 112-29-8 | 1-bromodecane | 12.3 | 142,018.3 |
| 608-27-5 | 2,3-dichloroaniline | 12.3 | 237,885.3 |
| 321-60-8 | 1-fluoro-2-phenylbenzene | 12.5 | 599,634.6 |
| 141-28-6 | diethyl hexanedioate | 12.6 | 261,867.7 |
| 1120-36-1 | tetradec-1-ene | 12.7 | 330,158.3 |
| 719-22-2 | 2,6-ditert-butylcyclohexa-2,5-diene-1,4-dione | 13.6 | 103,298.9 |
| 7283-69-4 | bis(2-methylpropyl) (E)-but-2-enedioate | 13.7 | 314,769.3 |
| 96-76-4 | 2,4-ditert-butylphenol | 13.9 | 576,435.9 |
| 128-37-0 | 2,6-ditert-butyl-4-methylphenol | 14.0 | 522,447.2 |
| 2162-98-3 | 1,10-dichlorodecane | 14.3 | 732,021.1 |
| 544-76-3 | hexadecane | 14.7 | 382,995.0 |
| 119-61-9 | diphenylmethanone | 15.0 | 490,375.2 |
| 636-09-9 | diethyl benzene-1,4-dicarboxylate | 15.2 | 332,441.9 |
| 131-58-8 | (2-methylphenyl)-phenylmethanone | 15.3 | 349,309.7 |
| 451-40-1 | 1,2-diphenylethanone | 15.8 | 814,712.9 |
| 1620-98-0 | 3,5-ditert-butyl-4-hydroxybenzaldehyde | 16.1 | 376,847.4 |
| 84-74-2 | dibutyl benzene-1,2-dicarboxylate | 17.4 | 866,586.2 |
| 301-02-0 | (Z)-octadec-9-enamide | 19.9 | 100,949.6 |
| 115-86-6 | triphenyl phosphate | 20.3 | 147,008.6 |
| 88-24-4 | 2-tert-butyl-6-[(3-tert-butyl-5-ethyl-2-hydroxyphenyl) methyl]-4-ethylphenol | 21.1 | 207,657.5 |
| 117-81-7 | bis(2-ethylhexyl) benzene-1,2-dicarboxylate | 21.3 | 694,013.2 |
| 112-84-5 | (Z)-docos-13-enamide | 22.9 | 214,488.5 |
| 111-02-4 | (6E,10E,14E,18E)-2,6,10,15,19,23-hexamethyltetracosa-2,6,10,14,18,22-hexaene | 23.2 | 442,697.1 |
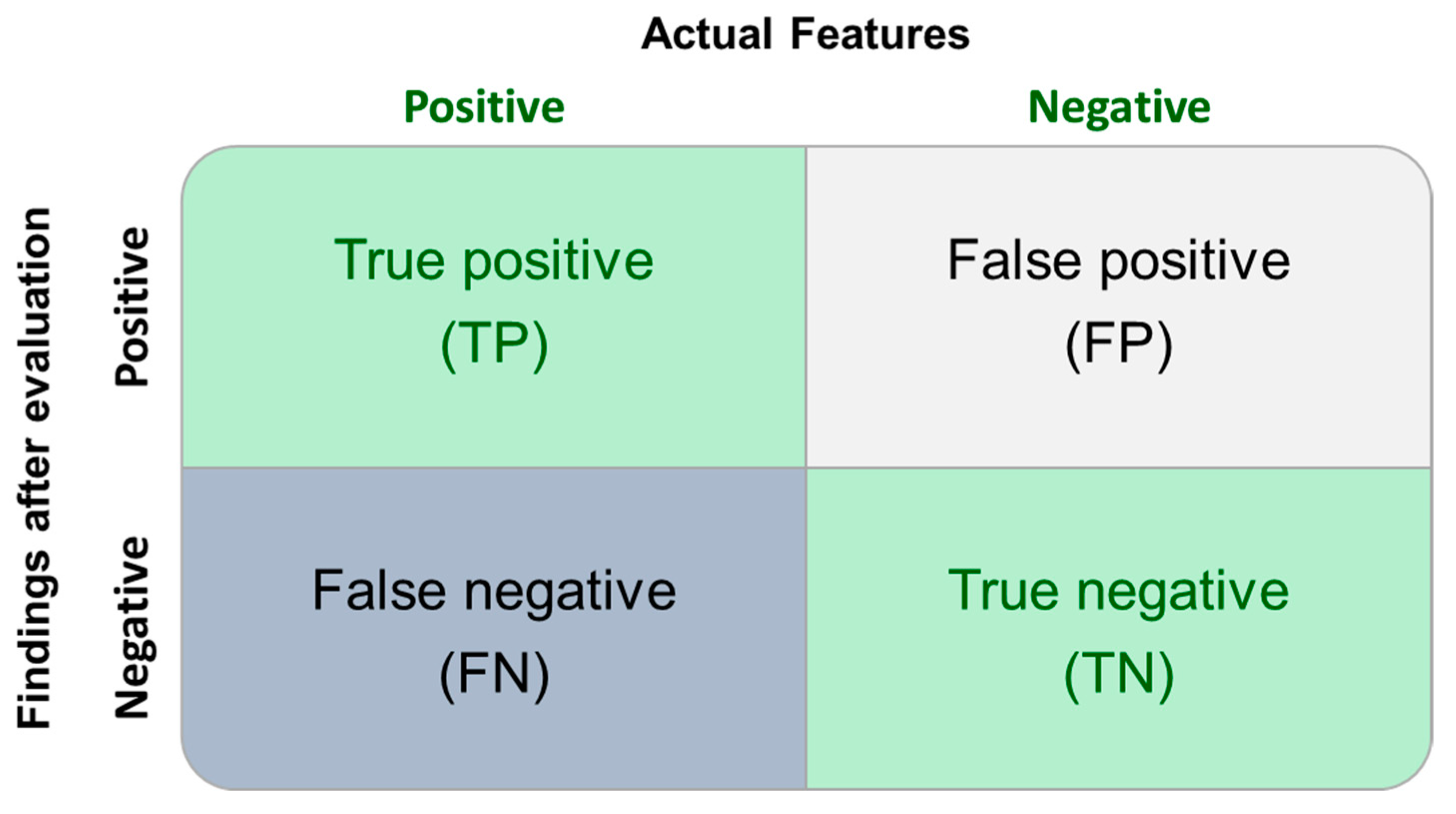
| Categories | In the Critical Population | Total Population | Considering the 0.5 UF Value | |
|---|---|---|---|---|
| True Positive | 16/21 | 16/26 | 20/21 | 20/26 |
| False Negative | 5/21 | 10/26 | 1/21 | 6/26 |
| Sensitivity | 76.2% | 61.5% * | 95.2% | 76.9% |
| Categories | Total Population | Considering the 0.5 UF Value |
|---|---|---|
| True Positive | 16/22 | 19/22 |
| False Negative | 6/22 | 3/22 |
| Sensitivity | 72.7% | 86.4% |
| Selected IS | Dodec-1-Ene (CAS: 112-41-4) | 1-Fluoro-2-Phenylbenzene (CAS: 321-60-8) | ||
|---|---|---|---|---|
| Scenarios | Non-Accounting for UF | With UF | Non-Accounting for UF | With UF |
| True Positive | 35/42 | 35/42 | 11/42 | 31/42 |
| False Negative | 7/42 | 7/42 | 31/42 | 11/42 |
| Sensitivity | 71.4% | 83.3% | 26.2% | 73.8% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kritikos, N.; Bletsou, A.; Konstantinou, C.; Neofotistos, A.-D.; Kousoulos, C.; Dotsikas, Y. Determination of Response Factors for Analytes Detected during Migration Studies, Strategy and Internal Standard Selection for Risk Minimization. Molecules 2023, 28, 5772. https://doi.org/10.3390/molecules28155772
Kritikos N, Bletsou A, Konstantinou C, Neofotistos A-D, Kousoulos C, Dotsikas Y. Determination of Response Factors for Analytes Detected during Migration Studies, Strategy and Internal Standard Selection for Risk Minimization. Molecules. 2023; 28(15):5772. https://doi.org/10.3390/molecules28155772
Chicago/Turabian StyleKritikos, Nikolaos, Anna Bletsou, Christina Konstantinou, Antonios-Dionysios Neofotistos, Constantinos Kousoulos, and Yannis Dotsikas. 2023. "Determination of Response Factors for Analytes Detected during Migration Studies, Strategy and Internal Standard Selection for Risk Minimization" Molecules 28, no. 15: 5772. https://doi.org/10.3390/molecules28155772
APA StyleKritikos, N., Bletsou, A., Konstantinou, C., Neofotistos, A.-D., Kousoulos, C., & Dotsikas, Y. (2023). Determination of Response Factors for Analytes Detected during Migration Studies, Strategy and Internal Standard Selection for Risk Minimization. Molecules, 28(15), 5772. https://doi.org/10.3390/molecules28155772