
Fibrillization Process of Human Amyloid-Beta Protein (1–40) under a Molecular Crowding Environment Mimicking the Interior of Living Cells Using Cell Debris


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
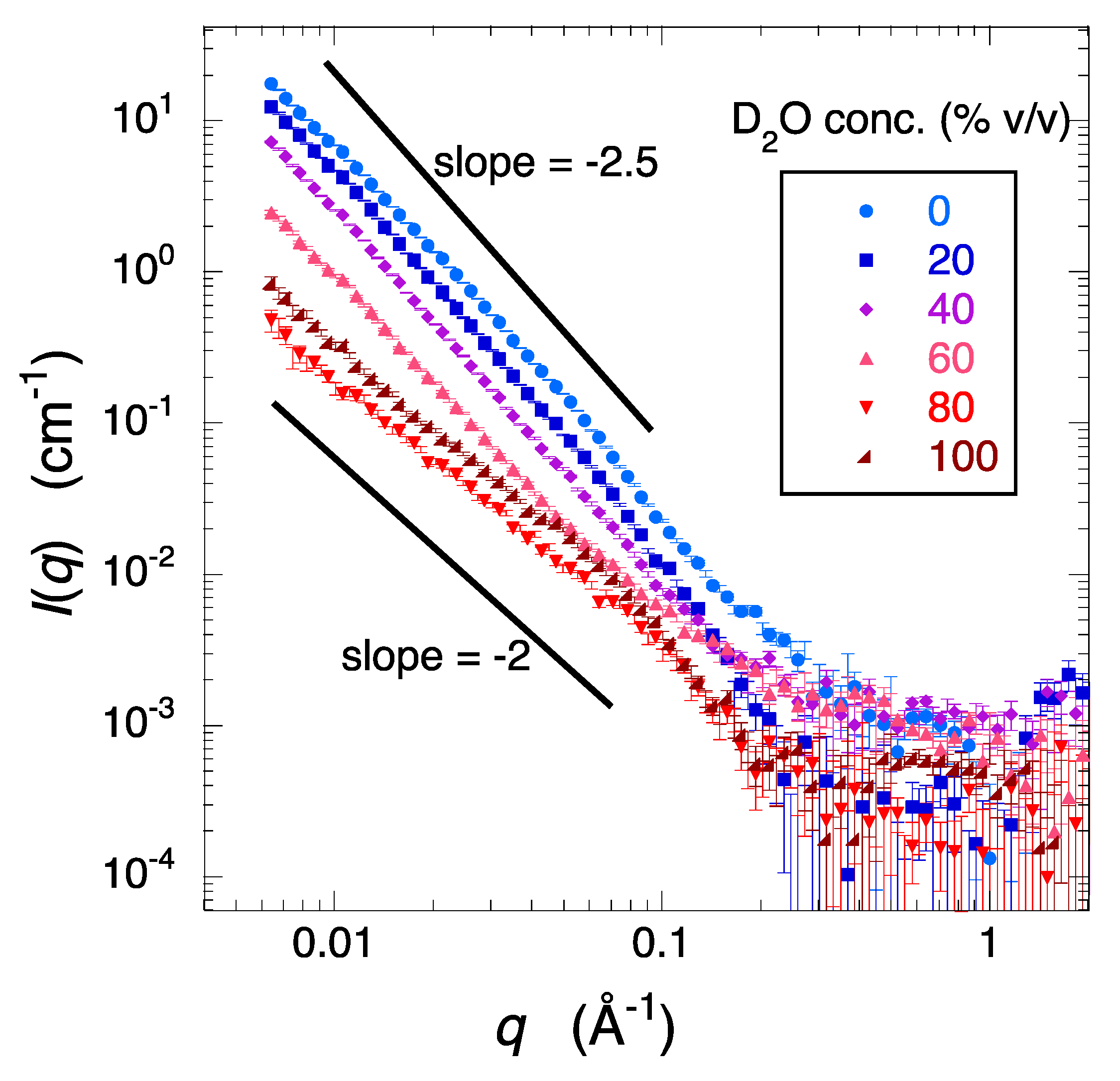
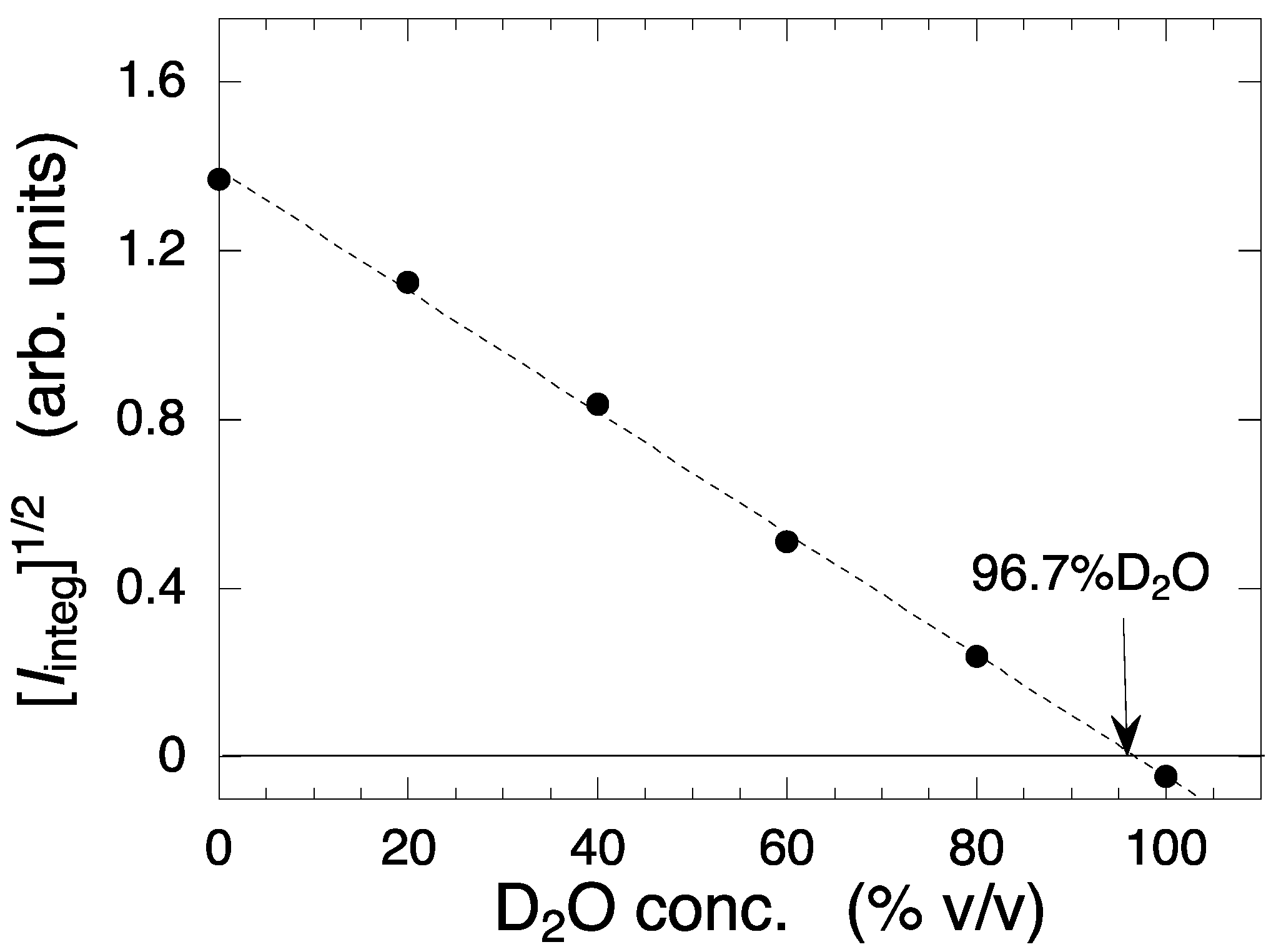
2.1. Observation and Structural Characterization of Deuterated Cell Debris Solution: A Model System Closely Mimicking the Intracellular Molecular Crowding Environment
2.2. Time Evolution of the Process of Fibril Formation of Human Amyloid-Beta (1–40) under a Molecular Crowded Environment Using Cell Debris Closely Mimicking the Interior of Living Cells
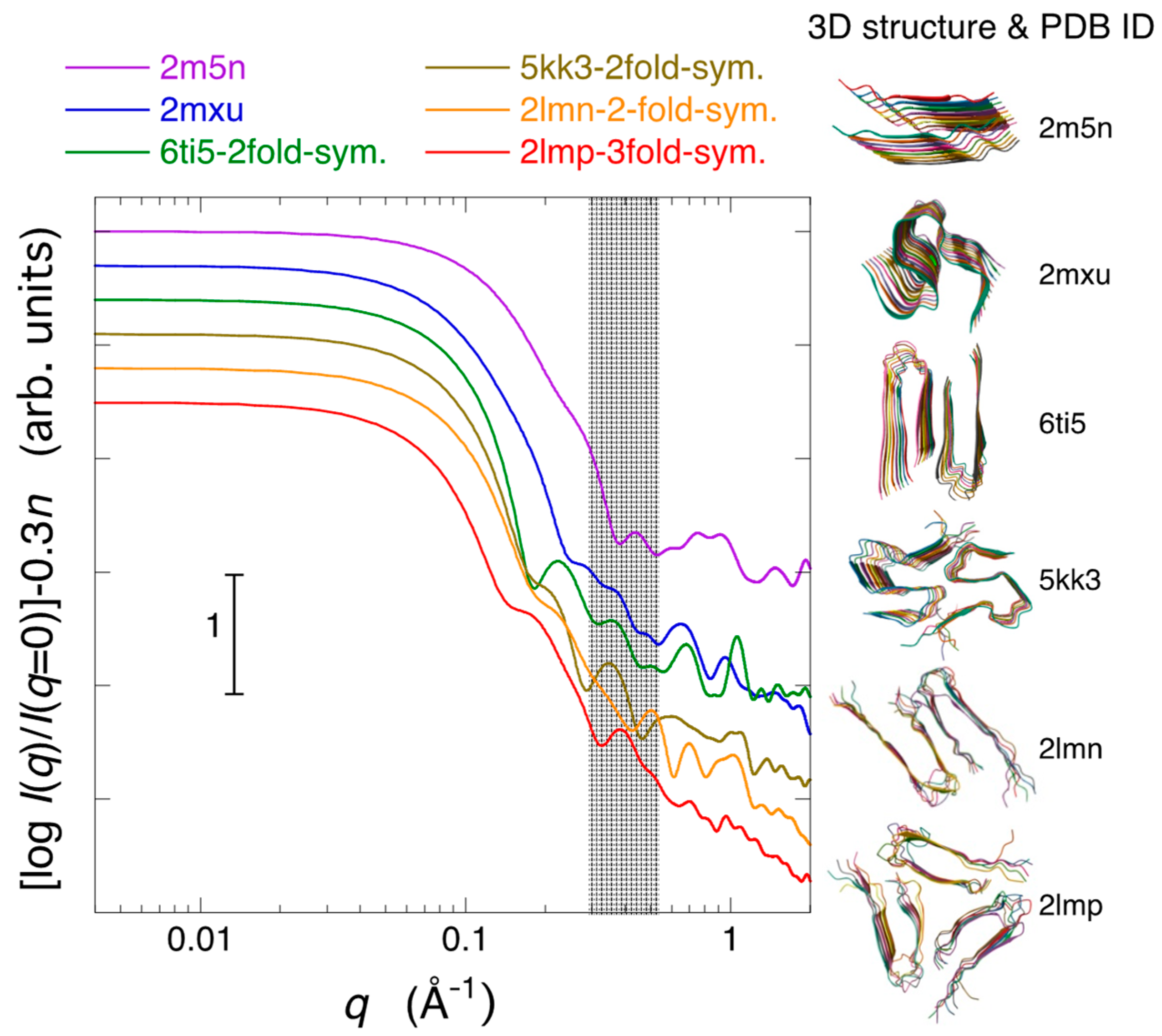
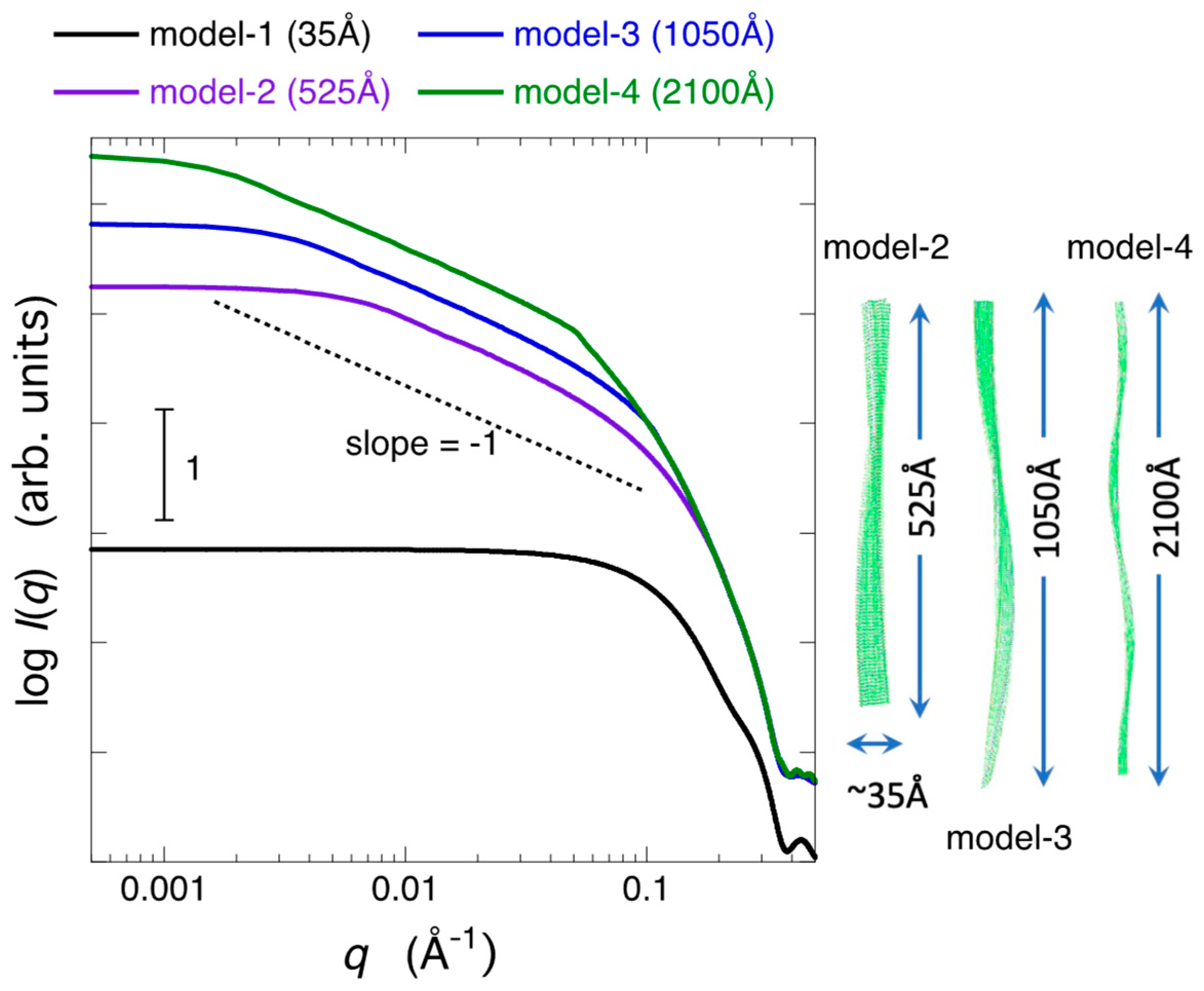
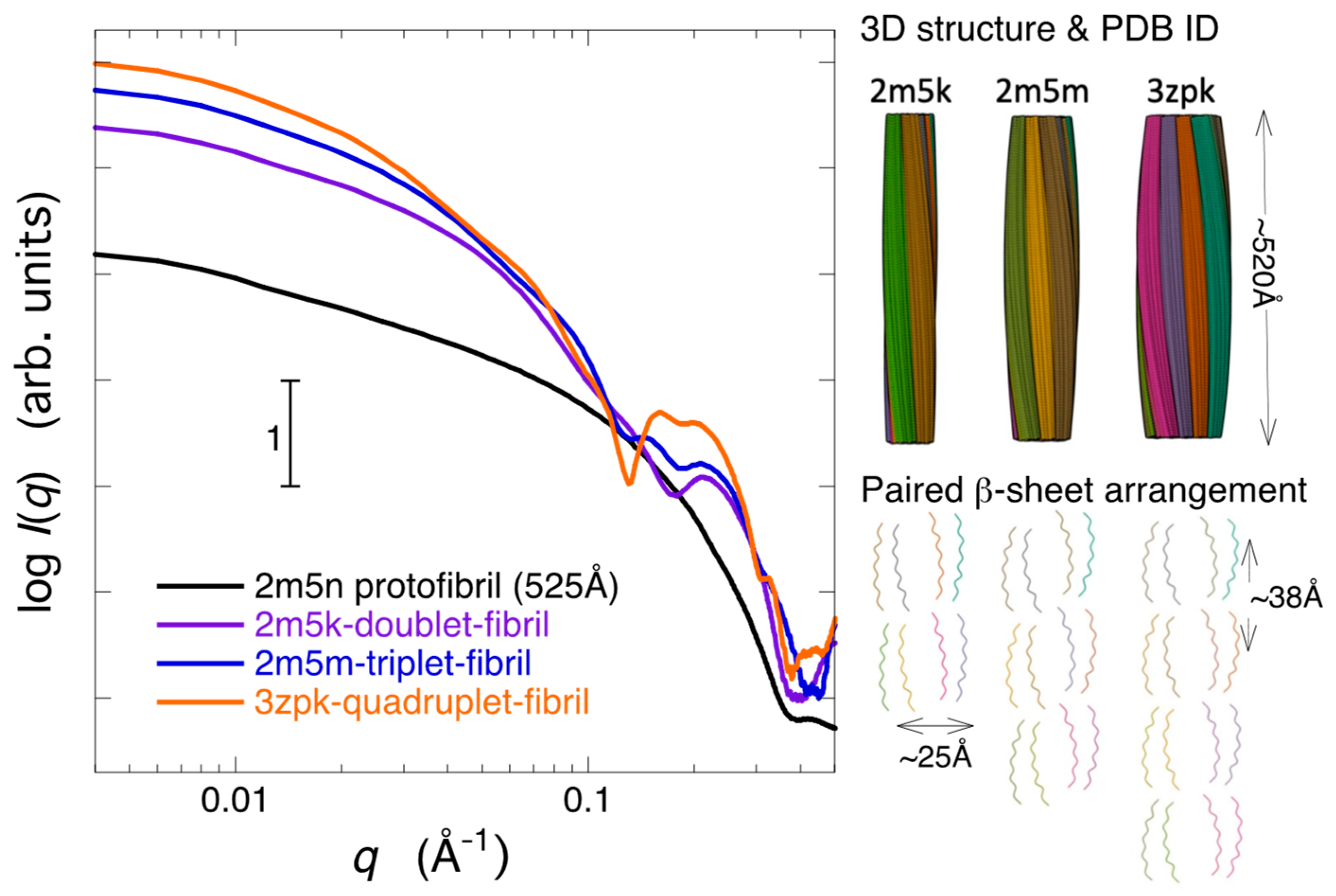
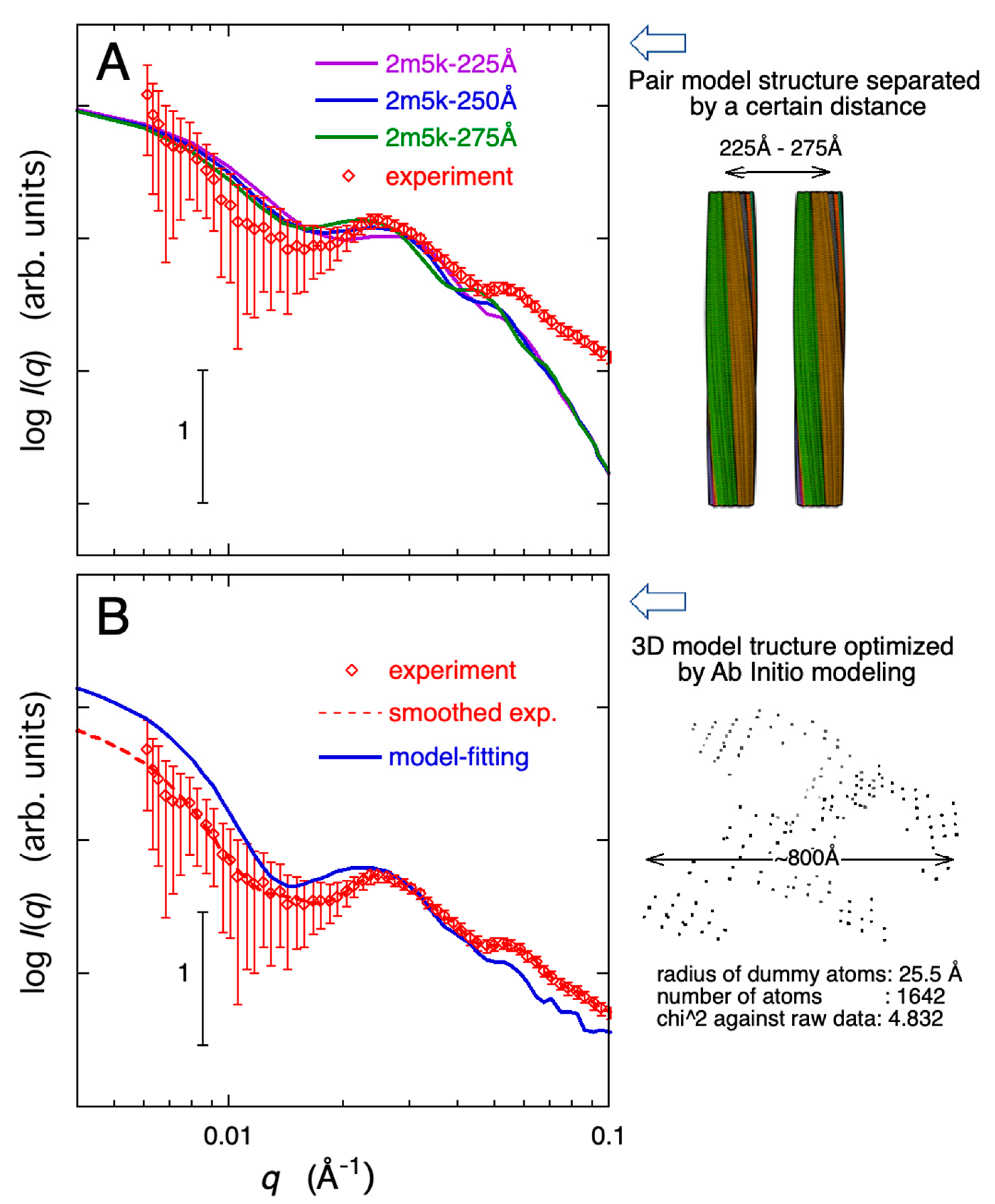
2.3. Modeling Analysis of the Aggregate Structure of Amyloid Fibrils in Cell Debris Solution: Possibility of the Formation of Higher-Ordered Fibril
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ellis, R.J.; Minton, A.P. Cell biology: Join the crowd. Nature 2003, 425, 27–28. [Google Scholar] [CrossRef] [PubMed]
- Rivas, G.; Ferrone, F.; Herzfeld, J. Life in a crowded world. EMBO Rep. 2004, 5, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Rivas, G.; Minton, A.P. Macromolecular-crowding in vitro, in vivo, and in between. Trends Biochem. Sci. 2016, 41, 970–981. [Google Scholar] [CrossRef]
- Gao, M.; Held, C.; Patra, S.; Arns, L.; Sadowski, G.; Winter, R. Crowders and Cosolvents—Major Contributors to the Cellular Milieu and Efficient Means to Counteract Environmental Stresses. Chem. Phys. Chem. 2017, 18, 2951–2972. [Google Scholar] [CrossRef] [PubMed]
- Rösgen, J.; Pettitt, B.M.; Bolen, D.W. Protein Folding, Stability, and Solvation Structure in Osmolyte Solutions. Biophys. J. 2005, 89, 2988–2997. [Google Scholar] [CrossRef] [PubMed]
- Rösgen, J.; Pettitt, B.M.; Bolen, D.W. An Analysis of the Molecular Origin of Osmolyte-dependent Protein Stability. Protein Sci. 2007, 16, 733–743. [Google Scholar] [CrossRef]
- Shahid, S.; Hassan, M.I.; Islam, A.; Ahmad, F. Size-Dependent Studies of Macromolecular-crowding on the Thermodynamic Stability, Structure and Functional Activity of Proteins: In Vitro and in Silico Approaches. Biochim. Biophys. Acta 2017, 1861, 178–197. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska, N.; Feig, M.; Trylska, J. Modeling Crowded Environment in Molecular Simulations. Front. Mol. Biosci. 2019, 6, 86. [Google Scholar] [CrossRef]
- Stuhrmann, H.B.; Miller, A. Small-Angle Scattering of Biological Structures. J. Appl. Cryst. 1978, 11, 325–345. [Google Scholar] [CrossRef]
- Stuhrmann, H.B. Unique aspects of neutron scattering for the study of biological systems. Rep. Prog. Phys. 2004, 67, 1073–1115. [Google Scholar] [CrossRef]
- Hirai, M. Neutrons in Soft Matter; Imae, T., Kanaya, T., Furusaka, M., Torikai, N., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 351–382. [Google Scholar]
- Ajito, S.; Hirai, M.; Iwase, H.; Shimizu, N.; Igarashi, N.; Ohta, N. Protective Action of Trehalose and Glucose on Protein Hydration Shell Clarified by Using X-ray and Neutron Scattering. Physica B Cond. Mat. 2018, 551, 249–255. [Google Scholar] [CrossRef]
- Ajito, S.; Iwase, H.; Takata, S.; Hirai, M. Sugar-Mediated Stabilization of Protein against Chemical or Thermal Denaturation. J. Phys. Chem. B 2018, 122, 8685–8697. [Google Scholar] [CrossRef] [PubMed]
- Hirai, M.; Ajito, S.; Sugiyama, M.; Iwase, H.; Takata, S.; Shimizu, N.; Igarashi, N.; Martel, A.; Porcar, L. Direct Evidence for the Effect of Glycerol on Protein Hydration and Thermal Structural Transition. Biophy. J. 2018, 115, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Hirai, M.; Ajito, S.; Sugiyama, M.; Iwase, H.; Takata, S.; Shimizu, N.; Igarashi, N.; Martel, A.; Porcar, L. Macromolecular-crowding Effect on Protein Structure and Hydration Clarified by Using X-ray and Neutron Scattering. Physica B Cond. Mat. 2018, 551, 212–217. [Google Scholar] [CrossRef]
- Harada, R.; Sugita, Y.; Feig, M. Protein Crowding Affects Hydration Structure and Dynamics. J. Am. Chem. Soc. 2012, 134, 4842–4849. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yu, I.; Feig, M.; Sugita, Y. Influence of Protein Crowder Size on Hydration Structure and Dynamics in Macromolecular-crowding. Chem. Phys. Lett. 2017, 671, 63–70. [Google Scholar] [CrossRef]
- Hirai, M.; Ajito, S.; Arai, S.; Adachi, M.; Shimizu, R.; Wakamatsu, K.; Takata, S.; Iwase, H. Observation of Protein and Lipid Membrane Structures in a Model Mimicking the Molecular-Crowding Environment of Cells Using Neutron Scattering and Cell Debris. J. Phys. Chem. B 2019, 123, 3189–3198. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Masters, C.L.; Selkoe, D.J. Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. Aβ oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.M.; Tycko, R. Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef] [PubMed]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef]
- Munishkina, L.A.; Cooper, E.M.; Uversky, V.N.; Fink, A.L. The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J. Mol. Recognit. 2004, 17, 456–464. [Google Scholar] [CrossRef]
- Assarsson, S.; Linse, S.; Cabaleiro-Lago, C. Effects of Polyamino Acids and Polyelectrolytes on Amyloid β Fibril. Langmuir 2014, 30, 8812–8818. [Google Scholar] [CrossRef]
- Theillet, F.X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef]
- Meyer, N.; Arroyo, N.; Janot, J.M.; Lepoitevin, M.; Stevenson, A.; Nemeir, I.A.; Perrier, V.; Bougard, D.; Belondrade, M.; Cot, D.; et al. Detection of Amyloid-β Fibrils Using Track-Etched Nanopores: Effect of Geometry and Crowding Formation. ACS Sens. 2021, 6, 3733–3743. [Google Scholar] [CrossRef]
- Li, X.; Mehler, E.L. Simulation of Molecular Crowding Effects on an Alzheimer’s β-Amyloid Peptide. Cell Biochem. Biophys. 2006, 46, 123–141. [Google Scholar] [CrossRef]
- Latshaw, D.C.; Cheon, M.; Hall, C.K. Effects of Macromolecular Crowding on Amyloid Beta (16–22) Aggregation Using Coarse-Grained Simulations. J. Phys. Chem. B 2014, 118, 13513–13526. [Google Scholar] [CrossRef]
- Bridstrup, J.; Yuan, J.M.; Schreck, J.S. Stochastic kinetic study of protein aggregation and molecular crowding effects of Aβ40 and Aβ42. J. Chin. Chem. Soc. 2023, 70, 579–588. [Google Scholar] [CrossRef]
- Voet, D.; Voet, J.D. Biochemistry; John Willey & Sons, Inc.: Hoboken, NJ, USA, 1995. [Google Scholar]
- Porod, G. Die Röntgenkleinwinkelstreuung von dichtgepackten kolloiden Systemen. Kolloid Z. 1951, 124, 83–114. [Google Scholar] [CrossRef]
- Feigin, L.A.; Svergun, D.I. Structure Analysis by Small-Angle X-ray and Neutron Scattering; Taylor, G.W., Ed.; Plenum Press: New York, NY, USA, 1987. [Google Scholar]
- Martin, J.E.; Hurd, A.J. Scattering from Fractals. J. Appl. Cryst. 1989, 20, 61–78. [Google Scholar] [CrossRef]
- Schaefer, D.W. Polymers, Fractals, and Ceramic Materials. Science 1989, 243, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.I.; Barberato, C.; Koch, M.H.J. CRYSOL-a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J. Appl. Cryst. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Svergun, D.I.; Richard, S.; Koch, M.H.J.; Sayers, S.; Kuprin, S.; Zaccai, G. Protein Hydration in Solution: Experimental Observation by X-ray and Neutron scattering. Proc. Natl. Acad. Sci. USA 1998, 95, 2267–2272. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Müller, S.A.; et al. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. A beta (1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef]
- Cerofolini, L.; Ravera, E.; Bologna, S.; Wiglenda, T.; Boddrich, A.; Purfurst, B.; Benilova, I.; Korsak, M.; Gallo, G.; Rizzo, D.; et al. Mixing A beta (1–40) and A beta (1–42) peptides generates unique amyloid fibrils. Chem. Commun. 2020, 56, 8830–8833. [Google Scholar] [CrossRef]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic A beta 42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef]
- Hirai, M.; Iwase, H.; Hayakawa, T.; Miura, K.; Inoue, K. Structural Hierarchy of Several Proteins Observed by Wide-Angle Solution Scattering. J. Synchrotron Rad. 2002, 9, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Hirai, M.; Koizumi, M.; Hayakawa, T.; Takahashi, H.; Abe, S.; Hirai, H.; Miura, K.; Inoue, K. Hierarchical Map of Protein Unfolding and Refolding at Thermal Equilibrium Revealed by Wide-Angle X-ray Scattering. Biochemistry 2004, 43, 9036–9049. [Google Scholar] [CrossRef] [PubMed]
- Sachse, C.; Fändrich, M.; Grigorieff, N. Paired β-sheet structure of an Aβ (1–40) amyloid fibril revealed by electron microscopy. Proc. Natl. Acad. Sci. USA 2008, 105, 7462–7466. [Google Scholar] [CrossRef]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Cryst. 2009, 42, 342–346. [Google Scholar] [CrossRef]
- Takata, S.; Suzuki, J.; Shinohara, T.; Oku, T.; Timinaga, T.; Ohishi, K.; Iwase, H.; Nakatani, T.; Inamura, Y.; Ito, T.; et al. The Design and q Resolution of the Small and Wide Angle Neutron Scattering Instrument (TAIKAN) in J-PARC. JPS Conf. Proc. 2015, 8, 036020. [Google Scholar] [CrossRef]
- Sugiyama, M.; Yagi, H.; Yamaguchi, T.; Kumoi, K.; Hirai, M.; Oba, Y.; Sato, N.; Porcar, L.; Martele, A.; Kato, K. Conformational characterization of a protein complex involving intrinsically disordered protein by small-angle neutron scattering using the inverse contrast matching method: A case study of interaction between α-synuclein and PbaB tetramer as a model chaperone. J. Appl. Cryst. 2014, 47, 430–435. [Google Scholar]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirai, M.; Arai, S.; Iwase, H. Fibrillization Process of Human Amyloid-Beta Protein (1–40) under a Molecular Crowding Environment Mimicking the Interior of Living Cells Using Cell Debris. Molecules 2023, 28, 6555. https://doi.org/10.3390/molecules28186555
Hirai M, Arai S, Iwase H. Fibrillization Process of Human Amyloid-Beta Protein (1–40) under a Molecular Crowding Environment Mimicking the Interior of Living Cells Using Cell Debris. Molecules. 2023; 28(18):6555. https://doi.org/10.3390/molecules28186555
Chicago/Turabian StyleHirai, Mitsuhiro, Shigeki Arai, and Hiroki Iwase. 2023. "Fibrillization Process of Human Amyloid-Beta Protein (1–40) under a Molecular Crowding Environment Mimicking the Interior of Living Cells Using Cell Debris" Molecules 28, no. 18: 6555. https://doi.org/10.3390/molecules28186555