Abstract
Imidazole was first synthesized by Heinrich Debus in 1858 and was obtained by the reaction of glyoxal and formaldehyde in ammonia, initially called glyoxaline. The current literature provides much information about the synthesis, functionalization, physicochemical characteristics and biological role of imidazole. Imidazole is a structure that, despite being small, has a unique chemical complexity. It is a nucleus that is very practical and versatile in its construction/functionalization and can be considered a rich source of chemical diversity. Imidazole acts in extremely important processes for the maintenance of living organisms, such as catalysis in enzymatic processes. Imidazole-based compounds with antibacterial, anti-inflammatory, antidiabetic, antiparasitic, antituberculosis, antifungal, antioxidant, antitumor, antimalarial, anticancer, antidepressant and many others make up the therapeutic arsenal and new bioactive compounds proposed in the most diverse works. The interest and importance of imidazole-containing analogs in the field of medicinal chemistry is remarkable, and the understanding from the development of the first blockbuster drug cimetidine explores all the chemical and biological concepts of imidazole in the context of research and development of new drugs.
1. The Chemistry of Imidazole
Imidazole 1 (Figure 1) was first synthesized by Heinrich Debus in 1858, but since the 1840s, several imidazole derivatives have been discovered. Its synthesis started from the use of glyoxal 2 and formaldehyde 3 in ammonia, producing imidazole 1 as a final product, which was initially called glyoxaline (Figure 2) [1,2]. This synthesis, despite producing relatively low yields, is still used to create C-substituted imidazoles.
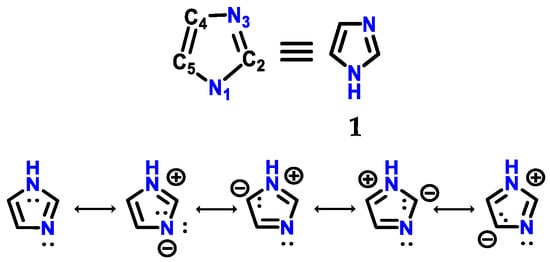
Figure 1.
Structure of imidazole 1 with its respective numbering and resonance hybrids.
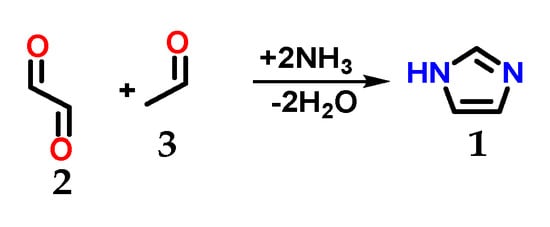
Figure 2.
Scheme of the synthesis of imidazole 1 using glyoxal 2 and formaldehyde 3 in ammonia.
Among the nitrogen-based heterocyclic compounds, imidazole 1 plays an important role in humans. It is included in chemical sciences, biological sciences and materials science, and used as a catalyst in compound synthesis and the process of developing new drugs (Figure 3) [3,4,5,6].
Figure 3.
Applications of imidazole 1 in different areas of knowledge.
With a 5-membered planar ring, imidazole 1 exhibits solubility in water and other polar solvents. Two equivalent tautomeric forms are observed because the hydrogen atom can be located on either of the two nitrogen atoms [2]. Imidazole 1 is a highly polar compound, as seen by a calculated dipole of 3.61D, and is completely soluble in water [2,7]. Imidazole 1 is classified as an amphoteric compound, acting as both an acid and a base. The compound is classified as aromatic due to the presence of a sextet of π electrons, consisting of a pair of nonbonding electrons from the nitrogen N-1 atom and one from each of the four remaining ring atoms [2,7].
Imidazole 1 can form stable crystalline salts with strong acids through the protonation of the sp2 nitrogen (N-3), known as imidazolium salts. Imidazole 1 has a pKaH of 7.1 (Figure 4), acting as a strong base [8]. The basicity of imidazole is above that of pyridine 4 (pKaH of 5.2) due to the amidine-like 5 resonance, which allows both nitrogens to participate equally in charge accommodation [7,8,9]. Comparatively, the basicity of imidazole 1 contrasts with the basicity of pyrrole 6 (pKaH of 0.4), which is an extremely weak base, because when pyrrole 6 is protonated, there is a loss of aromaticity which is built with the participation of the nonbonding electron pair of N-1 nitrogen [7].
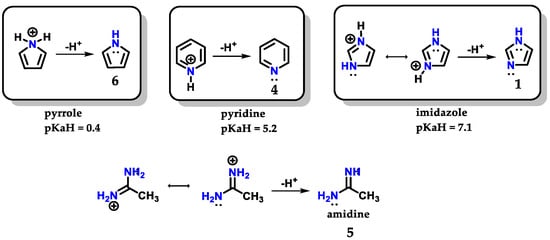
Figure 4.
Protonated structures of pyrrole 6, pyridine 4 and imidazole 1, with their respective pKaH values and the structure of protonated amidine 5 compared to imidazole.
Imidazole 1 is a good donor and acceptor of hydrogen bond interactions; the sp2 nitrogen (N-3) accepts a hydrogen interaction, while N-1 nitrogen, being relatively acidic, donates its hydrogen to an interaction (Figure 5A) [8]. This property is fundamental for the mode of action of several enzymes that use the imidazole ring, such as the histidine 7 amino acid residue, one of the 20 amino acids found in proteins [8]. These important interactions for living organisms are present not only in macromolecules but also in bioactive small molecules.
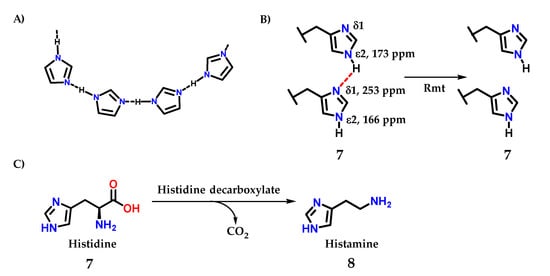
Figure 5.
(A) Intermolecular interactions through the hydrogen interactions that imidazole-containing derivatives can perform. (B) Intermolecular interactions through the hydrogen interactions in histidine residues. (C) Structure of histidine 7, precursor of histamine 8 biosynthesis.
A recent study demonstrating this important effect was carried out by Movellan and collaborators [10], where it was possible to analyze the hydrogen residues in histidine residues 7 in the M2 tetramer of influenza A, important for the process of endocytosis and maintenance of the virus life cycle be [1,2,3,4,11,12,13,14,15]. Using the M2 conduction domain construct in lipid bilayers, that the imidazole ring is hydrogen bonded even at a pH of 7.8 in the neutral charge state (Figure 5B). An intermolecular 2hJNN hydrogen bond of 8.9 ± 0.3 Hz was observed between H37 Nε and Nδ. However, this 2hJNN interaction could not be detected in the sample connected to the drug rimantadine (Rmt), with consequent modification in the proton chemical shifts value of 3 ppm for histidine residues [10].
Histamine 8 is an example of a small molecule with different actions in living organisms. It is biosynthesized from histidine 7 itself by the action of the enzyme histidine decarboxylase (Figure 5B) [16].
The presence of the N-1 nitrogen in the imidazole 1 structure makes it tautomer, which becomes evident in non-symmetrically substituted compounds, such as methyl imidazole 9 (Figure 6) [9]. This curious feature of imidazole chemistry means that simply writing “4-methylimidazole” would be incorrect, considering the rapid tautomeric equilibrium with the 5-methylimidazole structure [9,17].
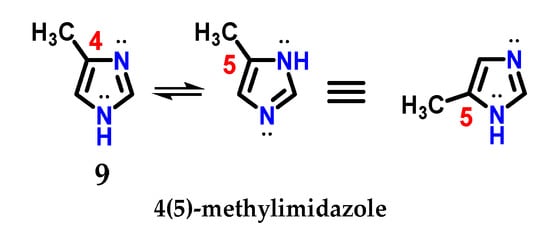
Figure 6.
Representation of the tautomeric forms of methyl imidazole 9.
Despite the existence of the tautomeric effect in imidazole 1, the ratio in terms of proportion of these tautomers varies according to the substituent added to the ring. The tautomer ratio observed in the 4(5)-nitroimidazole 10 derivative is approximately 400:1 for 4-nitroimidazole [9]. Reports in the literature suggest a relationship between tautomers 1.4 and 1.5 in the following proportion: log ([1.4]/[1.5]) = 4 × σm [17,18]. However, for any substituent, whether electron withdrawing (EWG) or electron donor (EDG), the 1,4 tautomer predominates since the meta substituent constant (σm) is governed by inductive effects (σm = 0.71, 0.37 and 0.10 for -NO2, -Br and -OCH3, respectively) [17]. The 4(5)-nitroimidazole 10 derivative has a pKa of 9.30 (different from the pKa of its imidazole 1 precursor, pKa = 14.5) with a predominance of tautomer 1.4 (Figure 7) [19,20]. It is possible to notice these effects in a quick energy calculation for a geometric equilibrium in water using the PM3 semiempirical method, using the Spartan 18 v1.2.0 program (Wavefunction, Inc & Q-Chem, Irvine, California, U.S.A). There is a significant difference in the energy obtained for tautomer 1.4 (15.21 kJ/mol) in comparison with that of corresponding tautomer 1.5 (25.36 kJ/mol).
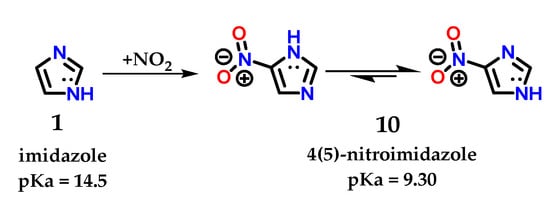
Figure 7.
Representation of imidazole 1 and tautomeric forms of 4(5)-nitroimidazole 10, with their respective pKa values.
Imidazole 1 is an electron-rich heterocycle, as mentioned above. Electrophilic substitution normally occurs at the C-4 or C-5 position; however, as previously mentioned, the predominance of a given tautomer is relative and follows the factors already discussed, while nucleophilic substitution usually occurs at C-2 [7]. Calculations in Spartan 18 v1.2.0 help us obtain a better view of these data. Figure 8 shows the electrostatic map with the values of natural charge for imidazole 1 (−0.290, −0.013, −0.217, −0.194 and −0.092, for N-1, C-2, N-3, C-4 and C-5 respectively), demonstrating the electronic density for each atom of the imidazole ring, suggesting the electronegativity (χ) of the structure, as well as the dipole moment of imidazole 1, which starts from N-1nitrogen toward sp2 nitrogen. With these results, we can consider the information taken from the literature, where C-4 and C-5 have higher electron densities, making them susceptible to electrophilic reactions, and C-2 has a lower density, making it susceptible to nucleophilic reactions.
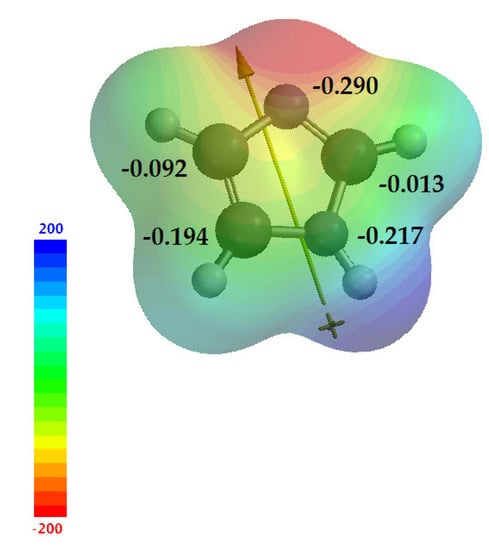
Figure 8.
Electrostatic map of imidazole 1, showing the natural charges of each atom and the dipole moment.
Although imidazole 1 and pyrrole 6 are π-excessive heterocycles (π donors), imidazoles do not establish η5(π)-complexes (η being the chemical hardness) with transition metals. Imidazole 1 behaves as a π-deficient ligand (π-acceptor) similar to pyridine 4, and its sp2 nitrogen atoms mainly form η1(σ,Npy) complexes (Figure 9), as does pyridine 4 [7].
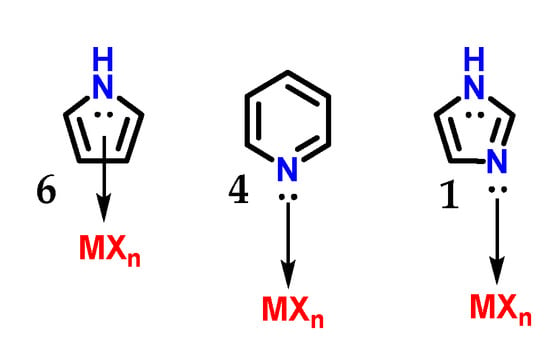
Figure 9.
Complex with transition metals η5(π) carried out by pyrrole 6 and the complexes η1(σ,Npy) carried out by imidazole 1 and pyridine 4.
Imidazole 1 has a very exciting physicochemical complexity. This makes it a target nucleus for the most diverse applications, which employ synthetic methodologies for its obtention and functionalization.
2. Synthesis and Functionalization of Imidazole
The synthesis of substituted imidazole 1 using heterogeneous catalysis has been widely exploited. These functionalized structures are useful building blocks for the synthesis of molecules of biological and pharmaceutical interest.
2.1. Mono-Substituted Derivatives
One-pot reactions using iodobenzene 11 and imidazole 1 in the presence of K3PO4 as the base, CuI as the catalyst and DMF as the solvent at 35–40 °C for 40 h, give the corresponding N-arylimidazoles 12 in quantitative yields (Figure 10) [21].
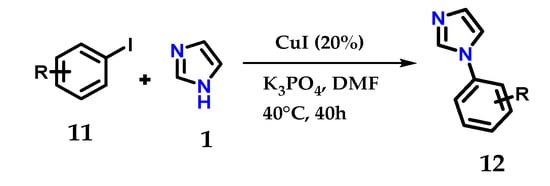
Figure 10.
Catalytic N-arylation of imidazole 1 with aryl iodides and CuI.
Exploring the bifunctionalization of 1,2-disubstituted acetylenes 13 by ruthenium carbonyl to form cis-enediol diacetates 14, followed by reaction with ammonium carbonate as a source of nitrogen and methanol for the C-2 carbon, permitted us to obtain monosubstituted imidazoles 15, as shown below (Figure 11). In reactions where (R) were aromatic rings, both substituents, withdrawers (EWG) and donors (EDG) were tolerated under the applied conditions [22].
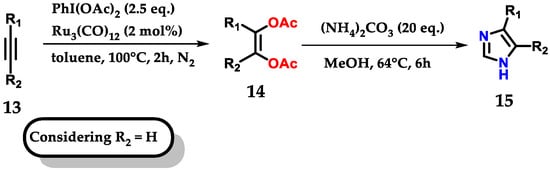
Figure 11.
Ruthenium-catalyzed oxidation of alkynes for mono-substituted imidazole 15 synthesis.
2.2. Disubstituted Derivatives
The work below shows the development of an efficient methodology for the synthesis of novel 2-aryl-4-benzoyl-imidazoles 16 by structural modification of 2-aryl-imidazole-4-carboxylic amide (AICA) 17 and 4-substituted methoxylbenzoyl-aryl-thiazoles (SMART) 18, presenting antiproliferative activity (Figure 12) [23].
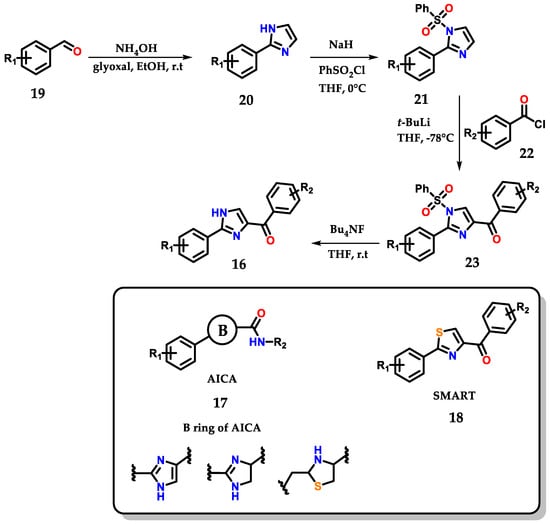
Figure 12.
Obtaining 2-aryl-4-benzoyl-imidazoles 16 based on AICA 17 and SMART 18.
Disubstituted imidazoles 24 could also be synthesized by cyclization of α-keto-aldehydes 25 obtained from the oxidation of aryl methyl-ketones 26 with selenium dioxide (SeO2) after treatment with ammonium acetate and ethanol. The synthesized derivatives were used as fluorogenic sensors for detection, selectivity and sensitivity to Fe3+ ions (Figure 13) [24].
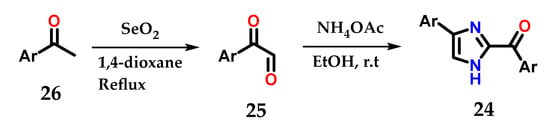
Figure 13.
Obtaining disubstituted imidazoles 24 from aryl-methylketones 26.
Another synthetic strategy used to construct disubstituted imidazole derivatives 27 from methyl ketones 28 consisted of exploiting its metal-free acid-catalyzed oxidation and coupling with aldehydes 29 and 30 in the presence of ammonium acetate (Figure 14) [25].
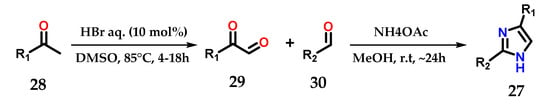
Figure 14.
Synthesis of disubstituted imidazoles 27 starting from the acid-catalyzed oxidation of methylketones 28.
Moreover, disubstituted imidazoles presenting a carbomethoxy group 31 at C-4 could be obtained from the coupling of functionalized amidoximes 32 and methyl propiolate 33 in the presence of a catalytic amount of 1,4-diazabicyclo[2.2]octane (DABCO) under microwave irradiation (Figure 15) [26].
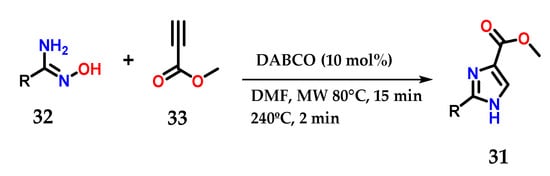
Figure 15.
Disubstituted imidazoles 31 were obtained from the base-catalyzed condensation of amidoximes 32 and methyl propiolate 33.
Amido-nitrile 34 cyclization mediated by functionalized boronic acids is also able to produce 2,4-disubstituted imidazoles 35. It was possible to explore a considerable diversity of substituents, considering the reaction conditions reported below (Figure 16) [27].
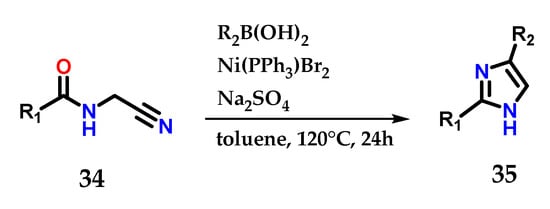
Figure 16.
Nickel-catalyzed cyclization of amido-nitriles 34 to obtain disubstituted imidazoles 35.
2.3. Trisubstituted Derivatives
On the other hand, 2,4,5-trisubstituted imidazoles 36 could be obtained by using 2,3-dioxo-3-substituted propanoates 37 as precursors after condensation using ammonium acetate and various aromatic aldehydes 38 in EtOH and AcOH as catalysts at room temperature (Figure 17) [28,29].
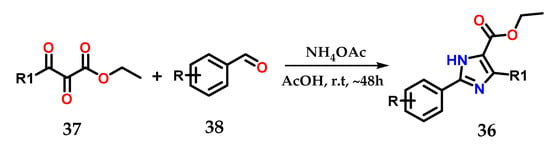
Figure 17.
Obtaining 2,4,5-trisubstituted imidazoles 36 from 2,3-dioxo-3-substituted propanoates 37.
2-Aryl-4,5-dicarbonitrile imidazole derivatives 39 could be obtained from the coupling of substituted aromatic aldehydes 40 and 2,3-diaminomaleonitriles 41 in the presence of a mixture of cerium (IV) ammonium nitrate/nitric acid (CAN: NA|0.05: 0.4 eq.) at 120 °C for less than 1 h without using solvents (Figure 18) [30].
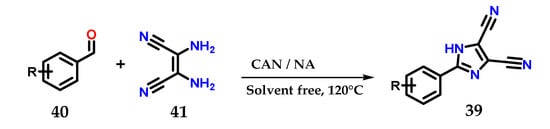
Figure 18.
Obtaining 2-aryl-4,5-dicarbonitrile imidazole derivatives 39 using a mixture of CAN:NA as catalysts.
The efficient combination of α-aminoketones 42 with formamide 43 in THF at 180 °C for 8 h is also able to provide 1,4,5-trisubstituted imidazoles 44 [31] (Figure 19).
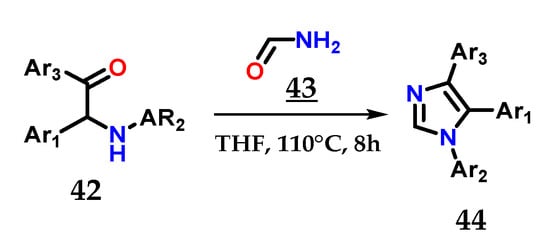
Figure 19.
Obtaining 1,4,5-trisubstituted imidazoles 44 from the coupling of α-aminoketones 42 with formamide 43.
Using the Van Leusen method, it was possible to synthesize 1,4,5-trisubstituted imidazoles containing a trifluoromethyl group 45, exploiting the coupling of N-aryltrifluoroacetimidoyl chloride 46 and tosyl-methylisocyanate (TosMIC) 47 using sodium hydride as a base in dry THF at room temperature (Figure 20) [32].
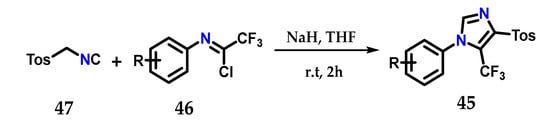
Figure 20.
Obtaining 1,4,5-trisubstituted imidazoles 45 using the Van Leusen method.
Methodologies reported in the literature show the use of substituted 1,2-diphenylethane-1,2-dione (benzyl) 48, substituted aldehydes 49 and ammonium acetate under various conditions, with the aim of optimizing the construction of 2,4,5-trisubstituted imidazole 50 with great structural diversity [33,34,35,36,37,38,39,40]. The reactions are simple and fast, as illustrated in the example below, which uses the system with EtOH and fluorinated graphene oxide (A-MFGO) as a catalyst at room temperature (Figure 21).
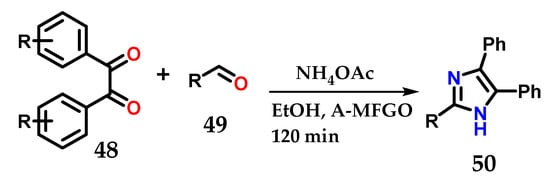
Figure 21.
Obtaining 2,4,5-trisubstituted imidazoles 50 from the coupling of substituted 1,2-diphenylethane-1,2-dione (benzyl) 48, substituted aldehydes 49 and NH4OAc using A-MFGO as a catalyst.
More complex heterocycles presenting the imidazole ring in their structure are described in the literature, such as benzo[d]imidazo[2,1-b]thiazoles 51, which could be synthesized by the condensation of aromatic ketones 52 and 5-(biphenyl-4-yl)-1,3,4-thiadiazol-2-amine 53 in the presence of N-bromosuccinimide 54, PEG-400 and water as solvent under microwave irradiation at 85 °C in quantitative yields after a few minutes of reaction (Figure 22) [41].
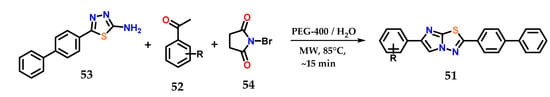
Figure 22.
Obtaining benzo[d]imidazo[2,1-b]thiazoles 51 from the condensation of aromatic ketones 52 and 5-(biphenyl-4-yl)-1,3,4-thiadiazol-2-amine 53.
2.4. Tetrasubstituted Derivatives
Using various aldehydes 55, benzyl 56, ammonium acetate and prop-2-ynylamine 57 in the presence of CuFe2O4NPs as a catalyst in H2O:EtOH under reflux for approximately 50 min, it was possible to obtain several tetrasubstituted imidazole derivatives 58 in a multicomponent synthesis. It was possible to reuse the catalyst for six reactions without losing its efficiency (Figure 23) [42].
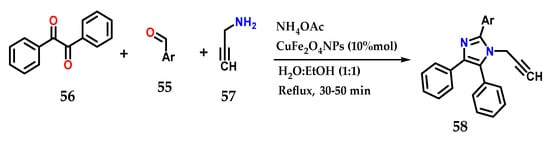
Figure 23.
Tetrasubstituted imidazoles 58 were obtained by the coupling of benzyl 56, functionalized aldehyde 55, prop-2-ynylamine 57 and ammonium acetate using CuFe2O4NPs as the catalyst.
In the presence of SO42−/Y2O3 as a catalyst, the multicomponent condensation of benzyl 56, aminoethylpiperazine 59, various aldehydes 60 and ammonium acetate in ethanol at 80 °C for 10 h was carried out to form tetrasubstituted 1,2,4,5-imidazole derivative 61. The catalyst was reused up to five times with no significant loss in catalytic efficiency (Figure 24) [43].
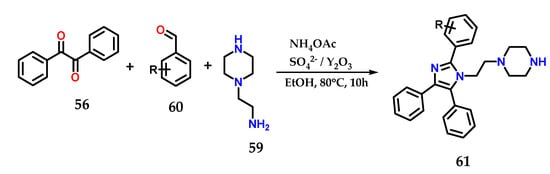
Figure 24.
Tetrasubstituted imidazoles 61 were obtained from the multicomponent condensation of benzyl 56, aminoethylpiperazine 59, various aldehydes 60 and ammonium acetate using SO42−/Y2O3 as a catalyst.
Alternatively, the synthesis of 1,2,4,5-tetrasubstituted imidazole derivatives 62 could be achieved through the condensation of benzyl 56, aldehydes 63 and anilines 64 in the presence of ammonium acetate under the solvent-free catalysis of Fe3O4@SiO2/bipyridinium nanocomposite (Fe3O4@SiO2/BNC) (Figure 25). The catalyst was reused until the fifth reaction without much change in catalytic efficiency. Methodologies using other catalysts and even solvents have also been reported [44,45,46,47].
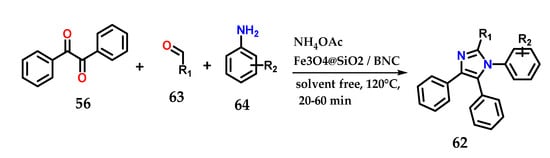
Figure 25.
Tetrasubstituted imidazoles 62 were obtained through the condensation of benzyl 56, aldehydes 63 and anilines 64 in the presence of ammonium acetate using Fe3O4@SiO2/BNC as a catalyst.
Another methodology used for obtaining hybrid imidazole derivatives was starting with hippuric acid 65 and 2-chloroquinoline-3-carbaldehyde 66 in acetic anhydride, and 4-((2-chloroquinolin-3-yl)methylene)-2-phenyloxazol-5(4H)-ones 67 could be obtained through Perkin condensation in the presence of anhydrous sodium acetate under microwave irradiation. Subsequently, the previously obtained derivatives were condensed with N-aminoarylcarboxamides 68 in pyridine under reflux to furnish the final desired azaheterocyclic acylhydrazides 69. For some derivatives of this series, antimicrobial properties were observed (Figure 26) [48].
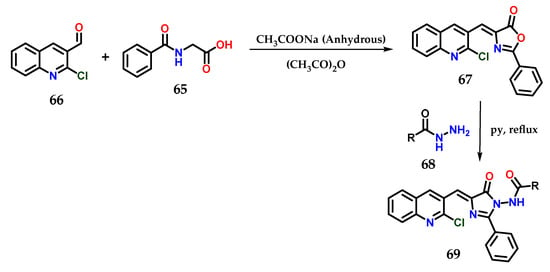
Figure 26.
Synthesis of N-(4-((2-chloroquinolin-3-yl)methylene)-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-yl)(aryl)amides 69.
Since its synthesis in 1858, the imidazole 1 ring has been exploited in different contexts, whether chemical or biological. The examples presented herein illustrate the more recent ways of obtaining this azaheterocyclic system through the use of a range of methodologies and chemical reagents, providing great chemical diversity.
3. Imidazole as a Privileged Structure in Medicinal Chemistry
As already mentioned, imidazole 1, in biological systems, is found in the form of the amino acid histidine 7, presenting an important role in the catalysis promoted by enzymatic systems [8]. Furthermore, the neurotransmitter histamine 8 induces immunological processes [8,16,49,50,51] and composes the structures of the guanine 70 and adenosine 71 bases of nucleic acids (Figure 27) [52].
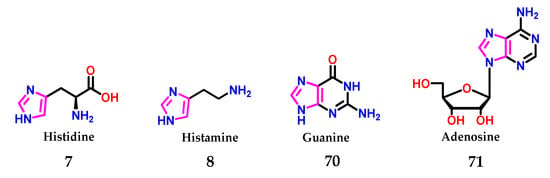
Figure 27.
Presence of the imidazole 1 nucleus in several biologically active compounds.
3.1. The Catalytic Potential of Imidazole in Biological Systems
The breakage of the P-O and C-O bonds are biological events of extreme importance and are estimated to be on the order of 5 to 13 million years [53] in the absence of enzymes and can reach the order of billions of years for DNA [54]. Imidazole 1 stands out for constituting numerous enzymatic active sites in the form of the histidine 7 amino acid residue and acting in catalytic processes, accelerating such unfavorable reactions [55].
The imidazole 1 group in biological systems generally acts in acid–base and nucleophilic catalysis (Figure 28). As an acid catalyst, protonated imidazole 1 acts as an acid facilitating the exit of the RO- group through hydrogen transfer [56]. In the neutral form, imidazole 1 can act as a nucleophilic catalyst by attacking the electrophilic center, leading to a phosphorylated or acylated intermediate, which is consecutively hydrolyzed, regenerating the imidazole 1 group [57]. Therefore, imidazole 1 catalyzes the cleavage of the X-O bond (for X = C or P). Finally, imidazole 1 can also act as a basic catalyst, assisting the attack of a nucleophile (Nü-H) on the electrophilic center of a substrate, abstracting a proton and thus increasing its nucleophilicity [58,59,60,61].
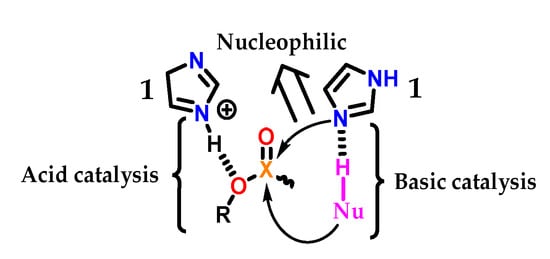
Figure 28.
Acidic, basic and nucleophilic catalysis promoted by an imidazole 1 subunit.
The amino acid histidine 7 plays a fundamental role in several enzymatic active sites, including ribonucleases, phosphotriesterases, kinases, chymotrypsins and histone deacetylases [62,63,64,65]. As illustrated in Figure 29, histidine 7 residues (H573/H574) participate in the catalytic process of deacetylation of the lysine residue by histone deacetylase 6 (HDAC6). We can also see the residue (H614) acting on the triad of amino acids responsible for sustaining the zinc atom (through the interaction η1(σ,Npy)) present in the catalytic site of the enzyme [7,65].
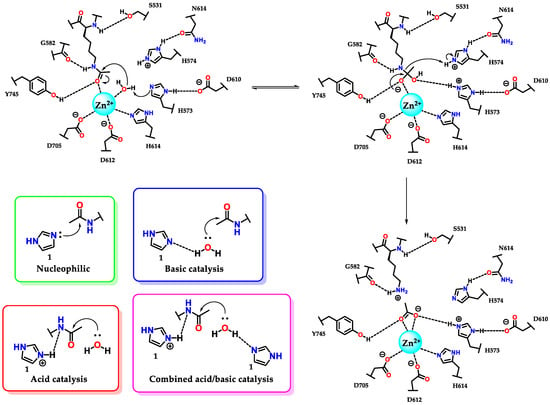
Figure 29.
Participation of histidine 7 residues in the deacetylation reaction of lysine residues promoted by enzyme histone deacetylase 6 (HDAC6) and the kinetic profiles of imidazole catalysis.
It is interesting to analyze in more detail the kinetic profiles considering the pH for an acid, basic, bifunctional acid–base and nucleophilic catalysis for the cases of deacylation reactions [66,67], but which similarly follow the same profiles in dephosphorylation [68,69]. In some pH ranges (considered pH 5-9), imidazole 1 has pronounced activity, with prevalence at pH values (considering pH 8-10) above the pKaH, for the neutral imidazole 1 species. This action makes it a basic and nucleophilic catalyst with similar kinetic profiles for both cases, where the rate constant is directly proportional to the increase in pH, capable of presenting a level where the amount of neutral and reactive species remains constant (pH ~9) (Figure 29) [49]. At the plateau, it is common to observe that the rate constant in nucleophilic catalysis is higher than that in basic catalysis, considering that nucleophilic processes are faster [55]. For pH values below the pKaH, where protonated imidazole 1 predominates, the catalysis is preferably acidic. As with basic and nucleophilic catalysis, acid catalysis has the same effect at the plateau (pH ~5) (Figure 29) [55].
Considering the above remarks, we can conclude that histidine 7 residues present in the active site of HDAC6 participate in catalysis by a combined acid/base mechanism, as this hydrolyses assists with the aid of the tyrosine residue (Y745) and the metal itself (Zn2+), which, when complexed with the carbonyl oxygen, further favors the reaction shown, considering the low reactivity of amide carbonyls (Figure 29).
3.2. Imidazole as a Building Block in the Structure of Bioactive Molecules and Drugs
Imidazole 1 is present in several chemical structures of pharmaceutical interest because its particular chemical properties could favor molecular recognition by different targets. Examples of the presence of imidazole 1 in the structure of bioactive substances include antibacterial [70,71,72,73], anti-inflammatory [74,75,76], antidiabetic [77], antiparasitic [78], antituberculosis [79], antifungal [80,81,82], antioxidant [83], antitumor [84,85,86], antimalarial [87,88], anticancer [89,90,91], antidepressant [92] and many other compounds (Figure 30).
Figure 30.
Presence of imidazole 1 in the most diverse classes of bioactive compounds.
Moreover, imidazole 1 is also present in several natural compounds with biological activity. As an example, pilocarpine 72 is used for the treatment of xerostomia and glaucoma [93], topsentin 73 shows anticancer activity [94] and isonaamine A 74 also shows anticancer activity through its action as an inhibitor of the epidermal growth factor receptor (EGFR) [95] (Figure 31).
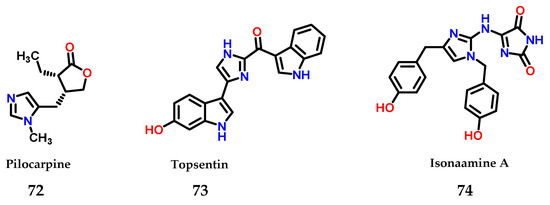
Figure 31.
Structures of imidazole-containing natural products pilocarpine 72, topsentin 73 and isonaamine A 74.
To date, a great diversity of imidazole-containing compounds with biological activity is known, whether of natural or synthetic origin. Among the compounds with imidazole-containing activity, azomycin 75 can be considered the one with the simplest structural complexity of all. The antibiotic azomycin was first isolated by Maeda in 1953 from a strain similar to Mesenteric nocardia [96]. In addition to azomycin 75, we can illustrate the structure of several more complex compounds that are even well known in current pharmacotherapy containing the imidazole nucleus, such as dacarbazine 76 [97], nafimidone 77 [98], flumizole 78 [99], cimetidine 79 [100], losartan 80 [101] and ketoconazole 81 [102] (Figure 32).
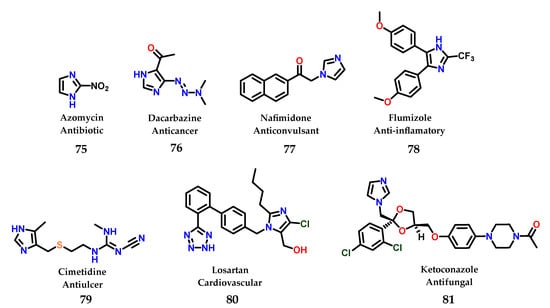
Figure 32.
Structures of azomycin 75 and other imidazole-containing drugs currently used in pharmacotherapy.
Imidazole 1 proves to be a very versatile structure for medicinal chemistry, not only for its ability to act directly as a pharmacophoric group but also for being able to act as a “guide” to other groups, directing them and favoring correct auxophoric/pharmacophoric interactions, allowing the exploration of a large number of possible substitutions. Imidazole 1 is considered a bioisostere of a carboxamide unit. Thus, it can be interpreted as a peptide backbone unit isostere [103]. Depending on the substituents and their substitution pattern, small mimetic oligopeptides with trans and cis conformations can be evidenced (Figure 33) [103,104]. The isosteric exchange of amides for imidazole can be considered a good strategy to overcome problems resulting from metabolic instability promoted by amidases [105].
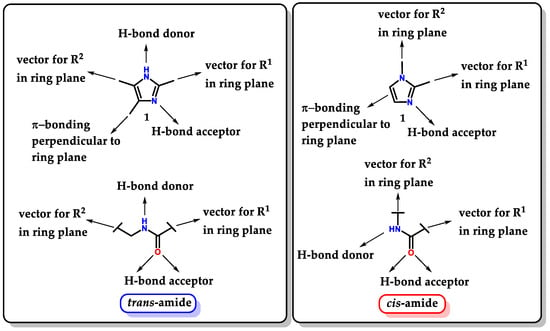
Figure 33.
Isosteric relationship between imidazole 1 and trans/cis-amides.
In a more recent work by Heppner and collaborators [106], the importance of the imidazole nucleus in compounds containing biological activity was highlighted. The aim of the study was the modulation of the mutated epidermal growth factor receptor (EGFR) target, in the context of non-small-cell lung cancer, which presents acquired drug resistance. Compounds with nanomolar potency against EGFR (L858R/T790M/C797S) were obtained in a reversible binding mechanism [106].
Analysis of the X-ray crystallographic results shows how the imidazole nucleus acts as a hydrogen bond acceptor for the catalytic residue of lysine (K745) in the “αC-helix out” inactive state of EGFR. Furthermore, selective N-methylation on the imidazole nucleus at the hydrogen bond acceptor position drastically reduces the potency, confirming the importance of the interaction of (K745) with the imidazole nucleus for modulating the EGFR variant (C797S) [106]. Additionally, it was observed that there is an intramolecular hydrogen interaction between the N-1 and the phenylacrylamide group 82 (Figure 34) [107]. The covalent bond with (C797) does not significantly change the mode of inhibitory interaction compared to the reversible compounds, indicating that the interaction of imidazole with (K757) is conserved in both covalent and non-covalent modes.
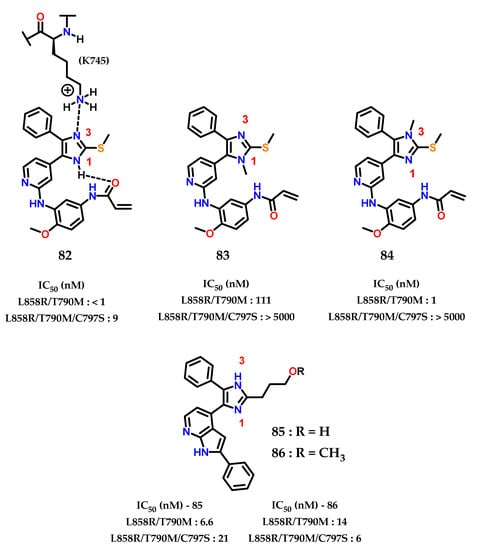
Figure 34.
EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) imidazole inhibitors.
Analyzing the activity values for 82, 83 and 84 (Figure 34), it was concluded that N-1 methylation 83 impacts activity against EGFR (L858R/T790M), indicating that the intramolecular interaction between imidazole and the phenylacrylamide group is required for the inhibitory mode of interaction. For N-3 methylation 84, the nitrogen involved in the interaction with (K745) in the inactive form “αC-helix out” of EGFR, does not significantly alter the inhibition activity towards EGFR (L858R/T790M), showing that the potency of 82 does not depend on the interaction with the residue (K745) but more on the covalent bond with (C797).
Finally, it was observed that, for EGFR (L858R/T790M/C797S), the presence of both unsubstituted nitrogens in the imidazole nucleus are extremely important for activity against this mutated form. Confirmed through the data already presented by 83 and 84, as well as the data for 85 and 86, showing excellent IC50 values with this interaction pattern, and acting as reversible inhibitors.
This study shows us as an interesting practical example how imidazole may be able to act as an important auxophoric group and act directing other auxophoric and pharmacophoric groups.
Another example of imidazole application was described by Lee et al. [108], where the activity-based sensing (ABS) strategy for detecting copper in living cells was presented, which preserves spatial information through a copper-dependent bio-conjugation reaction. Copper-targeted acyl imidazole dyes were designed that operate through copper-mediated activation of acyl imidazole electrophiles for subsequent labeling of proximal proteins at sites of high labile copper to provide a permanent color that resists washing and fixation (Figure 35).
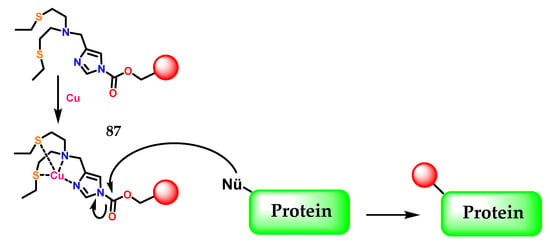
Figure 35.
The activity-based sensing (ABS) strategy using acyl imidazole electrophiles 87 for subsequent labeling of proximal proteins at sites of high labile copper.
Labile pools have been characterized using this strategy in three main types of brain cells: neurons, astrocytes and microglia. Exposure of each of these cell types to physiologically relevant stimuli distinct changes in these labile copper pools. Neurons exhibit translocation of labile copper from somatic cell bodies to peripheral processes after activation, while astrocytes and microglia exhibit global decreases and increases in intracellular labile copper pools, respectively, after exposure to inflammatory stimuli [108].
This work provides fundamental information on cell-type-dependent copper homeostasis, an essential metal in the brain, as well as a starting point for the design of new activity-transmitted probes for metals and other signal analytes and dynamic stress in biology, in the context of poor regulation of copper in inflammation and neurodegenerative process [109,110,111,112,113]. In Figure 35 the imidazole 1 present in 87 as a metal chelator, in this case copper, is demonstrated through the η1(σ,Npy) interaction, as previously commented.
In a recent background of compounds containing imidazole for anti-proliferative activity, a large number of di/triaryl imidazole-based derivatives (Figure 36) have been reported with p38α MAP and/or BRAF kinase inhibitory activities [114,115,116,117]. Considering these representatives, compound 88 (SB203580) was reported as a p38α MAP kinase inhibitor with IC50 value of 48 nM [114,115]. Furthermore, compounds 89 and 90 exhibited their inhibitory activities against p38α at IC50 values of 19 and 41 nM, respectively [116]. Compound 91 diaryl imidazole showed inhibitory activity against BRAF (IC50 = 900 nM) [117]. However, triaryl imidazole exhibited greater inhibitory activity against BRAF. In a series of triaryl-substituted imidazole-based derivatives, compound 92 was the most potent in inhibiting mutant BRAF in vitro [118].
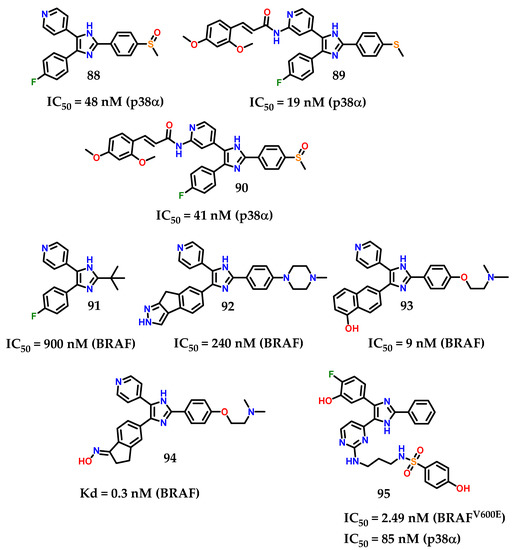
Figure 36.
Di/triaryl imidazole-based derivatives with p38α and/or BRAF inhibitory activity.
More studies about compound 92 show a weak inhibitory activity against mutant BRAF at the cellular level. Nevertheless, compound 93 showed potent BRAF inhibition (IC50 = 9 nM) and inhibited the growth of BRAF-dependent WM266.4 cells with GI50 of 0.22 µM [119]. Takle and coworkers [117] reported on SB-590885 94 among a series of derivatives based on triaryl imidazole with potent and selective inhibitory activity against BRAF kinase. The compound 94 was prepared to improve water solubility. Furthermore, it showed increased potency with more than 1000 times greater selectivity for p38α, GSK3-β and lck kinases compared to BRAF [117]. Recently, compound 95 (Figure 36) was reported as a derivative based on imidazol-5-yl-pyrimidine with BRAFV600E/p38α dual activity [120]. Compound 95 inhibited BRAFV600E and p38α MAP with IC50 values of 2.49 and 85 nM, respectively.
The designed compounds 96 and 97 were made by Youssif and collaborators [121] considering the proposed binding interactions of compound 95 with p38α and BRAFV600E (Figure 37). Investigation of compound 88’s binding interactions in p38α MAP revealed the presence of different types of binding interactions, including hydrogen bonding and hydrophobic interactions [121]. However, an unfavorable donor–donor interaction of the nitrogen N-1 on the imidazole ring of compound 88 with Lys53 at p38α was reported. To avoid this unfavorable interaction, the imidazole N-1 was alquilated with the hexyl group. Hexyl groups can also provide hydrophobic interactions with hydrophobic residues on p38α. The structure of the compounds 96 and 97 was also extended by a four-atom linker connecting the 1,3-benzodioxolo 96 and 4-methoxybenzyl 97 ring substituents with the triaryl imidazole scaffold [121]. The ligand includes three hydrogen bond acceptor atoms (CO-O-N=) and a hydrogen bond donor group (NH2) that can form hydrogen bonds with amino acids on the phosphate biding region at p38α [121].
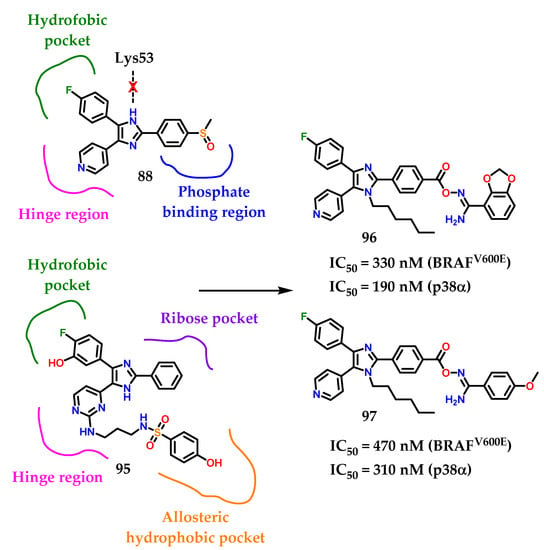
Figure 37.
Rational design of compounds 96 and 97.
Among the docking studies, compounds 96 and 97 were able to properly interact with Lys53 in the imidazole scaffold N-3. In conclusion, we can see that 96 and 97 are promising lead compounds with high potential for development as new dual p38α/BRAFV600E inhibitors based on the good activity values.
With these recent examples from the literature, we can see that imidazole presents itself as a privileged structure, being widely explored as a nucleus capable of efficiently participating and assisting in the modulation of several molecular targets and acting in other areas, such as probes for biological assessment.
3.3. The Origin of Cimetidine as an Imidazole-Containing Drug
To the best of our knowledge, there are four types of histamine receptors that belong to the G-protein-coupled receptor family: histamine H1 receptor, histamine H2 receptor, histamine H3 receptor and histamine H4 receptor, which could be stimulated by the endogenous messenger histamine 8 without distinction [122,123]. However, properly designed antagonists must be able to distinguish between them [124]. In the 1960s–1970s, selective antagonists capable of inhibiting the histamine receptors involved in the allergic process (H1 receptors) and antagonists capable of inhibiting histamine receptors responsible for gastric acid secretion (H2 receptors) were already known [124].
Variations were made in the structure of histamine 8 so that it was recognized by the receptor. Simultaneously, it was able to act as an antagonist. SAR studies on histamine analogs revealed that the requirements for histamine 8 to bind to the proposed H1 and H2 receptors were slightly different (Figure 38) [125]. On the H1 receptors, the essential requirements are as follows:
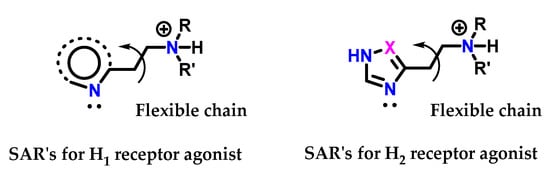
Figure 38.
SAR for histamine H1 and H2 receptor agonists.
The side chain must have a positively charged nitrogen atom with at least one hydrogen. Quaternary ammonium salts are not active.
There must be a flexible chain between the cation and the heteroaromatic ring.
The heteroaromatic ring does not have to be imidazole 1 but must have a nitrogen atom with a pair of electrons adjacent to the side chain.
On the other hand, the requirements for H2 and H1 receptors are the same, except that:
The heteroaromatic ring must have an amidine 5 unit (-HN-CH=N-) acting as a proton transfer agent.
Based on this information, it appears that the terminal amino group being protonated is involved in an ionic interaction with both types of receptors, while the nitrogen atoms of the heteroaromatic ring are linked through hydrogen bonds, and for H2 receptors, the ring participates in an extra interaction through proton transfer (Figure 39) [125].

Figure 39.
Scheme of the spatial arrangement for the interaction of histamine 8 on the H1 and H2 receptors.
Checking the changes in the structure of histamine 8, it was noted that 4(5)-methylhistamine 98 is a highly selective H2 receptor agonist, showing greater selectivity for H2 than for H1 [125,126]. 4(5)-Methylhistamine 98, similar to histamine 8, is a highly flexible molecule due to its side chain, but structural studies show that some of their conformations are less stable than others (Figure 40) [125,126]. In particular, conformation (A) is disadvantaged due to the steric interaction between the methyl group and the side ethylamine chain. The selectivity observed suggests that, for both receptors, 4(5)-methylhistamine 98 has to adopt two different conformations to bind to these receptors [125,126]. As 4(5)-methylhistamine 98 is more active as an agonist of the H2 receptor, this suggests that the favorable conformation (B) is required for the interaction with the H2 receptor, while the conformation required for the H1 receptor would be the sterically unfavorable conformation (A) [125,126].
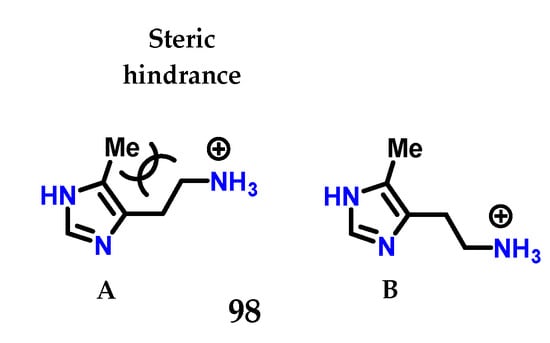
Figure 40.
Conformations of 4(5)-methylhistamine 98. Energetically unfavorable conformation A and the more stable conformation B.
In histamine 8, the exocyclic amino group is protonated at physiological pH, exerting a strong electron-withdrawing effect on the imidazole ring. This effect is more pronounced for the nitrogen closest to this side chain, so the hydrogen atom in the nitrogen Nπ will be more acidic than the one bound to the Nτ [125,126,127]. For this reason, the last tautomer (Nτ) is more stable than π (Nπ) in histamine 8, given that the favored ionization in the structure would lead to stabilization of the charge formed (Figure 41). On the other hand, the thiourea group of burimamide 99 exerts a less pronounced electron-donating effect, and therefore, the Nπ tautomer is favored [125,126,127]. Thus, the idea would be to remove electrons from the burimamide 99 side chain instead of donating electrons through the introduction of an electron withdrawing atom in this chain. The use of an isosteric group of the methylene group was proposed [125,126,127]. The sulfur atom is considered a good classical isostere of the methylene group, as their van der Waals radii and bond angles are similar [125,126,127,128]. The substitution site was also chosen based on synthetic reasons (Figure 41).
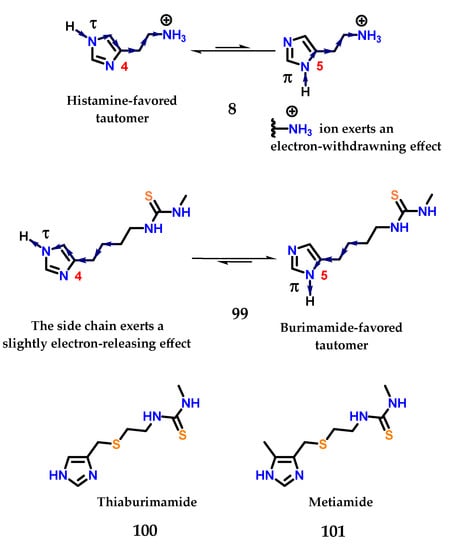
Figure 41.
Preferred tautomeric forms of histamine 8 and burimamide 99. Thiaburimamide 100 and metiamide 101 derivatives as H2 receptor antagonists.
The sulfur atom present in the side chain of thiaburimamide 100 carried out the removal of electrons, favoring the tau tautomer (τ). Furthermore, a hypothesis has been raised about the possibility of increasing the favoring of this tautomer through the insertion of a group in position 5 of the imidazole 1 ring [125,126,127]. In this position, the inductive effect would have a more pronounced impact on the neighboring nitrogen atom (Nτ). The methyl group was chosen because it was known that (4)-methylhistamine 98 was highly selective against the H2 receptor, in addition to influencing the pKa of hydrogen Nτ, with an electron donating effect [125,126,127,129]. Metiamide 101 was obtained, showing the highest antagonistic activity on H2 receptors (Figure 41).
Metiamide 101 was shown to be 10 times more potent than burimamide 99 [130] and showed great promise as an antiulcer agent. Unfortunately, several patients suffered from kidney problems and granulocytopenia, a disease that leads to a reduction in leukocytes and makes patients more susceptible to infection due to the metabolism of thiourea, which led to metiamide 101 not being approved in stage I clinical trials [131,132].
After the study of metiamide 101, several other studies were carried out with the objective of modifying the thiourea subunit, a region that is also responsible for important interactions with the target receptor, giving the compounds the profile of antagonists (region of binding of the antagonist). Figure 42 summarizes all the work and rationale used to design the first blockbuster drug cimetidine. Cimetidine, similar to other examples in the literature, is a drug that has an imidazole nucleus in its structure and is a beautiful and inspiring example of rational drug design carried out in the mid-1960s–1970s [133].
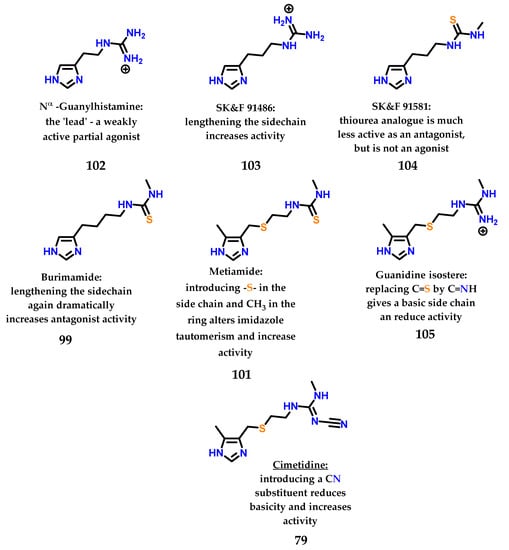
Figure 42.
Sequential analogues described by Black, Ganellin and coworkers at SK&F until the discovery of cimetidine 79.
Figueiredo and coworkers [134], who described the synthesis of methyl-imidazolyl N-acylhydrazone (NAH) derivatives showing antinociceptive activity, demonstrated another interesting study on imidazole 1 tautomerism, exploring the idea of cimetidine 79. Using the AM1 Hamiltonian [135] with unsubstituted derivative 106, the conformational behavior of the (NAH) motif around C-4 from the imidazole nucleus was investigated. Figueiredo and coworkers observed that the S-cis-like conformation (A) of the N-4 tautomer from compound 106 was ca. 6.0 kcal/mol more stable than the corresponding S-trans (A′) conformation (Figure 43) [134]. A five-member-like intramolecular hydrogen bond involving the hydrogen atom of the N-acylhydrazone moiety and the nitrogen atom N-3 of the imidazole 1 ring could be the reason for this favored conformation. Hydrogen bonding was also observed in the N-5 tautomer (B′) (Figure 43); however, in this case, a 3 kcal/mol decrease in stability was observed [134].
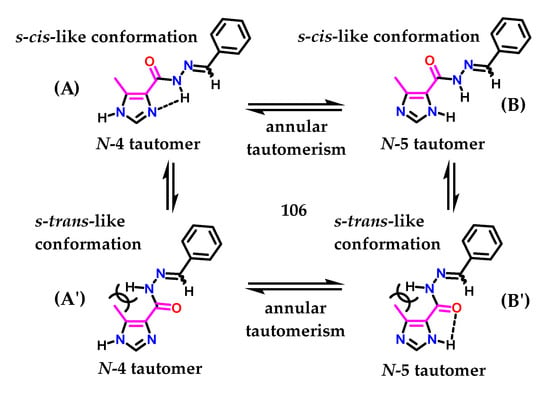
Figure 43.
Ring isomerism and conformers of N-acylhydrazone derivative 90.
We observed in this study [134] that for structure (A), there are several favorable factors to maintain the (4)-tautomer, i.e., the withdrawing group (EWG) in the C-4 position and the methyl group at the C-5 position, in addition to the possible intramolecular interaction due to the presence of the NAH subunit. Additionally, the methyl group at the C-5 position may contribute slightly through steric effects to the formation of (A) instead (A′), given the steric hindrance caused by methyl and the NAH substituent. A similar effect was observed for 4(5)-methylhistamine 98. Considering all the structures, (B′) demonstrates several unfavorable requirements to maintain (5)-tautomerism, even presenting an intramolecular interaction. Given that several of these structural factors play against the pKa value of N-1 present in the imidazole ring, its acidity is increased.
4. Conclusions
To date, much work has been done on the synthesis, functionalization, description of physicochemical characteristics and biological application of imidazole heterocycles. This work highlighted the special interest and importance of imidazole-containing derivatives in the field of medicinal chemistry and drug discovery.
As demonstrated, imidazole 1 is a structure that, despite being small, presents unique chemical complexity. It is a nucleus that proves to be very practical and versatile in its construction/functionalization and can be considered a rich source of chemical diversity. The role and importance of imidazole 1 in processes for the maintenance of living organisms, such as catalytic participation in enzymatic processes, were also reported. We observed examples of imidazole-based compounds with antibacterial, anti-inflammatory, antidiabetic, antiparasitic, antituberculosis, antifungal, antioxidant, antitumor, antimalarial, anticancer, antidepressant and many other activities in the literature. Finally, the role of imidazole 1 in drug research and development was briefly demonstrated through the discussion of the discovery of cimetidine 79. It was possible to explore several chemical and biological phenomena of this important drug, which certainly served as a rich base for knowledge and inspiration for several other bioactive imidazole-based drug candidates. This was also demonstrated in the structure of methyl-imidazolyl N-acylhydrazone derivative 106, which was produced after cimetidine and presented a nociceptive effect, and those that continue to currently be developed.
Author Contributions
All authors have written, read and agreed to the published version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001. The authors would like to thank INCT-INOFAR (BR, grant number 465.249/2014-0), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ, grant numbers E-26/010.001273/2016 and SEI-260003/003613/2022, fellowship to CAMF), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, grant number 304394/2017-3) and Departamento de Ciência e Tecnologia (DECIT, Ministry of Heath, Brazil).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Debus, H. Ueber Die Einwirkung Des Ammoniaks Auf Glyoxal. Ann. Chem. Pharm. 1858, 107, 199–208. [Google Scholar] [CrossRef]
- Verma, A.; Joshi, S.; Singh, D. Imidazole: Having Versatile Biological Activities. J. Chem. 2013, 1, 1–12. [Google Scholar] [CrossRef]
- Asif, M. A Mini Review: Biological Significances of Nitrogen Hetero Atom Containing Heterocyclic Compounds. Int. J. Bioorg. Chem. 2017, 2, 146–152. [Google Scholar]
- Zhao, C.; Qiao, X.; Yi, Z.; Guan, Q.; Li, W. Active Centre and Reactivity Descriptor of a Green Single Component Imidazole Catalyst for Acetylene Hydrochlorination. Phys. Chem. Chem. Phys. 2020, 22, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Olofson, A.; Yakushijin, K.; Horne, D.A. Synthesis of Marine Sponge Alkaloids Oroidin, Clathrodin, and Dispacamides. Preparation and Transformation of 2-Amino-4, 5–Dialkoxy-4, 5–Dihydroimidazolines from 2-Aminoimidazoles. J. Org. Chem. 1998, 63, 1248–1253. [Google Scholar] [CrossRef]
- Wan, Y.; Hur, W.; Cho, C.Y.; Liu, Y.; Adrian, F.J.; Lozach, O.; Bach, S.; Mayer, T.; Fabbro, D.; Meijer, L. Synthesis and Target Identification of Hymenialdisine Analogs. Chem. Biol. 2004, 11, 247–259. [Google Scholar] [CrossRef]
- Richaud, A.; Barba-Behrens, N.; Méndez, F. Chemical Reactivity of the Imidazole: A Semblance of Pyridine and Pyrrole? Org. Lett. 2011, 13, 972–975. [Google Scholar] [CrossRef]
- Mullins, R.J.; Azman, A.M. Imidazoles. In Tetrahedron Organic Chemistry Series; Elsevier: Amsterdam, The Netherlands, 2007; Volume 26, pp. 407–433. [Google Scholar] [CrossRef]
- Stockman, R.A. Heterocyclic Chemistry. Annu. Rep. Prog. Chem. Sect. B Org. Chem. 2007, 103, 107–124. [Google Scholar] [CrossRef]
- Movellan, K.T.; Wegstroth, M.; Overkamp, K.; Leonov, A.; Becker, S.; Andreas, L.B. Imidazole–Imidazole Hydrogen Bonding in the PH-Sensing Histidine Side Chains of Influenza A M2. J. Am. Chem. Soc. 2020, 142, 2704–2708. [Google Scholar] [CrossRef]
- Kochendoerfer, G.G.; Salom, D.; Lear, J.D.; Wilk-Orescan, R.; Kent, S.B.H.; DeGrado, W.F. Total Chemical Synthesis of the Integral Membrane Protein Influenza A Virus M2: Role of Its C–Terminal Domain in Tetramer Assembly. Biochemistry 1999, 38, 11905–11913. [Google Scholar] [CrossRef]
- Sakaguchi, T.; Tu, Q.; Pinto, L.H.; Lamb, R.A. The Active Oligomeric State of the Minimalistic Influenza Virus M2 Ion Channel Is a Tetramer. Proc. Nat. Acad. Sci. USA 1997, 94, 5000–5005. [Google Scholar] [CrossRef]
- Sugrue, R.J.; Hay, A.J. Structural Characteristics of the M2 Protein of Influenza a Viruses: Evidence That It Forms a Tetrameric Channe. Virology 1991, 180, 617–624. [Google Scholar] [CrossRef]
- Sugrue, R.J.; Bahadur, G.; Zambon, M.C.; Hall-Smith, M.; Douglas, A.R.; Hay, A.J. Specific Structural Alteration of the Influenza Haemagglutinin by Amantadine. EMBO J. 1990, 9, 3469–3476. [Google Scholar] [CrossRef]
- Colvin, M.T.; Andreas, L.B.; Chou, J.J.; Griffin, R.G. Proton Association Constants of His 37 in the Influenza–A M218–60 Dimer–of-Dimers. Biochemistry 2014, 53, 5987–5994. [Google Scholar] [CrossRef]
- Bylund, D.B. Histamine. In Reference Module in Biomedical Sciences. 2017. Available online: https://scitechconnect.elsevier.com/resources/reference-module-biomedical-sciences/ (accessed on 31 December 2022). [CrossRef]
- Arnau, N.; Arredondo, Y.; Moreno-Mañas, M.; Pleixats, R.; Villarroya, M. Palladium (0)-catalyzed Allylation of 4 (5)-substituted Imidazoles, 5 (6)-substituted Benzimidazoles, Benzotriazole and 5 (6)-methylbenzotriazole. J. Heterocycl. Chem. 1995, 32, 1325–1334. [Google Scholar] [CrossRef]
- Kuzuya, M.; Noguchi, A.; Mano, E.; Okuda, T. The Structure–Reactivity–Chemoselectivity Relationship on the Reactions of 1–Unsubstituted Tautomeric 2-Pyridones with Benzyne. Bull. Chem. Soc. Jpn. 1985, 58, 1149–1155. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Lagowski, J.M. Prototropic Tautomerism of Heteroaromatic Compounds: IV. Five–Membered Rings with Two or More Hetero Atoms. Adv. Heterocycl. Chem. 1963, 2, 27–81. [Google Scholar]
- Walba, H.; Isensee, R.W. Acidity Constants of Some Arylimidazoles and Their Cations. J. Org. Chem. 1961, 26, 2789–2791. [Google Scholar] [CrossRef]
- Zhu, L.; Li, G.; Luo, L.; Guo, P.; Lan, J.; You, J. Highly Functional Group Tolerance in Copper-Catalyzed N-Arylation of Nitrogen-Containing Heterocycles under Mild Conditions. J. Org. Chem. 2009, 74, 2200–2202. [Google Scholar] [CrossRef]
- Ruan, Y.; Chen, Y.; Gu, L.; Luo, Y.; Yang, Z.; He, L. Preparation of Imidazole Derivatives via Bisfunctionalization of Alkynes Catalyzed by Ruthenium Carbonyl. Synthesis 2019, 51, 3520–3528. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Z.; Li, C.-M.; Lu, Y.; Vaddady, P.K.; Meibohm, B.; Dalton, J.T.; Miller, D.D.; Li, W. Discovery of Novel 2-Aryl–4–Benzoyl–Imidazoles Targeting the Colchicines Binding Site in Tubulin as Potential Anticancer Agents. J. Med. Chem. 2010, 53, 7414–7427. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, B.; Tan, M.; Ekmekci, Z.; Menges, N. A Novel Fluorescent Sensor Based on Imidazole Derivative for Fe3+ Ions. J. Lumin. 2017, 192, 1096–1103. [Google Scholar] [CrossRef]
- de Toledo, I.; Grigolo, T.A.; Bennett, J.M.; Elkins, J.M.; Pilli, R.A. Modular Synthesis of Di-and Trisubstituted Imidazoles from Ketones and Aldehydes: A Route to Kinase Inhibitors. J. Org. Chem. 2019, 84, 14187–14201. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, D.A.; Dunsford, J.J.; Ngwerume, S.; Saunders, A.R.; Gill, D.M.; Camp, J.E. Synthesis of 2, 4–Disubstituted Imidazoles via Nucleophilic Catalysis. Synlett. 2020, 31, 797–800. [Google Scholar]
- Fang, S.; Yu, H.; Yang, X.; Li, J.; Shao, L. Nickel-Catalyzed Construction of 2, 4-Disubstituted Imidazoles via C–C Coupling and C− N Condensation Cascade Reactions. Adv. Synth. Catal. 2019, 361, 3312–3317. [Google Scholar] [CrossRef]
- Wong, L.C.; Gehre, A.; Stanforth, S.P.; Tarbit, B. Convenient Synthesis of Highly Substituted Imidazole Derivatives. Synth. Commun. 2013, 43, 80–84. [Google Scholar] [CrossRef]
- Noriega-Iribe, E.; Díaz-Rubio, L.; Estolano-Cobián, A.; Barajas-Carrillo, V.W.; Padrón, J.M.; Salazar-Aranda, R.; Díaz–Molina, R.; García-González, V.; Chávez–Santoscoy, R.A.; Chávez, D. In Vitro and in Silico Screening of 2, 4, 5–Trisubstituted Imidazole Derivatives as Potential Xanthine Oxidase and Acetylcholinesterase Inhibitors, Antioxidant, and Antiproliferative Agents. Appl. Sci. 2020, 10, 2889. [Google Scholar] [CrossRef]
- Kalhor, M.; Seyedzade, Z.; Zarnegar, Z. (NH4) 2Ce (NO3) 6/HNO3 as a High-Performance Oxidation Catalyst for the One–Step, Solvent–Free Synthesis of Dicyano Imidazoles. Polycycl. Aromat. Compd. 2021, 41, 1506–1514. [Google Scholar] [CrossRef]
- Takashima, R.; Tsunekawa, K.; Shinozaki, M.; Suzuki, Y. Selective Synthesis of 1, 4, 5–Trisubstituted Imidazoles from α-Imino Ketones Prepared by N-Heterocyclic–Carbene–Catalyzed Aroylation. Tetrahedron 2018, 74, 2261–2267. [Google Scholar] [CrossRef]
- Bunev, A.S.; Vasiliev, M.A.; Statsyuk, V.E.; Ostapenko, G.I.; Peregudov, A.S. Synthesis of 1–Aryl-4-Tosyl-5-(Trifluoromethyl)-1H–Imidazoles. J. Fluor. Chem. 2014, 163, 34–37. [Google Scholar] [CrossRef]
- Hanoon, H.D.; Kowsari, E.; Abdouss, M.; Ghasemi, M.H.; Zandi, H. Highly Efficient and Simple Protocol for Synthesis of 2, 4, 5–Triarylimidazole Derivatives from Benzil Using Fluorinated Graphene Oxide as Effective and Reusable Catalyst. Res. Chem. Intermed. 2017, 43, 4023–4041. [Google Scholar] [CrossRef]
- Tan, J.; Li, J.R.; Hu, Y.L. Novel and Efficient Multifunctional Periodic Mesoporous Organosilica Supported Benzotriazolium Ionic Liquids for Reusable Synthesis of 2, 4, 5-Trisubstituted Imidazoles. J. Saudi Chem. Soc. 2020, 24, 777–784. [Google Scholar] [CrossRef]
- Hilal, D.A.; Hanoon, H.D. Bronsted Acidic Ionic Liquid Catalyzed an Eco-Friendly and Efficient Procedure for Synthesis of 2, 4, 5-Trisubstituted Imidazole Derivatives under Ultrasound Irradiation and Optimal Conditions. Res. Chem. Intermed. 2020, 46, 1521–1538. [Google Scholar] [CrossRef]
- Alinezhad, H.; Tajbakhsh, M.; Maleki, B.; Pourshaban Oushibi, F. Acidic Ionic Liquid [H-NP] HSO4 Promoted One-Pot Synthesis of Dihydro–1H-Indeno [1, 2–b] Pyridines and Polysubstituted Imidazoles. Polycycl. Aromat. Compd. 2020, 40, 1485–1500. [Google Scholar] [CrossRef]
- Maleki, A.; Rahimi, J.; Valadi, K. Sulfonated Fe3O4@ PVA Superparamagnetic Nanostructure: Design, in-Situ Preparation, Characterization and Application in the Synthesis of Imidazoles as a Highly Efficient Organic–Inorganic Bronsted Acid Catalyst. Nano–Struct. Nano–Objects. 2019, 18, 100264. [Google Scholar] [CrossRef]
- Gupta, S.; Lakshman, M. Magnetic Nano Cobalt Ferrite: An Efficient Recoverable Catalyst for Synthesis of 2, 4, 5-Trisubstituted Imidazoles. J. Med. Chem. Sci. 2019, 2, 51–54. [Google Scholar]
- Kalhor, M.; Zarnegar, Z. Fe3O4/SO3H@ Zeolite-Y as a Novel Multi–Functional and Magnetic Nanocatalyst for Clean and Soft Synthesis of Imidazole and Perimidine Derivatives. RSC Adv. 2019, 9, 19333–19346. [Google Scholar] [CrossRef]
- Singh, H.; Rajput, J.K. Co (II) Anchored Glutaraldehyde Crosslinked Magnetic Chitosan Nanoparticles (MCS) for Synthesis of 2, 4, 5-trisubstituted and 1, 2, 4, 5-tetrasubstituted Imidazoles. Appl. Organomet. Chem. 2018, 32, e3989. [Google Scholar] [CrossRef]
- Wagare, D.S.; Sonone, A.; Farooqui, M.; Durrani, A. An Efficient and Green Microwave-Assisted One Pot Synthesis of Imidazothiadiazoles in PEG–400 and Water. Polycycl. Aromat. Compd. 2021, 41, 1749–1754. [Google Scholar] [CrossRef]
- Ali, M.A.E.A.A.; Abu-Dief, A.M. CuFe2O4 Nanoparticles: An Efficient Heterogeneous Magnetically Separable Catalyst for Synthesis of Some Novel Propynyl-1H–Imidazoles Derivatives. Tetrahedron 2015, 71, 2579–2584. [Google Scholar]
- Rajkumar, R.; Kamaraj, A.; Krishnasamy, K. Multicomponent, One–Pot Synthesis and Spectroscopic Studies of 1-(2–(2, 4, 5-Triphenyl-1 H-Imidazol–1–Yl) Ethyl) Piperazine Derivatives. J. Taibah Uni. Sci. 2015, 9, 498–507. [Google Scholar] [CrossRef]
- Wang, S.; Kirillova, K.; Lehto, X. Travelers’ Food Experience Sharing on Social Network Sites. J. Travel Tour. Mark. 2017, 34, 680–693. [Google Scholar] [CrossRef]
- Hosseini, S.; Kiasat, A.R.; Farhadi, A. Fe3O4@ SiO2/Bipyridinium Nanocomposite as a Magnetic and Recyclable Heterogeneous Catalyst for the Synthesis of Highly Substituted Imidazoles Via Multi–Component Condensation Strategy. Polycycl. Aromat. Compd. 2021, 41, 761–771. [Google Scholar] [CrossRef]
- Davoodnia, A.; Heravi, M.M.; Safavi–Rad, Z.; Tavakoli–Hoseini, N. Green, One-Pot, Solvent–Free Synthesis of 1, 2, 4, 5-Tetrasubstituted Imidazoles Using a Brønsted Acidic Ionic Liquid as Novel and Reusable Catalyst. Synth. Commun. 2010, 40, 2588–2597. [Google Scholar] [CrossRef]
- Safa, K.D.; Feyzi, A.; Allahvirdinesbat, M.; Sarchami, L.; Panahi, P.N. Synthesis of Novel Organosilicon Compounds Possessing Fully Substituted Imidazole Nucleus Sonocatalyzed by Fe–Cu/ZSM-5 Bimetallic Oxides. Synth. Commun. 2015, 45, 382–390. [Google Scholar] [CrossRef]
- Desai, N.C.; Maheta, A.S.; Rajpara, K.M.; Joshi, V.; Vaghani, H.V.; Satodiya, H.M. Green Synthesis of Novel Quinoline Based Imidazole Derivatives and Evaluation of Their Antimicrobial Activity. J. Saudi Chem. Soc. 2014, 18, 963–971. [Google Scholar] [CrossRef]
- Green, J.P.; Prell, G.D.; Khandelwal, J.K.; Blandina, P. Aspects of Histamine Metabolism. Agents Actions 1987, 22, 1–15. [Google Scholar] [CrossRef]
- Haas, H.L.; Sergeeva, O.A.; Selbach, O. Histamine in the Nervous System. Physiol. Rev. 2008, 88, 1183–1241. [Google Scholar] [CrossRef]
- Fox, S.W. Chemistry of the Biologically Important Imidazoles. Chem. Rev. 1943, 32, 47–71. [Google Scholar] [CrossRef]
- Gupta, R.R.; Kumar, M.; Gupta, V. Heterocyclic Chemistry: Volume II: Five-Membered Heterocycles; Springer Science & Business Media: Berlin, German, 2013; ISBN 3662077574. [Google Scholar]
- Wolfenden, R.; Snider, M.J. The Depth of Chemical Time and the Power of Enzymes as Catalysts. Acc. Chem. Res. 2001, 34, 938–945. [Google Scholar] [CrossRef]
- Liu, C.; Wang, M.; Zhang, T.; Sun, H. DNA Hydrolysis Promoted by Di–and Multi–Nuclear Metal Complexes. Coord. Chem. Rev. 2004, 248, 147–168. [Google Scholar] [CrossRef]
- Silva, V.B.; Orth, E.S. Imidazole and Catalysis: A Perffect Match. Quim. Nova. 2021, 44, 318–333. [Google Scholar]
- Schowen, K.B.; Limbach, H.-H.; Denisov, G.S.; Schowen, R.L. Hydrogen Bonds and Proton Transfer in General–Catalytic Transition-State Stabilization in Enzyme Catalysis. Biochim. Biophys. Bioenerg. 2000, 1458, 43–62. [Google Scholar] [CrossRef]
- Attwood, P.; Piggott, M.J.; Zu, X.L.; Besant, P.G. Focus on Phosphohistidine. Amino Acids 2007, 32, 145–156. [Google Scholar] [CrossRef]
- Raines, R.T. Ribonuclease a. Chem. Rev. 1998, 98, 1045–1066. [Google Scholar] [CrossRef]
- Anderson, V.E.; Ruszczycky, M.W.; Harris, M.E. Activation of Oxygen Nucleophiles in Enzyme Catalysis. Chem. Rev. 2006, 106, 3236–3251. [Google Scholar] [CrossRef]
- Matuszak, C.A.; Matuszak, A.J. Imidazole-Versatile Today, Prominent Tomorrow. J. Chem. Educ. 1976, 53, 280. [Google Scholar] [CrossRef]
- Orth, E.S.; Brandao, T.A.S.; Milagre, H.M.S.; Eberlin, M.N.; Nome, F. Intramolecular Acid− Base Catalysis of a Phosphate Diester: Modeling the Ribonuclease Mechanism. J. Am. Chem. Soc. 2008, 130, 2436–2437. [Google Scholar] [CrossRef]
- Raushel, F.M.; Holden, H.M. Phosphotriesterase: An Enzyme in Search of Its Natural Substrate. Adv. Enzymol. Relat. Areas Mol. Biol. 2000, 74, 51–93. [Google Scholar]
- Yang, W. Nucleases: Diversity of Structure, Function and Mechanism. Q. Rev. Biophys. 2011, 44, 1–93. [Google Scholar] [CrossRef]
- Lehninger, A.L.; Nelson, D.L.; Cox, M.M. Overhead Transparency Set for Lehninger Principles of Biochemistry, 4th ed.; Palgrave Mcmillan: London, UK, 2004; ISBN 071675956X. [Google Scholar]
- Hai, Y.; Christianson, D.W. Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-S.; Ueno, A. A New Feature of Bifunctional Catalysis. Cyclodextrins Bearing Two Imidazole Moieties as Hydrolysis Enzyme Model. Chem. Lett. 2000, 29, 258–259. [Google Scholar] [CrossRef]
- Brown, R.S.; Clewley, R.G. Intramolecular Imidazole-Promoted General–Base Catalysis of the Hydrolysis of an Acetylimidazole. J. Org. Chem. 1987, 52, 1216–1218. [Google Scholar] [CrossRef]
- Orth, E.S.; Almeida, T.G.; Silva, V.B.; Oliveira, A.R.M.; Ocampos, F.M.M.; Barison, A. Mechanistic Insight on the Catalytic Detoxification of Paraoxon Mediated by Imidazole: Furnishing Optimum Scaffolds for Scavenging Organophosphorus Agents. J. Mol. Catal. A Chem. 2015, 403, 93–98. [Google Scholar] [CrossRef]
- Campos, R.B.; Menezes, L.R.A.; Barison, A.; Tantillo, D.J.; Orth, E.S. The Importance of Methyl Positioning and Tautomeric Equilibria for Imidazole Nucleophilicity. Eur. J. Chem. 2016, 22, 15521–15528. [Google Scholar] [CrossRef]
- Khabnadideh, S.; Rezaei, Z.; Khalafi–Nezhad, A.; Bahrinajafi, R.; Mohamadi, R.; Farrokhroz, A.A. Synthesis of N-Alkylated Derivatives of Imidazole as Antibacterial Agents. Bioorg. Med. Chem. Lett. 2003, 13, 2863–2865. [Google Scholar] [CrossRef]
- Kalaria, P.N.; Satasia, S.P.; Avalani, J.R.; Raval, D.K. Ultrasound–Assisted One-Pot Four–Component Synthesis of Novel 2-Amino-3-Cyanopyridine Derivatives Bearing 5-Imidazopyrazole Scaffold and Their Biological Broadcast. Eur. J. Med. Chem. 2014, 83, 655–664. [Google Scholar] [CrossRef]
- Moraski, G.C.; Thanassi, J.A.; Podos, S.D.; Pucci, M.J.; Miller, M.J. One–Step Syntheses of Nitrofuranyl Benzimidazoles That Are Active against Multidrug–Resistant Bacteria. J. Antibiot. 2011, 64, 667–671. [Google Scholar] [CrossRef]
- Lu, B.; Lu, F.; Ran, L.; Yu, K.; Xiao, Y.; Li, Z.; Dai, F.; Wu, D.; Lan, G. Self-Assembly of Natural Protein and Imidazole Molecules on Gold Nanoparticles: Applications in Wound Healing against Multi–Drug Resistant Bacteria. Int. J. Biol. Macromol. 2018, 119, 505–516. [Google Scholar] [CrossRef]
- Toja, E.; Selva, D.; Schiatti, P. 3-Alkyl-2-Aryl–3H-Naphth [1, 2–d] Imidazoles, a Novel Class of Nonacidic Antiinflammatory Agents. J. Med. Chem. 1984, 27, 610–616. [Google Scholar] [CrossRef]
- Bender, P.E.; Hill, D.; Offen, P.H.; Razgaitis, K.; Lavanchy, P.; Stringer, O.D.; Sutton, B.M.; Griswold, D.E.; DiMartino, M. 5, 6–Diaryl-2, 3–Dihydroimidazo [2, 1-b] Thiazoles: A New Class of Immunoregulatory Antiinflammatory Agents. J. Med. Chem. 1985, 28, 1169–1177. [Google Scholar] [CrossRef]
- de Gaetano, M.; Butler, E.; Gahan, K.; Zanetti, A.; Marai, M.; Chen, J.; Cacace, A.; Hams, E.; Maingot, C.; McLoughlin, A. Asymmetric Synthesis and Biological Evaluation of Imidazole-and Oxazole-Containing Synthetic Lipoxin A4 Mimetics (SLXms). Eur. J. Med. Chem. 2019, 162, 80–108. [Google Scholar] [CrossRef]
- Koh, A.; Molinaro, A.; Ståhlman, M.; Khan, M.T.; Schmidt, C.; Mannerås–Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through MTORC1. Cell 2018, 175, 947–961. [Google Scholar] [CrossRef]
- Adeyemi, O.S.; Eseola, A.O.; Plass, W.; Atolani, O.; Sugi, T.; Han, Y.; Batiha, G.E.; Kato, K.; Awakan, O.J.; Olaolu, T.D. Imidazole Derivatives as Antiparasitic Agents and Use of Molecular Modeling to Investigate the Structure–Activity Relationship. Parasitol. Res. 2020, 119, 1925–1941. [Google Scholar] [CrossRef]
- Gupta, P.; Hameed, S.; Jain, R. Ring-Substituted Imidazoles as a New Class of Anti-Tuberculosis Agents. Eur. J. Med. Chem. 2004, 39, 805–814. [Google Scholar] [CrossRef]
- Jeanmart, S.; Gagnepain, J.; Maity, P.; Lamberth, C.; Cederbaum, F.; Rajan, R.; Jacob, O.; Blum, M.; Bieri, S. Synthesis and Fungicidal Activity of Novel Imidazole–Based Ketene Dithioacetals. Bioorg. Med. Chem. 2018, 26, 2009–2016. [Google Scholar] [CrossRef]
- B’bhatt, H.; Sharma, S. Synthesis, Characterization, and Biological Evaluation of Some Tri-Substituted Imidazole/Thiazole Derivatives. J. Heterocycl. Chem. 2015, 52, 1126–1131. [Google Scholar] [CrossRef]
- Park, N. –H.; Shin, K.-H.; Kang, M.K. 34—Antifungal and Antiviral Agents. Pharmacol. Ther. 2017, 10, 488–503. [Google Scholar]
- Hu, D.-C.; Chen, L.-W.; Yang, Y.-X.; Liu, J.-C. Syntheses, Structures and Antioxidant Activities of Two New Cu (II) Complexes with a Benzimidazole Schiff Base Ligand. Inorg. Nano-Met. Chem. 2017. (just accepted). [Google Scholar] [CrossRef]
- Keppler, B.K.; Wehe, D.; Endres, H.; Rupp, W. Synthesis, Antitumor Activity, and x–Ray Structure of Bis (Imidazolium)(Imidazole) Pentachlororuthenate (III),(ImH) 2 (RuImCl5). Inorg. Chem. 1987, 26, 844–846. [Google Scholar] [CrossRef]
- Antonini, I.; Claudi, F.; Cristalli, G.; Franchetti, P.; Grifantini, M.; Martelli, S. Heterocyclic Quinones with Potential Antitumor Activity. 2. Synthesis and Antitumor Activity of Some Benzimidazole-4, 7–Dione Derivatives. J. Med. Chem. 1988, 31, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Shimozato, O.; Matsuo, N.; Mori, Y.; Shinozaki, Y.; Lin, J.; Watanabe, T.; Takatori, A.; Koshikawa, N.; Ozaki, T. Hydrophobic Structure of Hairpin Ten-Ring Pyrrole–Imidazole Polyamides Enhances Tumor Tissue Accumulation/Retention in Vivo. Bioorg. Med. Chem. 2018, 26, 2337–2344. [Google Scholar] [CrossRef] [PubMed]
- Shalmali, N.; Ali, M.R.; Bawa, S. Imidazole: An Essential Edifice for the Identification of New Lead Compounds and Drug Development. Mini Rev. Med. Chem. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Kondaparla, S.; Manhas, A.; Dola, V.R.; Srivastava, K.; Puri, S.K.; Katti, S.B. Design, Synthesis and Antiplasmodial Activity of Novel Imidazole Derivatives Based on 7-Chloro–4–Aminoquinoline. Bioorg. Chem. 2018, 80, 204–211. [Google Scholar] [CrossRef] [PubMed]
- James, D.A.; Koya, K.; Li, H.; Chen, S.; Xia, Z.; Ying, W.; Wu, Y.; Sun, L. Conjugated Indole-Imidazole Derivatives Displaying Cytotoxicity against Multidrug Resistant Cancer Cell Lines. Bioorg. Med. Chem. Lett. 2006, 16, 5164–5168. [Google Scholar] [CrossRef]
- Özkay, Y.; Işıkdağ, İ.; İncesu, Z.; Akalın, G. Synthesis of 2–Substituted–N–[4-(1–Methyl-4, 5–Diphenyl-1H–Imidazole–2–Yl) Phenyl] Acetamide Derivatives and Evaluation of Their Anticancer Activity. Eur. J. Med. Chem. 2010, 45, 3320–3328. [Google Scholar] [CrossRef]
- Bellina, F.; Guazzelli, N.; Lessi, M.; Manzini, C. Imidazole Analogues of Resveratrol: Synthesis and Cancer Cell Growth Evaluation. Tetrahedron 2015, 71, 2298–2305. [Google Scholar] [CrossRef]
- Seo, H.J.; Park, E.-J.; Kim, M.J.; Kang, S.Y.; Lee, S.H.; Kim, H.J.; Lee, K.N.; Jung, M.E.; Lee, M.; Kim, M.-S. Design and Synthesis of Novel Arylpiperazine Derivatives Containing the Imidazole Core Targeting 5-HT2A Receptor and 5-HT Transporter. J. Med. Chem. 2011, 54, 6305–6318. [Google Scholar] [CrossRef]
- Panarese, V.; Moshirfar, M. Pilocarpine. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2021. [Google Scholar]
- Hwang, J.; Kim, D.; Park, J.S.; Park, H.J.; Shin, J.; Lee, S.K. Photoprotective Activity of Topsentin, A Bis (Indole) Alkaloid from the Marine Sponge Spongosorites Genitrix, by Regulation of COX–2 and Mir–4485 Expression in UVB-Irradiated Human Keratinocyte Cells. Mar. Drugs. 2020, 18, 87. [Google Scholar] [CrossRef]
- Gibbons, J.B.; Salvant, J.M.; Vaden, R.M.; Kwon, K.-H.; Welm, B.E.; Looper, R.E. Synthesis of Naamidine A and Selective Access to N2-Acyl–2–Aminoimidazole Analogues. J. Org. Chem. 2015, 80, 10076–10085. [Google Scholar] [CrossRef]
- Maeda, K. A New Antibiotic, Azomycin. J. Antibiot. 1953, 6, 182. [Google Scholar]
- Al–Badr, A.A.; Alodhaib, M.M. Dacarbazine. Profiles Drug Subst. Excip. Relat. Methodol. 2016, 41, 323–377. [Google Scholar] [CrossRef]
- Hempel, A.; Camerman, N.; Camerman, A.; Mastropaolo, D. Nafimidone Monohydrate: An Imidazole Anticonvulsant. Acta Crystallogr. Sect. E Struct. Rep. 2005, 61, 1387–1389. [Google Scholar] [CrossRef]
- McIlhenny, H.M.; Bettis, J.W.; Wiseman, E.H. Flumizole, a New Nonsteroidal Anti-inflammatory Agent. J. Pharm. Sci. 1975, 64, 1469–1475. [Google Scholar] [CrossRef]
- Finkelstein, W.; Isselbacher, K.J. Cimetidine. N. Engl. J. Med. 1978, 299, 992–996. [Google Scholar]
- Goa, K.L.; Wagstaff, A.J. Losartan Potassium. Drugs 1996, 51, 820–845. [Google Scholar] [CrossRef]
- Sohn, C.A. Evaluation of Ketoconazole. Clin. Pharm. 1982, 1, 217–224. [Google Scholar]
- Petit, S.; Fruit, C.; Bischoff, L. New Family of Peptidomimetics Based on the Imidazole Motif. Org. Lett. 2010, 12, 4928–4931. [Google Scholar] [CrossRef]
- Bräse, S. Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation; Royal Society of Chemistry: London, UK, 2015; ISBN 1782622241. [Google Scholar]
- Busby, R.W.; Cai, X.; Yang, S.; Ramos, L.; Venkatarangan, L.; Shen, H.; Wax, S.; Sadeque, A.J.M.; de Colle, C. Metopimazine Is Primarily Metabolized by a Liver Amidase in Humans. Pharmacol. Res. Perspect. 2022, 10, 00903. [Google Scholar] [CrossRef]
- Heppner, D.E.; Günther, M.; Wittlinger, F.; Laufer, S.A.; Eck, M.J. Structural Basis for EGFR Mutant Inhibition by Trisubstituted Imidazole Inhibitors. J. Med. Chem. 2020, 63, 4293–4305. [Google Scholar] [CrossRef]
- Günther, M.; Juchum, M.; Kelter, G.; Fiebig, H.; Laufer, S. Lung Cancer: EGFR Inhibitors with Low Nanomolar Activity against a Therapy-resistant L858R/T790M/C797S Mutant. Angew. Chem. Int. Ed. 2016, 55, 10890–10894. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Chung, C.Y.S.; Liu, P.; Craciun, L.; Nishikawa, Y.; Bruemmer, K.J.; Hamachi, I.; Saijo, K.; Miller, E.W.; Chang, C.J. Activity–Based Sensing with a Metal-Directed Acyl Imidazole Strategy Reveals Cell Type–Dependent Pools of Labile Brain Copper. J. Am. Chem. Soc. 2020, 142, 14993–15003. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.J.; New, E.J.; de Jonge, M.D.; McColl, G. Imaging Metals in Biology: Balancing Sensitivity, Selectivity and Spatial Resolution. Chem. Soc. Rev. 2015, 44, 5941–5958. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Fan, Q.; Wang, X.; Zhou, B. Huntington Disease Arises from a Combinatory Toxicity of Polyglutamine and Copper Binding. Proc. Nati. Acad. Sci. USA 2013, 110, 14995–15000. [Google Scholar] [CrossRef]
- Fox, J.H.; Kama, J.A.; Lieberman, G.; Chopra, R.; Dorsey, K.; Chopra, V.; Volitakis, I.; Cherny, R.A.; Bush, A.I.; Hersch, S. Mechanisms of Copper Ion Mediated Huntington’s Disease Progression. PLoS ONE 2007, 2, e334. [Google Scholar] [CrossRef] [PubMed]
- Desai, V.; Kaler, S.G. Role of Copper in Human Neurological Disorders. Am. J. Clin. Nutr. 2008, 88, 855S–858S. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of Multifunctional Molecules as Potential Therapeutic Candidates for Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis in the Last Decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef]
- Tong, L.; Pav, S.; White, D.M.; Rogers, S.; Crane, K.M.; Cywin, C.L.; Brown, M.L.; Pargellis, C.A. A Highly Specific Inhibitor of Human P38 MAP Kinase Binds in the ATP Pocket. Nat. Struct. Biol. 1997, 4, 311–316. [Google Scholar] [CrossRef]
- Wang, Z.; Canagarajah, B.J.; Boehm, J.C.; Kassisà, S.; Cobb, M.H.; Young, P.R.; Abdel–Meguid, S.; Adams, J.L.; Goldsmith, E.J. Structural Basis of Inhibitor Selectivity in MAP Kinases. Structure 1998, 6, 1117–1128. [Google Scholar] [CrossRef]
- Peifer, C.; Abadleh, M.; Bischof, J.; Hauser, D.; Schattel, V.; Hirner, H.; Knippschild, U.; Laufer, S. 3, 4–Diaryl-Isoxazoles and–Imidazoles as Potent Dual Inhibitors of P38α Mitogen Activated Protein Kinase and Casein Kinase 1δ. J. Med. Chem. 2009, 52, 7618–7630. [Google Scholar] [CrossRef]
- Takle, A.K.; Brown, M.J.B.; Davies, S.; Dean, D.K.; Francis, G.; Gaiba, A.; Hird, A.W.; King, F.D.; Lovell, P.J.; Naylor, A. The Identification of Potent and Selective Imidazole–Based Inhibitors of B–Raf Kinase. Bioorg. Med. Chem. Lett. 2006, 16, 378–381. [Google Scholar] [CrossRef]
- Niculescu-Duvaz, D.; Niculescu–Duvaz, I.; Suijkerbuijk, B.M.J.M.; Ménard, D.; Zambon, A.; Nourry, A.; Davies, L.; Manne, H.A.; Friedlos, F.; Ogilvie, L. Novel Tricyclic Pyrazole BRAF Inhibitors with Imidazole or Furan Central Scaffolds. Bioorg. Med. Chem. 2010, 18, 6934–6952. [Google Scholar] [CrossRef]
- Niculescu–Duvaz, D.; Niculescu-Duvaz, I.; Suijkerbuijk, B.M.J.M.; Ménard, D.; Zambon, A.; Davies, L.; Pons, J.-F.; Whittaker, S.; Marais, R.; Springer, C.J. Potent BRAF Kinase Inhibitors Based on 2, 4, 5–Trisubstituted Imidazole with Naphthyl and Benzothiophene 4–Substituents. Bioorg. Med. Chem. 2013, 21, 1284–1304. [Google Scholar] [CrossRef]
- Ali, E.M.H.; El-Telbany, R.F.A.; Abdel-Maksoud, M.S.; Ammar, U.M.; Mersal, K.I.; Zaraei, S.-O.; El-Gamal, M.I.; Choi, S.-I.; Lee, K.-T.; Kim, H.-K. Design, Synthesis, Biological Evaluation, and Docking Studies of Novel (Imidazol-5-Yl) Pyrimidine–Based Derivatives as Dual BRAFV600E/P38α Inhibitors. Eur. J. Med. Chem. 2021, 215, 113277. [Google Scholar] [CrossRef]
- Youssif, B.G.M.; Gouda, A.M.; Moustafa, A.H.; Abdelhamid, A.A.; Gomaa, H.A.M.; Kamal, I.; Marzouk, A.A. Design and Synthesis of New Triarylimidazole Derivatives as Dual Inhibitors of BRAFV600E/P38α with Potential Antiproliferative Activity. J. Mol. Struct. 2022, 1253, 132218. [Google Scholar] [CrossRef]
- Akdis, C.A.; Simons, F.E.R. Histamine Receptors Are Hot in Immunopharmacology. Eur J. Pharmacol. 2006, 533, 69–76. [Google Scholar] [CrossRef]
- Thangam, E.B.; Jemima, E.A.; Singh, H.; Baig, M.S.; Khan, M.; Mathias, C.B.; Church, M.K.; Saluja, R. The Role of Histamine and Histamine Receptors in Mast Cell–Mediated Allergy and Inflammation: The Hunt for New Therapeutic Targets. Front. Immunol. 2018, 9, 1873. [Google Scholar] [CrossRef]
- Hill, S.J.; Ganellin, C.R.; Timmerman, H.; Schwartz, J.C.; Shankley, N.P.; Young, J.M.; Schunack, W.; Levi, R.; Haas, H.L. International Union of Pharmacology. XIII. Classification of Histamine Receptors. Pharmacol. Rev. 1997, 49, 253–278. [Google Scholar]
- Durant, G.J.; Ganellin, C.R.; Parsons, M.E. Chemical Differentiation of Histamine H1–and H2-Receptor Agonists. J. Med. Chem. 1975, 18, 905–909. [Google Scholar] [CrossRef]
- Reggio, P.; Topiol, S.; Weinstein, H. Molecular Determinants for the Agonist Activity of 2-Methylhistamine and 4-Methylhistamine at H2-Receptors. J. Med. Chem. 1986, 29, 2412–2415. [Google Scholar] [CrossRef]
- Ganellin, R. 1980 Award in medicinal chemistry. Medicinal chemistry and dynamic structure-activity analysis in the discovery of drugs acting at histamine H2 receptors. J. Med. Chem. 1981, 24, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.M.; Barreiro, E.J. Bioisosterism: A Useful Strategy for Molecular Modification and Drug Design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.J.; Kümmerle, A.E.; Fraga, C.A.M. The Methylation Effect in Medicinal Chemistry. Chem. Rev. 2011, 111, 5215–5246. [Google Scholar] [CrossRef] [PubMed]
- Prout, K.; Critchley, S.R.; Ganellin, C.R.; Mitchell, R.C. Crystal and Molecular Structure of the Histamine H 2–Receptor Antagonists, N–Methyl-N′–{2-[(5-Methylimidazol–4–Yl) Methylthio] Ethyl} Thiourea (Metiamide) and N–{2-[(Imidazol–4–Yl) Methylthio] Ethyl}-N′–Methylthiourea (Thiaburimamide). J. Chem. Soc. Perkin. Trans. 2 1977, 68–75. [Google Scholar] [CrossRef]
- Wermuth, C.G. The Practice of Medicinal Chemistry; Academic Press: Cambridge, MA, USA, 2011; ISBN 0080568777. [Google Scholar]
- Patrick, G.L. An Introduction to Medicinal Chemistry; Oxford University Press: New York, NY, USA, 2013; ISBN 0199697396. [Google Scholar]
- Brimblecombe, R.W.; Duncan, W.A.M.; Durant, G.J.; Emmett, J.C.; Ganellin, C.R.; Leslie, G.B.; Parsons, M.E. Characterization and Development of Cimetidine as a Histamine H2-Receptor Antagonist. Gastroenterology 1978, 74, 339–347. [Google Scholar] [CrossRef]
- Figueiredo, J.M.; de A Câmara, C.; Amarante, E.G.; Miranda, A.L.P.; Santos, F.M.; Rodrigues, C.R.; Fraga, C.A.M.; Barreiro, E.J. Design and Synthesis of Novel Potent Antinociceptive Agents: Methyl-Imidazolyl N–Acylhydrazone Derivatives. Bioorg. Med. Chem. 2000, 8, 2243–2248. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and Use of Quantum Mechanical Molecular Models. 76. AM1: A New General Purpose Quantum Mechanical Molecular Model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]











































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).