
A Theoretical Study on the Underlying Factors of the Difference in Performance of Organic Solar Cells Based on ITIC and Its Isomers

Abstract
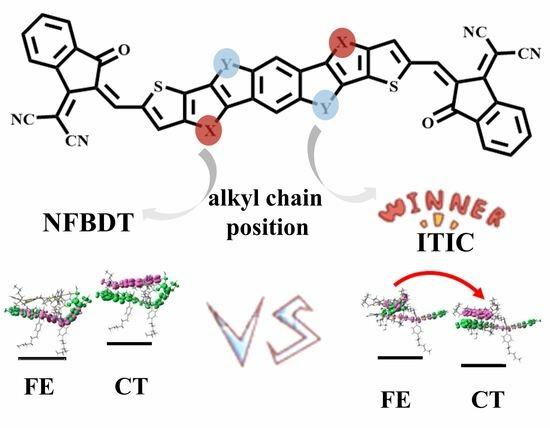
:
1. Introduction
2. Computational Methods
2.1. MD Simulations
2.2. Quantum Chemical Calculations
3. Results and Discussion
3.1. Properties Related to Ground State
3.2. Properties Related to Excited State
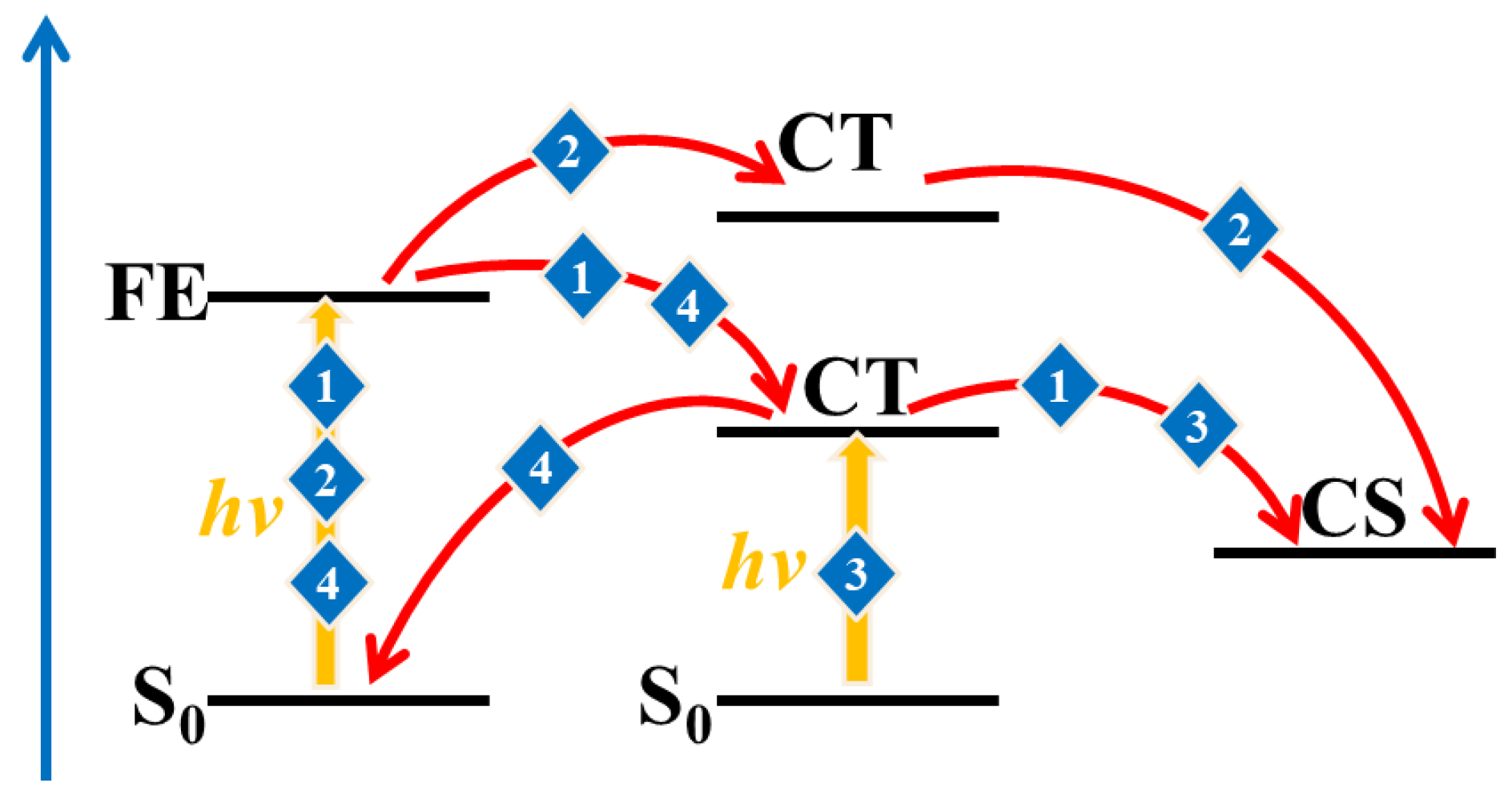
3.3. The Charge Separation and Recombination at the D/A Interface
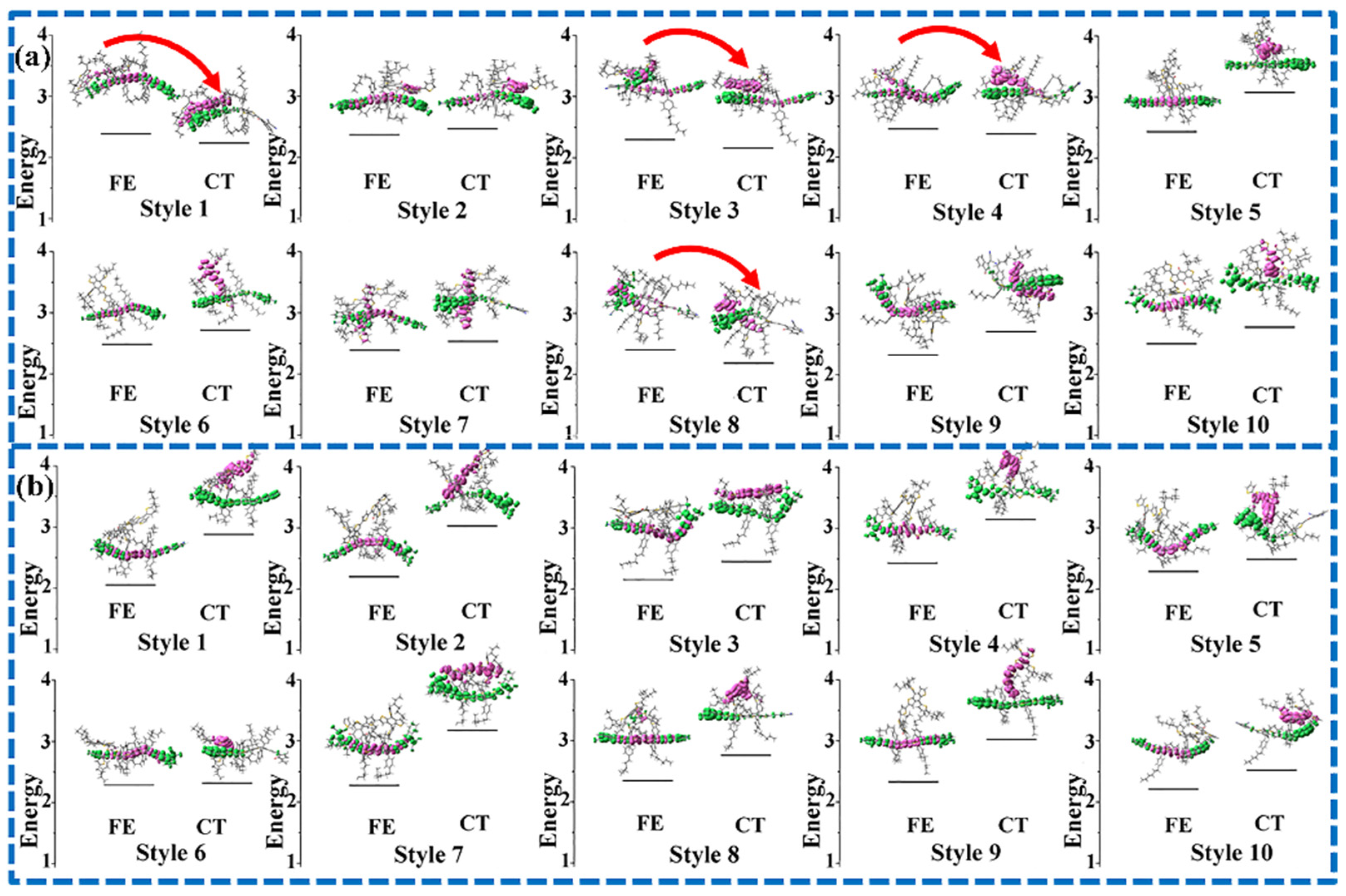
3.3.1. The Frenkel Exciton States and CT States
3.3.2. Charge Separation and Recombination Rate
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, R.; Jiang, Z.; Yang, C.; Yu, J.; Feng, J.; Adil, M.A.; Deng, D.; Zou, W.; Zhang, J.; Lu, K.; et al. All-small-molecule organic solar cells with over 14% efficiency by optimizing hierarchical morphologies. Nat. Commun. 2019, 10, 5393. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Lv, J.; Yang, K.; He, Y.; Yang, Q.; Chen, H.; Chen, Q.; Liao, Z.; Kan, Z.; Duan, T.; et al. 15.8% efficiency binary all-small-molecule organic solar cells enabled by a selenophene substituted sematic liquid crystalline donor. Energy Environ. Sci. 2021, 14, 5366–5376. [Google Scholar] [CrossRef]
- Markina, A.; Lin, K.H.; Liu, W.; Poelking, C.; Firdaus, Y.; Villalva, D.R.; Khan, J.I.; Paleti SH, K.; Harrison, G.T.; Gorenflot, J.; et al. Chemical Design Rules for Non-Fullerene Acceptors in Organic Solar Cells. Adv. Energy Mater. 2021, 11, 2102363. [Google Scholar] [CrossRef]
- Arredondo, B.; Carlos Pérez-Martínez, J.; Muñoz-Díaz, L.; López-González, M.d.C.; Martín-Martín, D.; del Pozo, G.; Hernández-Balaguera, E.; Romero, B.; Lamminaho, J.; Turkovic, V.; et al. Influence of solvent additive on the performance and aging behavior of non-fullerene organic solar cells. Sol. Energy 2022, 232, 120–127. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, J.; Bi, P.; Ren, J.; Wang, Y.; Yang, Y.; Liu, X.; Zhang, S.; Hou, J. Tandem organic solar cell with 20.2% efficiency. Joule 2022, 6, 171–184. [Google Scholar] [CrossRef]
- Jiang, Z.-Q.; Wang, T.-T.; Wu, F.-P.; Lin, J.-D.; Liao, L.-S. Recent advances in electron acceptors with ladder-type backbone for organic solar cells. J. Mater. Chem. A 2018, 6, 17256–17287. [Google Scholar] [CrossRef]
- Liu, W.; Xu, X.; Yuan, J.; Leclerc, M.; Zou, Y.; Li, Y. Low-bandgap non-fullerene acceptors enabling high-performance organic solar cells. ACS Energy Lett. 2021, 6, 598–608. [Google Scholar] [CrossRef]
- Zhou, D.; Wang, J.; Xu, Z.; Xu, H.; Quan, J.; Deng, J.; Li, Y.; Tong, Y.; Hu, B.; Chen, L. Recent advances of nonfullerene acceptors in organic solar cells. Nano Energy 2022, 103, 107802. [Google Scholar] [CrossRef]
- Zhang, Y.; Lang, Y.; Li, G. Recent advances of non-fullerene organic solar cells: From materials and morphology to devices and applications. Ecomat 2023, 5, e12281. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, Z.; Ye, L.; Zhan, C.; Hou, J.; Zhang, S.; Jiang, B.; Zhao, Y.; Huang, J.; Zhang, S.; et al. A potential perylene diimide dimer-based acceptor material for highly efficient solution-processed non-fullerene organic solar cells with 4.03% efficiency. Adv. Mater. 2013, 25, 5791–5797. [Google Scholar] [CrossRef]
- Meng, D.; Fu, H.; Xiao, C.; Meng, X.; Winands, T.; Ma, W.; Wei, W.; Fan, B.; Huo, L.; Doltsinis, N.L.; et al. Three-bladed rylene propellers with three-dimensional network assembly for organic electronics. J. Am. Chem. Soc. 2016, 138, 10184–10190. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, J.; Zhang, Z.-G.; Bai, H.; Li, Y.; Zhu, D.; Zhan, X. An electron acceptor challenging fullerenes for efficient polymer solar cells. Adv. Mater. 2015, 27, 1170–1174. [Google Scholar] [CrossRef] [PubMed]
- Bin, H.; Gao, L.; Zhang, Z.G.; Yang, Y.; Zhang, Y.; Zhang, C.; Chen, S.; Xue, L.; Yang, C.; Xiao, M.; et al. 11.4% Efficiency non-fullerene polymer solar cells with trialkylsilyl substituted 2D-conjugated polymer as donor. Nat. Commun. 2016, 7, 13651. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Qian, D.; Zhang, S.; Li, S.; Inganas, O.; Gao, F.; Hou, J. Fullerene-free polymer solar cells with over 11% efficiency and excellent thermal stability. Adv. Mater. 2016, 28, 4734–4739. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ye, L.; Zhao, W.; Zhang, S.; Mukherjee, S.; Ade, H.; Hou, J. Energy-level modulation of small-molecule electron acceptors to achieve over 12% efficiency in polymer solar cells. Adv. Mater. 2016, 28, 9423–9429. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ye, L.; Zhao, W.; Zhang, S.; Ade, H.; Hou, J. Significant influence of the methoxyl substitution position on optoelectronic properties and molecular packing of small-molecule electron acceptors for photovoltaic cells. Adv. Energy Mater. 2017, 7, 1700183. [Google Scholar] [CrossRef]
- Yao, H.; Cui, Y.; Qian, D.; Ponseca, C.S., Jr.; Honarfar, A.; Xu, Y.; Xin, J.; Chen, Z.; Hong, L.; Gao, B.; et al. 14.7% efficiency organic photovoltaic cells enabled by active materials with a large electrostatic potential difference. J. Am. Chem. Soc. 2019, 141, 7743–7750. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Fan, Q.; Xiong, M.; Wang, Q.; Chen, K.; Liu, H.; Gao, M.; Ye, L.; Guo, X.; Fang, J.; et al. Carboxylate substituted pyrazine: A simple and low-cost building block for novel wide bandgap polymer donor enables 15.3% efficiency in organic solar cells. Nano Energy 2021, 82, 105679. [Google Scholar] [CrossRef]
- Kan, B.; Feng, H.; Wan, X.; Liu, F.; Ke, X.; Wang, Y.; Wang, Y.; Zhang, H.; Li, C.; Hou, J.; et al. Small-molecule acceptor based on the heptacyclic benzodi (cyclopentadithiophene) unit for highly efficient nonfullerene organic solar cells. J. Am. Chem. Soc. 2017, 139, 4929–4934. [Google Scholar] [CrossRef]
- Xin, Y.; Zeng, G.; OuYang, J.; Zhao, X.; Yang, X. Enhancing thermal stability of nonfullerene organic solar cells via fluoro-side-chain engineering. J. Mater. Chem. C 2019, 7, 9513–9522. [Google Scholar] [CrossRef]
- Yang, J.; Li, Q.S.; Li, Z.S. End-capped group manipulation of indacenodithienothiophene-based non-fullerene small molecule acceptors for efficient organic solar cells. Nanoscale 2020, 12, 17795–17804. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Guo, Y.; Ning, L.; Yi, Y. Improving the electron mobility of ITIC by end-group modulation: The role of fluorination and π-extension. Solar RRL 2019, 3, 1800251. [Google Scholar] [CrossRef]
- Fan, Q.; Su, W.; Zhang, M.; Wu, J.; Jiang, Y.; Guo, X.; Liu, F.; Russell, T.P.; Zhang, M.; Li, Y. Synergistic Effects of Side-Chain Engineering and Fluorination on Small Molecule Acceptors to Simultaneously Broaden Spectral Response and Minimize Voltage Loss for 13.8% Efficiency Organic Solar Cells. Solar RRL 2019, 3, 1900169. [Google Scholar] [CrossRef]
- Clarke, T.M.; Durrant, J.R. Charge photogeneration in organic solar cells. Chem. Rev. 2010, 110, 6736–6767. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Fox, T.; Kollman, P.A. Application of the RESP methodology in the parametrization of organic solvents. J. Phys. Chem. B 1998, 102, 8070–8079. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Dapprich, S.; Komáromi, I.; Byun, K.S.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct. THEOCHEM 1999, 461, 1–21. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Qiu, M.; Zhu, D.; Yan, L.; Wang, N.; Han, L.; Bao, X.; Du, Z.; Niu, Y.; Yang, R. Strategy to manipulate molecular orientation and charge mobility in D–A type conjugated polymer through rational fluorination for improvements of photovoltaic performances. J. Phys. Chem. C 2016, 120, 22757–22765. [Google Scholar] [CrossRef]
- Tang, X.-D.; Liao, Y.; Gao, H.-Z.; Geng, Y.; Su, Z.-M. Theoretical study of the bridging effect on the charge carrier transport properties of cyclooctatetrathiophene and its derivatives. J. Mater. Chem. 2012, 22, 6907–6918. [Google Scholar] [CrossRef]
- Sarkar, R.; Boggio-Pasqua, M.; Loos, P.F.; Jacquemin, D. Benchmarking TD-DFT and wave function methods for oscillator strengths and excited-state dipole moments. J. Chem. Theory Comput. 2021, 17, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A. Electron transfer reactions in chemistry: Theory and experiment (Nobel lecture). Angew. Chem. Int. Ed. 1993, 32, 1111–1121. [Google Scholar] [CrossRef]
- Burquel, A.; Lemaur, V.; Beljonne, D.; Lazzaroni, R.; Cornil, J. Pathways for photoinduced charge separation and recombination at donor−acceptor heterojunctions: The case of oligophenylenevinylene−perylene bisimide complexes. J. Phys. Chem. A 2006, 110, 3447. [Google Scholar] [CrossRef]
- Kavarnos, G.J.; Turro, N.J. Photosensitization by reversible electron transfer: Theories, experimental evidence, and examples. Chem. Rev. 1986, 86, 401–449. [Google Scholar] [CrossRef]
- Lemaur, V.; Steel, M.; Beljonne, D.; Brédas, J.L.; Cornil, J. Photoinduced charge generation and recombination dynamics in model donor/acceptor pairs for organic solar cell applications: A full quantum-chemical treatment. J. Am. Chem. Soc. 2005, 127, 6077–6086. [Google Scholar] [CrossRef]
- Pan, Q.-Q.; Li, S.-B.; Wu, Y.; Zhang, J.; Li, H.-B.; Geng, Y.; Zhang, M.; Su, Z.-M. Theoretical design of three-dimensional non-fullerene acceptor materials based on an arylenediimide unit towards high efficiency organic solar cells. New J. Chem. 2017, 41, 3857–3864. [Google Scholar] [CrossRef]
- Krylov, A.I.; Gill, P.M. Q-Chem: An engine for innovation. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 317–326. [Google Scholar] [CrossRef]
- Voityuk, A.A. Estimation of electronic coupling in π-stacked donor-bridge-acceptor systems: Correction of the two-state model. J. Chem. Phys. 2006, 124, 64505. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Han, G.; Yi, Y. Molecular insight into efficient charge generation in low-driving-force nonfullerene organic solar cells. Acc. Chemcial Res. 2022, 55, 869–877. [Google Scholar] [CrossRef]
- Zheng, W.; Liu, J.; Guo, Y.; Han, G.; Yi, Y. Regulation of Molecular Orientations of A–D–A Nonfullerene Acceptors for Organic Photovoltaics: The Role of End-Group π–π Stacking. Adv. Funct. Mater. 2022, 32, 2108551. [Google Scholar] [CrossRef]
- Yang, J.; Ding, W.L.; Li, Q.S.; Li, Z.S. Theoretical study of non-fullerene acceptors using end-capped groups with different electron-withdrawing abilities toward efficient organic solar cells. J. Phys. Chem. Lett. 2022, 13, 916–922. [Google Scholar] [CrossRef]
- Han, G.; Hu, T.; Yi, Y. Reducing the singlet− triplet energy gap by end-group π−π stacking toward high-efficiency organic photovoltaics. Adv. Mater. 2020, 32, 2000975. [Google Scholar] [CrossRef]
- Li, M.-Y.; Ren, Y.; Huang, J.-C.; Sui, M.-Y.; Sun, G.-Y.; Su, Z.-M. Charge transfer regulated by domain differences between host and guest donors in ternary organic solar cells. J. Mater. Chem. A 2022, 10, 22477–22487. [Google Scholar] [CrossRef]
- Han, G.; Yi, Y. Local excitation/charge-transfer hybridization simultaneously promotes charge generation and reduces nonradiative voltage loss in nonfullerene organic solar cells. J. Phys. Chem. Lett. 2019, 10, 2911–2918. [Google Scholar] [CrossRef]
- Pan, Q.-Q.; Li, S.-B.; Wu, Y.; Geng, Y.; Zhang, M.; Su, Z.-M. Exploring more effective polymer donors for the famous non-fullerene acceptor ITIC in organic solar cells by increasing electron-withdrawing ability. Org. Electron. 2018, 53, 308–314. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λi-CS | λi-CR | λs | λCS | λCR | ∆GCS | ∆GCR | |
| PBDB-T/ITIC | 0.241 | 0.191 | 0.330 | 0.571 | 0.521 | −0.954 | −1.701 |
| PBDB-T/NFBDT | 0.270 | 0.220 | 0.329 | 0.599 | 0.549 | −0.967 | −1.687 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.-Q.; Wang, L.-L.; Pan, Q.-Q.; Zhao, Z.-W.; Gao, Y.; Su, Z.-M. A Theoretical Study on the Underlying Factors of the Difference in Performance of Organic Solar Cells Based on ITIC and Its Isomers. Molecules 2023, 28, 6968. https://doi.org/10.3390/molecules28196968
Huang S-Q, Wang L-L, Pan Q-Q, Zhao Z-W, Gao Y, Su Z-M. A Theoretical Study on the Underlying Factors of the Difference in Performance of Organic Solar Cells Based on ITIC and Its Isomers. Molecules. 2023; 28(19):6968. https://doi.org/10.3390/molecules28196968
Chicago/Turabian StyleHuang, Si-Qi, Li-Li Wang, Qing-Qing Pan, Zhi-Wen Zhao, Ying Gao, and Zhong-Min Su. 2023. "A Theoretical Study on the Underlying Factors of the Difference in Performance of Organic Solar Cells Based on ITIC and Its Isomers" Molecules 28, no. 19: 6968. https://doi.org/10.3390/molecules28196968
APA StyleHuang, S.-Q., Wang, L.-L., Pan, Q.-Q., Zhao, Z.-W., Gao, Y., & Su, Z.-M. (2023). A Theoretical Study on the Underlying Factors of the Difference in Performance of Organic Solar Cells Based on ITIC and Its Isomers. Molecules, 28(19), 6968. https://doi.org/10.3390/molecules28196968