
Exploring the Solubility Limits of Edaravone in Neat Solvents and Binary Mixtures: Experimental and Machine Learning Study


Abstract
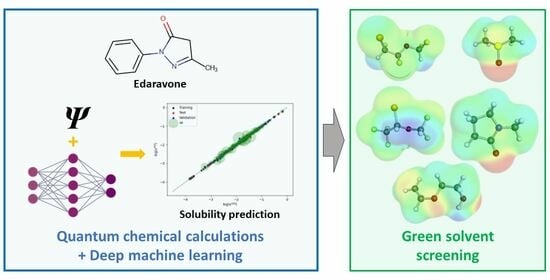
:
1. Introduction
2. Results and Discussion
2.1. Solubility Dataset
2.2. Extension of EDA Solubility Space with Neat Solvents
2.3. Extension of EDA Solubility Space with Aqueous Binary Solvents
2.4. Machine Learning Solubility Model
2.5. The Solubility Space Characteristics
3. Materials and Methods
3.1. Materials
3.2. Solubility Measurements
3.3. Instrumental Analysis of Solid Residues
3.4. Solubility Data Curation
3.5. Model Development
3.6. Molecular Descriptors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Drugbank Edaravone. Available online: https://go.drugbank.com/drugs/DB12243 (accessed on 28 November 2022).
- Watanabe, T.; Tahara, M.; Todo, S. The Novel Antioxidant Edaravone: From Bench to Bedside. Cardiovasc. Ther. 2008, 26, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, R.; Kuhad, A.; Kuhad, A. Edaravone: A new hope for deadly amyotrophic lateral sclerosis. Drugs Today 2018, 54, 349. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.-F.; Yan, N.; Xu, H.; Sun, J.-H.; Xiong, Y.-C.; Deng, X.-M. Edaravone, a free radical scavenger, is effective on neuropathic pain in rats. Brain Res. 2009, 1248, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Katsumura, Y.; Hata, K.; Muroya, Y.; Nakagawa, K. Pulse radiolysis study on free radical scavenger edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one). J. Photochem. Photobiol. B Biol. 2007, 89, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Martínez, F.; Jouyban, A.; Acree, W.E. Pharmaceuticals solubility is still nowadays widely studied everywhere. Pharm. Sci. 2017, 23, 1–2. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Govender, R.; Abrahmsén-Alami, S.; Folestad, S.; Olsson, M.; Larsson, A. Enabling modular dosage form concepts for individualized multidrug therapy: Expanding the design window for poorly water-soluble drugs. Int. J. Pharm. 2021, 602, 120625. [Google Scholar] [CrossRef]
- Lu, W.; Chen, H. Application of deep eutectic solvents (DESs) as trace level drug extractants and drug solubility enhancers: State-of-the-art, prospects and challenges. J. Mol. Liq. 2022, 349, 118105. [Google Scholar] [CrossRef]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef]
- Ting, J.M.; Porter, W.W.; Mecca, J.M.; Bates, F.S.; Reineke, T.M. Advances in Polymer Design for Enhancing Oral Drug Solubility and Delivery. Bioconjug. Chem. 2018, 29, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, S.R.; Savadkouhi, N.; Ebrahimzadeh, M.A. Drug design strategies that aim to improve the low solubility and poor bioavailability conundrum in quercetin derivatives. Expert Opin. Drug Discov. 2023, 18, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H. Biological assay challenges from compound solubility: Strategies for bioassay optimization. Drug Discov. Today 2006, 11, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Popa-Burke, I.; Russell, J. Compound Precipitation in High-Concentration DMSO Solutions. SLAS Discov. 2014, 19, 1302–1308. [Google Scholar] [CrossRef]
- Papaneophytou, C.P.; Mettou, A.K.; Rinotas, V.; Douni, E.; Kontopidis, G.A. Solvent Selection for Insoluble Ligands, a Challenge for Biological Assay Development: A TNF-α/SPD304 Study. ACS Med. Chem. Lett. 2013, 4, 137–141. [Google Scholar] [CrossRef]
- Hussain, A.; Alshehri, S.; Ramzan, M.; Afzal, O.; Altamimi, A.S.A.; Alossaimi, M.A. Biocompatible solvent selection based on thermodynamic and computational solubility models, in-silico GastroPlus prediction, and cellular studies of ketoconazole for subcutaneous delivery. J. Drug Deliv. Sci. Technol. 2021, 65, 102699. [Google Scholar] [CrossRef]
- Cysewski, P.; Jeliński, T.; Przybyłek, M.; Nowak, W.; Olczak, M. Solubility Characteristics of Acetaminophen and Phenacetin in Binary Mixtures of Aqueous Organic Solvents: Experimental and Deep Machine Learning Screening of Green Dissolution Media. Pharmaceutics 2022, 14, 2828. [Google Scholar] [CrossRef]
- Wu, X.; Yin, X.; Tang, T.; Zheng, H.; Xu, W.; Lin, Z.; Chen, X.; Li, R.; Zhao, J.; Han, D. Solubility of Edaravone in Four Mixed Solvents at 273.15-313.15 K and Correlation of Jouyban-Acree and CNIBS/R-K Models. J. Chem. Eng. Data 2020, 65, 1460–1467. [Google Scholar] [CrossRef]
- Li, R.; Yao, L.; Khan, A.; Zhao, B.; Wang, D.; Zhao, J.; Han, D. Co-solvence phenomenon and thermodynamic properties of edaravone in pure and mixed solvents. J. Chem. Thermodyn. 2019, 138, 304–312. [Google Scholar] [CrossRef]
- Acree, W.E.; Howard Rytting, J. Solubility in Binary Solvent Systems I: Specific versus Nonspecific Interactions. J. Pharm. Sci. 1982, 71, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.; Rengaswamy, R. Machine Learning Derived Quantitative Structure Property Relationship (QSPR) to Predict Drug Solubility in Binary Solvent Systems. Ind. Eng. Chem. Res. 2019, 58, 3082–3092. [Google Scholar] [CrossRef]
- Jouyban-Gharamaleki, A. Solubility correlation of structurally related drugs in binary solvent mixtures. Int. J. Pharm. 1998, 166, 205–209. [Google Scholar] [CrossRef]
- Acree, W.E. Mathematical representation of thermodynamic properties. Part 2. Derivation of the combined nearly ideal binary solvent (NIBS)/Redlich-Kister mathematical representation from a two-body and three-body interactional mixing model. Thermochim. Acta 1992, 198, 71–79. [Google Scholar] [CrossRef]
- Qiu, J.; Huang, H.; He, H.; Liu, H.; Hu, S.; Han, J.; Yi, D.; An, M.; Guo, Y.; Wang, P. Solubility Determination and Thermodynamic Modeling of Edaravone in Different Solvent Systems and the Solvent Effect in Pure Solvents. J. Chem. Eng. Data 2020, 65, 3240–3251. [Google Scholar] [CrossRef]
- Hatefi, A.; Jouyban, A.; Mohammadian, E.; Acree, W.E.; Rahimpour, E. Prediction of paracetamol solubility in cosolvency systems at different temperatures. J. Mol. Liq. 2019, 273, 282–291. [Google Scholar] [CrossRef]
- Cysewski, P.; Jeliński, T.; Przybyłek, M. Finding the Right Solvent: A Novel Screening Protocol for Identifying Environmentally Friendly and Cost-Effective Options for Benzenesulfonamide. Molecules 2023, 28, 5008. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Guo, Y.; He, H.; Huang, H.; Qiu, J.; Han, J.; Hu, S.; Liu, H.; Zhao, Y. Solubility determination and thermodynamic modeling of n-acetylglycine in different solvent systems. J. Chem. Eng. Data 2021, 66, 1344–1355. [Google Scholar]
- Cysewski, P.; Przybyłek, M.; Rozalski, R. Experimental and Theoretical Screening for Green Solvents Improving Sulfamethizole Solubility. Materials 2021, 14, 5915. [Google Scholar] [CrossRef]
- Li, Z. Equilibrium solubility of edaravone in some binary aqueous and non-aqueous solutions reconsidered: Extended Hildebrand solubility approach, transfer property and preferential solvation. J. Mol. Liq. 2021, 331, 115794. [Google Scholar] [CrossRef]
- Cysewski, P.; Przybyłek, M.; Kowalska, A.; Tymorek, N. Thermodynamics and intermolecular interactions of nicotinamide in neat and binary solutions: Experimental measurements and COSMO-RS concentration dependent reactions investigations. Int. J. Mol. Sci. 2021, 22, 7365. [Google Scholar] [CrossRef] [PubMed]
- Kolář, P.; Shen, J.-W.; Tsuboi, A.; Ishikawa, T. Solvent selection for pharmaceuticals. Fluid Phase Equilib. 2002, 194, 771–782. [Google Scholar] [CrossRef]
- Modarresi, H.; Conte, E.; Abildskov, J.; Gani, R.; Crafts, P. Model-Based Calculation of Solid Solubility for Solvent Selection—A Review. Ind. Eng. Chem. Res. 2008, 47, 5234–5242. [Google Scholar] [CrossRef]
- Constable, D.J.C.; Jimenez-Gonzalez, C.; Henderson, R.K. Perspective on Solvent Use in the Pharmaceutical Industry. Org. Process Res. Dev. 2007, 11, 133–137. [Google Scholar] [CrossRef]
- Baumann, M.; Baxendale, I.R. The synthesis of active pharmaceutical ingredients (APIs) using continuous flow chemistry. Beilstein J. Org. Chem. 2015, 11, 1194–1219. [Google Scholar] [CrossRef] [PubMed]
- Simić, S.; Zukić, E.; Schmermund, L.; Faber, K.; Winkler, C.K.; Kroutil, W. Shortening Synthetic Routes to Small Molecule Active Pharmaceutical Ingredients Employing Biocatalytic Methods. Chem. Rev. 2022, 122, 1052–1126. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, E.; Tula, A.K.; Gani, R. Solvent selection methodology for pharmaceutical processes: Solvent swap. Chem. Eng. Res. Des. 2016, 115, 443–461. [Google Scholar] [CrossRef]
- Laboukhi-Khorsi, S.; Daoud, K.; Chemat, S. Efficient Solvent Selection Approach for High Solubility of Active Phytochemicals: Application for the Extraction of an Antimalarial Compound from Medicinal Plants. ACS Sustain. Chem. Eng. 2017, 5, 4332–4339. [Google Scholar] [CrossRef]
- Ottoboni, S.; Wareham, B.; Vassileiou, A.; Robertson, M.; Brown, C.J.; Johnston, B.; Price, C.J. A Novel Integrated Workflow for Isolation Solvent Selection Using Prediction and Modeling. Org. Process Res. Dev. 2021, 25, 1143–1159. [Google Scholar] [CrossRef]
- DeSimone, J.M. Practical approaches to green solvents. Science 2002, 297, 799–803. [Google Scholar] [CrossRef]
- Cvjetko Bubalo, M.; Vidović, S.; Radojčić Redovniković, I.; Jokić, S. Green solvents for green technologies. J. Chem. Technol. Biotechnol. 2015, 90, 1631–1639. [Google Scholar] [CrossRef]
- Becker, J.; Manske, C.; Randl, S. Green chemistry and sustainability metrics in the pharmaceutical manufacturing sector. Curr. Opin. Green Sustain. Chem. 2022, 33, 100562. [Google Scholar] [CrossRef]
- Castiello, C.; Junghanns, P.; Mergel, A.; Jacob, C.; Ducho, C.; Valente, S.; Rotili, D.; Fioravanti, R.; Zwergel, C.; Mai, A. GreenMedChem: The challenge in the next decade toward eco-friendly compounds and processes in drug design. Green Chem. 2023, 25, 2109–2169. [Google Scholar] [CrossRef]
- Cysewski, P.; Jeliński, T.; Przybyłek, M. Intermolecular Interactions of Edaravone in Aqueous Solutions of Ethaline and Glyceline Inferred from Experiments and Quantum Chemistry Computations. Molecules 2023, 28, 629. [Google Scholar] [CrossRef]
- König-Mattern, L.; Komarova, A.O.; Ghosh, A.; Linke, S.; Rihko-Struckmann, L.K.; Luterbacher, J.; Sundmacher, K. High-throughput computational solvent screening for lignocellulosic biomass processing. Chem. Eng. J. 2023, 452, 139476. [Google Scholar] [CrossRef]
- Gupta, Y.; Bhattacharyya, S.; Vlachos, D.G. Extraction of valuable chemicals from food waste via computational solvent screening and experiments. Sep. Purif. Technol. 2023, 316, 123719. [Google Scholar] [CrossRef]
- Vilas-Boas, S.M.; Cordova, I.W.; Kurnia, K.A.; Almeida, H.H.S.; Gaschi, P.S.; Coutinho, J.A.P.; Pinho, S.P.; Ferreira, O. Comparison of two computational methods for solvent screening in countercurrent and centrifugal partition chromatography. J. Chromatogr. A 2022, 1666, 462859. [Google Scholar] [CrossRef]
- González-Miquel, M.; Díaz, I. Green solvent screening using modeling and simulation. Curr. Opin. Green Sustain. Chem. 2021, 29, 100469. [Google Scholar] [CrossRef]
- Vermeire, F.H.; Chung, Y.; Green, W.H. Predicting Solubility Limits of Organic Solutes for a Wide Range of Solvents and Temperatures. J. Am. Chem. Soc. 2022, 144, 10785–10797. [Google Scholar] [CrossRef]
- Cysewski, P.; Jeliński, T.; Przybyłek, M. Application of COSMO-RS-DARE as a Tool for Testing Consistency of Solubility Data: Case of Coumarin in Neat Alcohols. Molecules 2022, 27, 5274. [Google Scholar] [CrossRef]
- Chen, X.; Fadda, H.M.; Aburub, A.; Mishra, D.; Pinal, R. Cosolvency approach for assessing the solubility of drugs in poly(vinylpyrrolidone). Int. J. Pharm. 2015, 494, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Harten, P.; Martin, T.; Gonzalez, M.; Young, D. The software tool to find greener solvent replacements, PARIS III. Environ. Prog. Sustain. Energy 2020, 39, e13331. [Google Scholar] [CrossRef]
- Galaon, T.; David, V. Deviation from van’t Hoff dependence in RP-LC induced by tautomeric interconversion observed for four compounds. J. Sep. Sci. 2011, 34, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.A.; Holste, J.C.; Hall, K.R.; Ali Mansoori, G. A simple relation to predict or to correlate the excess functions of multicomponent mixtures. Fluid Phase Equilib. 1991, 62, 173–189. [Google Scholar] [CrossRef]
- Jouyban, A. Handbook of Solubility Data for Pharmaceuticals; CRC Press: Boca Raton, FL, USA, 2009; ISBN 9781439804889. [Google Scholar]
- Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. Optuna: A Next-generation Hyperparameter Optimization Framework. In Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 25 July 2019; pp. 2623–2631. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Dassault Systèmes. COSMOtherm, Version 22.0.0. Dassault Systèmes. Biovia: San Diego, CA, USA, 2022.
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799. [Google Scholar] [CrossRef]
- Cysewski, P.; Jeliński, T.; Cymerman, P.; Przybyłek, M. Solvent Screening for Solubility Enhancement of Theophylline in Neat, Binary and Ternary NADES Solvents: New Measurements and Ensemble Machine Learning. Int. J. Mol. Sci. 2021, 22, 7347. [Google Scholar] [CrossRef]
- Cramer, R.D.; Bunce, J.D.; Patterson, D.E.; Frank, I.E. Crossvalidation, Bootstrapping, and Partial Least Squares Compared with Multiple Regression in Conventional QSAR Studies. Quant. Struct. Relatsh. 1988, 7, 18–25. [Google Scholar] [CrossRef]
- Acree, W.; Chickos, J.S. Phase Transition Enthalpy Measurements of Organic and Organometallic Compounds. Sublimation, Vaporization and Fusion Enthalpies from 1880 to 2015. Part 1. C1–C10. J. Phys. Chem. Ref. Data 2016, 45, 33101. [Google Scholar] [CrossRef]
- Acree, W.; Chickos, J.S. Phase Transition Enthalpy Measurements of Organic and Organometallic Compounds and Ionic Liquids. Sublimation, Vaporization, and Fusion Enthalpies from 1880 to 2015. Part 2. C11–C192. J. Phys. Chem. Ref. Data 2017, 46, 013104. [Google Scholar] [CrossRef]
- Mojtahedi, M.M.; Javadpour, M.; Abaee, M.S. Convenient ultrasound mediated synthesis of substituted pyrazolones under solvent-free conditions. Ultrason. Sonochem. 2008, 15, 828–832. [Google Scholar] [CrossRef]
- Christian, S.D. Regular and Related Solutions: The Solubility of Gases, Liquids, and Solids (Hildebrand, Joel H.; Prausnitz, John M.). J. Chem. Educ. 1971, 48, A562. [Google Scholar] [CrossRef]
- Hildebrand, J.H.; Prausnitz, J.M.; Scott, R.L.; Robert, L. Regular and Related Solutions: The Solubility of Gases, Liquids, and Solids; Van Nostrand Reinhold Co.: New York, NY, USA, 1970. [Google Scholar]
- Nordström, F.L.; Rasmuson, Å.C. Determination of the activity of a molecular solute in saturated solution. J. Chem. Thermodyn. 2008, 40, 1684–1692. [Google Scholar] [CrossRef]
- Neau, S.H.; Bhandarkar, S.V.; Hellmuth, E.W. Differential molar heat capacities to test ideal solubility estimations. Pharm. Res. 1997, 14, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Przybyłek, M.; Kowalska, A.; Tymorek, N.; Dziaman, T.; Cysewski, P. Thermodynamic Characteristics of Phenacetin in Solid State and Saturated Solutions in Several Neat and Binary Solvents. Molecules 2021, 26, 4078. [Google Scholar] [CrossRef] [PubMed]
- Svärd, M.; Rasmuson, Å.C. (Solid + liquid) solubility of organic compounds in organic solvents–Correlation and extrapolation. J. Chem. Thermodyn. 2014, 76, 124–133. [Google Scholar] [CrossRef]
- Dassault Systèmes. COSMOconf, Version 22.0.0. Dassault Systèmes. Biovia: San Diego, CA, USA, 2022.
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- TURBOMOLE GmbH. TURBOMOLE, Version 7.6.0; TURBOMOLE GmbH: Karlsruhe, Germany, 2021.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent [CAS number] | Structure | Log (xEest) | EI (PCOP = 0) |
|---|---|---|---|
| enflurane [13838-16-9] |  | –1.20 ± 0.42 (–1.29) | 0.47 |
| DMSO [67-68-5] |  | –1.22 ± 0.20 (–1.28) | 0.26 |
| isoflurane [26675-46-7] |  | –1.29 ± 0.46 (–1.05) | 0.56 |
| NMP [872-50-4] |  | –1.40 ± 0.09 (–0.92) | 0.97 |
| 2-ethenoxyethanol [764-48-7] |  | –1.41 ± 0.09 (–1.35) | 0.97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Przybyłek, M.; Jeliński, T.; Mianowana, M.; Misiak, K.; Cysewski, P. Exploring the Solubility Limits of Edaravone in Neat Solvents and Binary Mixtures: Experimental and Machine Learning Study. Molecules 2023, 28, 6877. https://doi.org/10.3390/molecules28196877
Przybyłek M, Jeliński T, Mianowana M, Misiak K, Cysewski P. Exploring the Solubility Limits of Edaravone in Neat Solvents and Binary Mixtures: Experimental and Machine Learning Study. Molecules. 2023; 28(19):6877. https://doi.org/10.3390/molecules28196877
Chicago/Turabian StylePrzybyłek, Maciej, Tomasz Jeliński, Magdalena Mianowana, Kinga Misiak, and Piotr Cysewski. 2023. "Exploring the Solubility Limits of Edaravone in Neat Solvents and Binary Mixtures: Experimental and Machine Learning Study" Molecules 28, no. 19: 6877. https://doi.org/10.3390/molecules28196877