
Reduction of Hydrogen Peroxide by Human Mitochondrial Amidoxime Reducing Component Enzymes

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
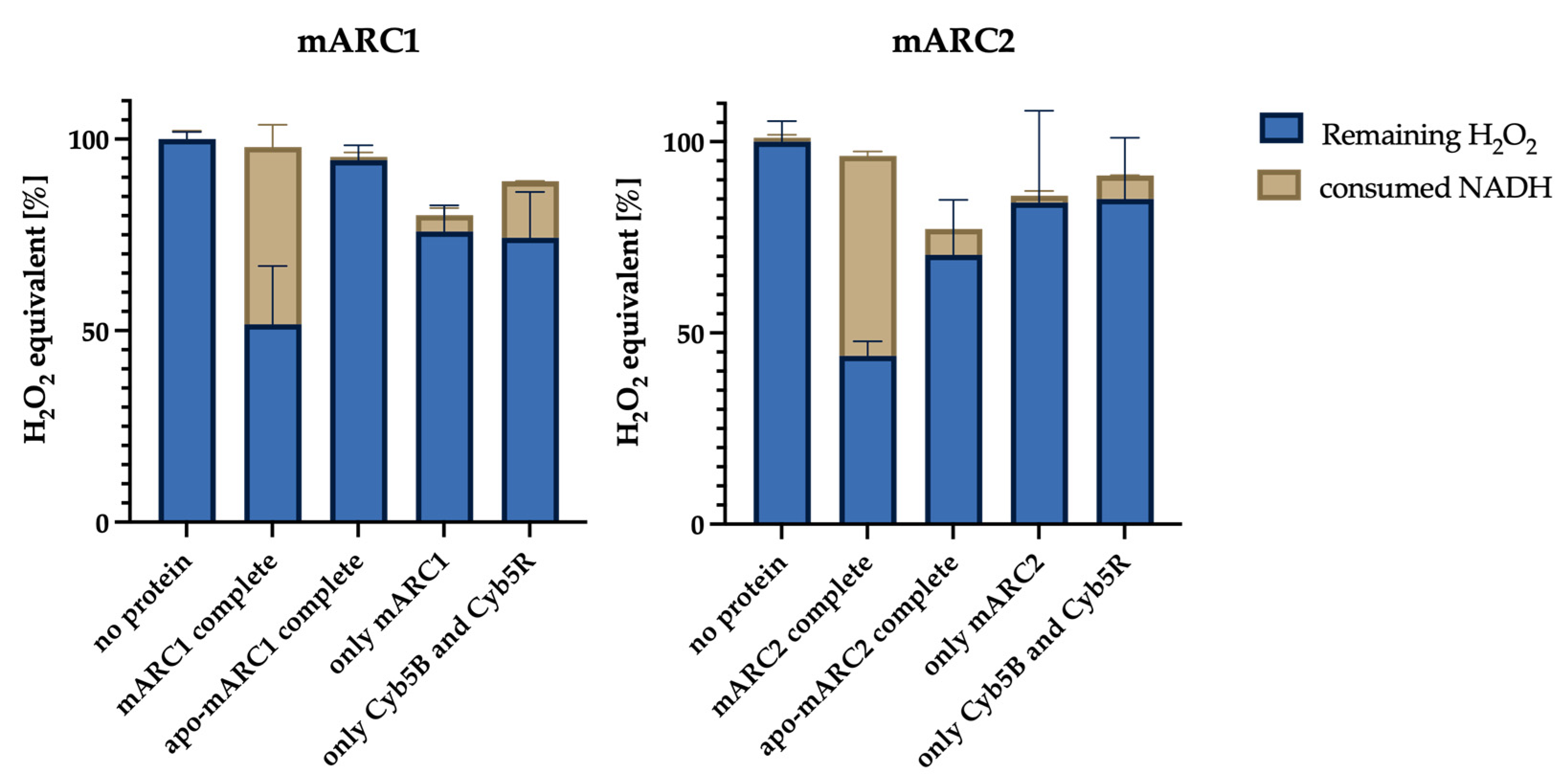
2.1. Molybdenum-Containing mARC1 and mARC2 Both Reduce H2O2
2.2. Kinetics of mARC-Dependent H2O2 Reduction
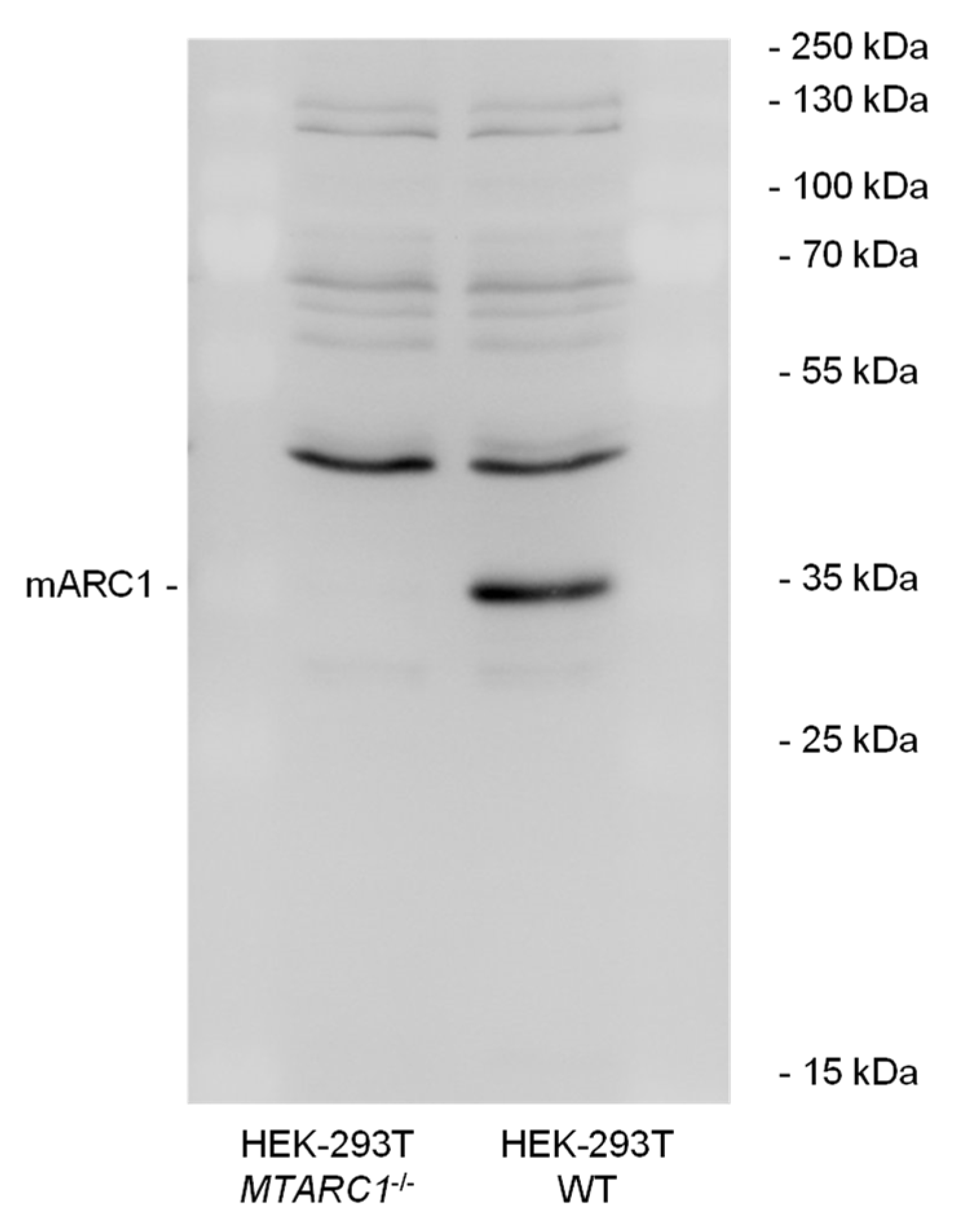
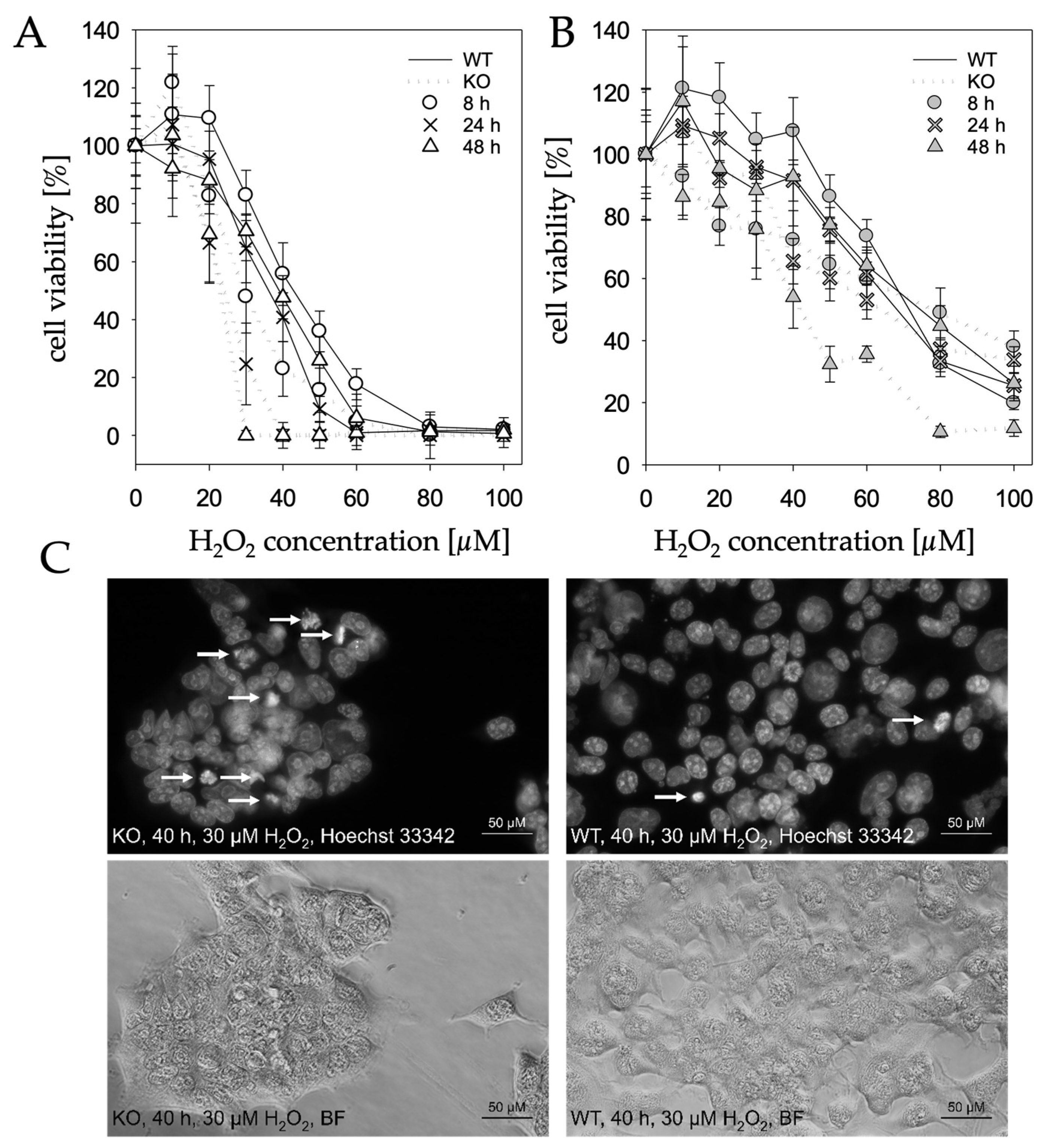
2.3. MTARC1 Knockout Decreases Cell Viability in Presence of H2O2
3. Discussion
4. Materials and Methods
4.1. Protein Sources
4.2. Photometric Assay
4.3. Fluorometric Activity Assay
4.4. Peroxide Assay
4.5. Molecular Biology
4.6. Mammalian Cell Culture
4.7. SDS-PAGE and Immunoblotting
4.8. Viability Assay
4.9. Proliferation Assay
4.10. Microscopy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Havemeyer, A.; Bittner, F.; Wollers, S.; Mendel, R.; Kunze, T.; Clement, B. Identification of the missing component in the mitochondrial benzamidoxime prodrug-converting system as a novel molybdenum enzyme. J. Biol. Chem. 2006, 281, 34796–34802. [Google Scholar] [CrossRef] [PubMed]
- Indorf, P.; Kubitza, C.; Scheidig, A.; Kunze, T.; Clement, B. Drug metabolism by the Mitochondrial Amidoxime Reducing Component (mARC): Rapid assay and identification of new substrates. J. Med. Chem. 2019, 63, 6538–6546. [Google Scholar] [CrossRef] [PubMed]
- Rajapakshe, A.; Astashkin, A.V.; Klein, E.L.; Reichmann, D.; Mendel, R.R.; Bittner, F.; Enemark, J.H. Structural studies of the molybdenum center of mitochondrial amidoxime reducing component (mARC) by pulsed EPR spectroscopy and 17O-labeling. Biochemistry 2011, 50, 8813–8822. [Google Scholar] [CrossRef] [PubMed]
- Ott, G.; Havemeyer, A.; Clement, B. The mammalian molybdenum enzymes of mARC. J. Biol. Inorg. Chem. 2015, 20, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Kubitza, C.; Bittner, F.; Ginsel, C.; Havemeyer, A.; Clement, B.; Scheidig, A.J. Crystal structure of human mARC1 reveals its exceptional position among eukaryotic molybdenum enzymes. Proc. Natl. Acad. Sci. USA 2018, 115, 11958–11963. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Zhang, Y. Abundance, Ubiquity and Evolution of Molybdoenzymes. In Molybdenum and Tungsten Enzymes: Biochemistry; Hille, R., Schulzke, C., Kirk, M.L., Eds.; The Royal Society of Chemistry: London, UK, 2016. [Google Scholar]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Kelley, E.E.; Khoo, N.K.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010, 48, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Leimkühler, S. The Inactivation of Human Aldehyde Oxidase 1 by Hydrogen Peroxide and Superoxide. Drug Metab. Dispos. 2021, 49, 729–735. [Google Scholar] [CrossRef]
- Hänsch, R.; Lang, C.; Riebeseel, E.; Lindigkeit, R.; Gessler, A.; Rennenberg, H.; Mendel, R.R. Plant sulfite oxidase as novel producer of H2O2: Combination of enzyme catalysis with a subsequent non-enzymatic reaction step. J. Biol. Chem. 2006, 281, 6884–6888. [Google Scholar] [CrossRef]
- But, P.G.; Fomina, V.A.; Murav’ev, R.A.; Rogovin, V.V. Myeloperoxidase from Neutrophil Peroxisomes. Biol. Bull. Russ. Acad. Sci. 2003, 30, 207–211. [Google Scholar] [CrossRef]
- Deisseroth, A.; Dounce, A.L. Catalase: Physical and chemical properties, mechanism of catalysis, and physiological role. Physiol. Rev. 1970, 50, 319–375. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Pearson, A.G.; Pullar, J.M.; Jönsson, T.J.; Lowther, W.T.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin 3 is more resilient to hyperoxidation than cytoplasmic peroxiredoxins. Biochem. J 2009, 421, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.M.; Busch, J.D.; Potting, C.; Baker, M.J.; Langer, T.; Schwarz, G. The Mitochondrial Amidoxime-Reducing Component (mARC1) is a novel signal-anchored protein of the outer mitochondrial membrane. J. Biol. Chem. 2012, 287, 42795–42803. [Google Scholar] [CrossRef] [PubMed]
- Janik, M.K.; Smyk, W.; Kruk, B.; Szczepankiewicz, B.; Gornicka, B.; Lebiedzinska-Arciszewska, M.; Potes, Y.; Simoes, I.C.M.; Weber, S.N.; Lammert, F.; et al. MARC1 p.A165T variant is associated with decreased markers of liver injury and enhanced antioxidant capacity in autoimmune hepatitis. Sci. Rep. 2021, 11, 24407. [Google Scholar] [CrossRef] [PubMed]
- Klopp, C.; Struwe, M.A.; Plieth, C.; Clement, B.; Scheidig, A.J. New Design of an Activity Assay Suitable for High-Throughput Screening of Substrates and Inhibitors of the Mitochondrial Amidoxime Reducing Component (mARC). Anal. Chem. 2023, 95, 12452–12458. [Google Scholar] [CrossRef] [PubMed]
- Clement, B.; Struwe, M.A. The History of mARC. Molecules 2023, 28, 4713. [Google Scholar] [CrossRef]
- Chamizo-Ampudia, A.; Galvan, A.; Fernandez, E.; Llamas, A. The Chlamydomonas reinhardtii molybdenum cofactor enzyme crARC has a Zn-dependent activity and protein partners similar to those of its human homologue. Eukaryot. Cell 2011, 10, 1270–1282. [Google Scholar] [CrossRef]
- Cordeiro, J.V.; Jacinto, A. The role of transcription-independent damage signals in the initiation of epithelial wound healing. Nat. Rev. Mol. Cell Biol. 2013, 14, 249–262. [Google Scholar] [CrossRef]
- Burdon, R.H.; Rice-Evans, C. Free radicals and the regulation of mammalian cell proliferation. Free Radic. Res. Commun. 1989, 6, 345–358. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Heininger, Y.M.; Mossman, B.T.; Heintz, N.H.; Forman, H.J.; Kalyanaraman, B.; Finkel, T.; Stamler, J.S.; Rhee, S.G.; van der Vliet, A. Redox-based regulation of signal transduction: Principles, pitfalls, and promises. Free Radic. Biol. Med. 2008, 45, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiù, V.; Maccarrone, M. Chronic inflammatory disorders and their redox control: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 2605–2641. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Liang, J.J.; Zhuang, X.; Chen, S.W.; Ng, T.K.; Chen, H. Intravitreal Injection of Hydrogen Peroxide Induces Acute Retinal Degeneration, Apoptosis, and Oxidative Stress in Mice. Oxid. Med. Cell. Longev. 2018, 2018, 5489476. [Google Scholar] [CrossRef] [PubMed]
- Kehrer, J.P.; Robertson, J.D.; Smith, C.V. 1.14—Free Radicals and Reactive Oxygen Species. In Comprehensive Toxicology, 2nd ed.; McQueen, C.A., Ed.; Elsevier: Oxford, UK, 2010; pp. 277–307. [Google Scholar]
- Chandrashekar, R.; Tsuji, N.; Morales, T.H.; Carmody, A.B.; Ozols, V.O.; Welton, J.; Tang, L. Removal of hydrogen peroxide by a 1-cysteine peroxiredoxin enzyme of the filarial parasite Dirofilaria immitis. Parasitol. Res. 2000, 86, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Riemer, J.; Schwarzländer, M.; Conrad, M.; Herrmann, J.M. Thiol switches in mitochondria: Operation and physiological relevance. Biol. Chem. 2015, 396, 465–482. [Google Scholar] [CrossRef] [PubMed]
- Florian, S.; Wingler, K.; Schmehl, K.; Jacobasch, G.; Kreuzer, O.J.; Meyerhof, W.; Brigelius-Flohé, R. Cellular and subcellular localization of gastrointestinal glutathione peroxidase in normal and malignant human intestinal tissue. Free Radic. Res. 2001, 35, 655–663. [Google Scholar] [CrossRef]
- Frederick, S.E.; Newcomb, E.H. Cytochemical localization of catalase in leaf microbodies (peroxisomes). J. Cell Biol. 1969, 43, 343–353. [Google Scholar] [CrossRef]
- Chatzi, A.; Manganas, P.; Tokatlidis, K. Oxidative folding in the mitochondrial intermembrane space: A regulated process important for cell physiology and disease. Biochim. Biophys. Acta 2016, 1863 Pt A, 1298–1306. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Horne, W.; Kamimura, S.; Niemelä, O.; Parkkila, S.; Ylä-Herttuala, S.; Brittenham, G.M. Experimental liver cirrhosis induced by alcohol and iron. J. Clin. Investig. 1995, 96, 620–630. [Google Scholar] [CrossRef]
- Islinger, M.; Luöers, G.H.; Li, K.W.; Loos, M.; Voölkl, A. Rat Liver Peroxisomes after Fibrate Treatment: A Survey using quantitative mass spectrometry. J. Biol. Chem. 2007, 282, 23055–23069. [Google Scholar] [CrossRef] [PubMed]
- Wiese, S.; Gronemeyer, T.; Ofman, R.; Kunze, M.; Grou, C.P.; Almeida, J.A.; Eisenacher, M.; Stephan, C.; Hayen, H.; Schollenberger, L.; et al. Proteomics Characterization of Mouse Kidney Peroxisomes by Tandem Mass Spectrometry and Protein Correlation Profiling. Mol. Cell. Proteom. 2007, 6, 2045–2057. [Google Scholar] [CrossRef] [PubMed]
- Wahl, B.; Reichmann, D.; Niks, D.; Krompholz, N.; Havemeyer, A.; Clement, B.; Messerschmidt, T.; Rothkegel, M.; Biester, H.; Hille, R.; et al. Biochemical and spectroscopic characterization of the human Mitochondrial Amidoxime Reducing Components hmARC-1 and hmARC-2 suggests the existence of a new molybdenum enzyme family in eukaryotes. J. Biol. Chem. 2010, 285, 37847–37859. [Google Scholar] [CrossRef] [PubMed]
- Palmer, T.; Santini, C.-L.; lobbi-Nivol, C.; Eave, D.J.; Boxer, D.H.; Giordano, G. Involvement of the narJ and mob gene products in distinct steps in the biosynthesis of the molybdoenzyme nitrate reductase in Escherichia coli. Mol. Biol. 1996, 20, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Stewart, V.; MacGregor, C.H. Nitrate Reductase in Escherichia Coli K-12: Involvement of chlC, chlE, and chlG Loci. J. Bacteriol. 1982, 151, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Guilbault, G.G.; Brignac, P.J., Jr.; Juneau, M. New substrates for the fluorometric determination of oxidative enzymes. Anal. Chem. 1968, 40, 1256–1263. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Struhl, K. Subcloning of DNA Fragments. Curr. Protoc. Mol. Biol. 1991, 13, 3.16.1–3.16.2. [Google Scholar] [CrossRef]
- Ladner, C.L.; Yang, J.; Turner, R.J.; Edwards, R.A. Visible fluorescent detection of proteins in polyacrylamide gels without staining. Anal. Biochem. 2004, 326, 13–20. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rixen, S.; Indorf, P.M.; Kubitza, C.; Struwe, M.A.; Klopp, C.; Scheidig, A.J.; Kunze, T.; Clement, B. Reduction of Hydrogen Peroxide by Human Mitochondrial Amidoxime Reducing Component Enzymes. Molecules 2023, 28, 6384. https://doi.org/10.3390/molecules28176384
Rixen S, Indorf PM, Kubitza C, Struwe MA, Klopp C, Scheidig AJ, Kunze T, Clement B. Reduction of Hydrogen Peroxide by Human Mitochondrial Amidoxime Reducing Component Enzymes. Molecules. 2023; 28(17):6384. https://doi.org/10.3390/molecules28176384
Chicago/Turabian StyleRixen, Sophia, Patrick M. Indorf, Christian Kubitza, Michel A. Struwe, Cathrin Klopp, Axel J. Scheidig, Thomas Kunze, and Bernd Clement. 2023. "Reduction of Hydrogen Peroxide by Human Mitochondrial Amidoxime Reducing Component Enzymes" Molecules 28, no. 17: 6384. https://doi.org/10.3390/molecules28176384
APA StyleRixen, S., Indorf, P. M., Kubitza, C., Struwe, M. A., Klopp, C., Scheidig, A. J., Kunze, T., & Clement, B. (2023). Reduction of Hydrogen Peroxide by Human Mitochondrial Amidoxime Reducing Component Enzymes. Molecules, 28(17), 6384. https://doi.org/10.3390/molecules28176384