
New 1,2,3-Triazole/1,2,4-triazole Hybrids as Aromatase Inhibitors: Design, Synthesis, and Apoptotic Antiproliferative Activity

 , and
, and
Abstract
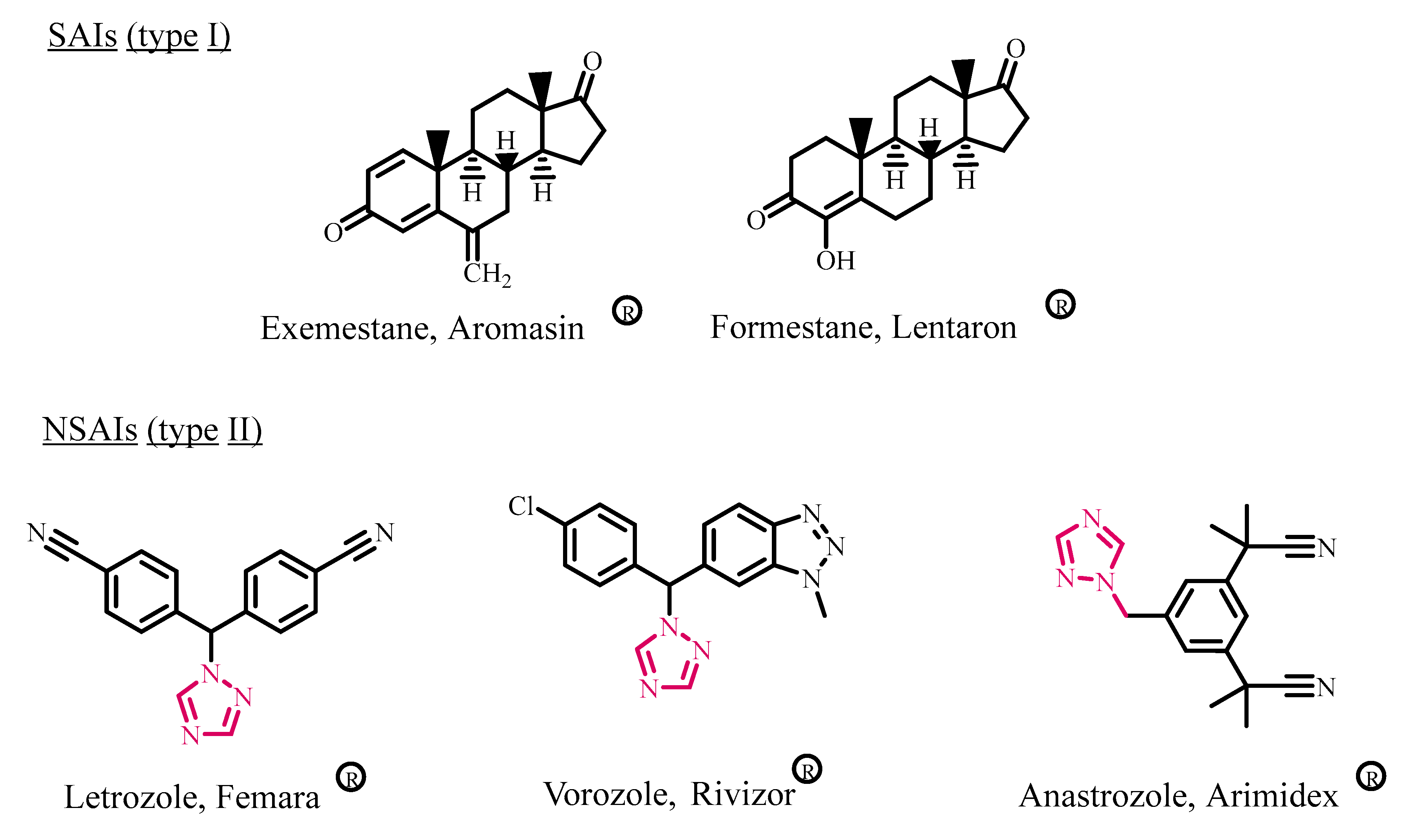
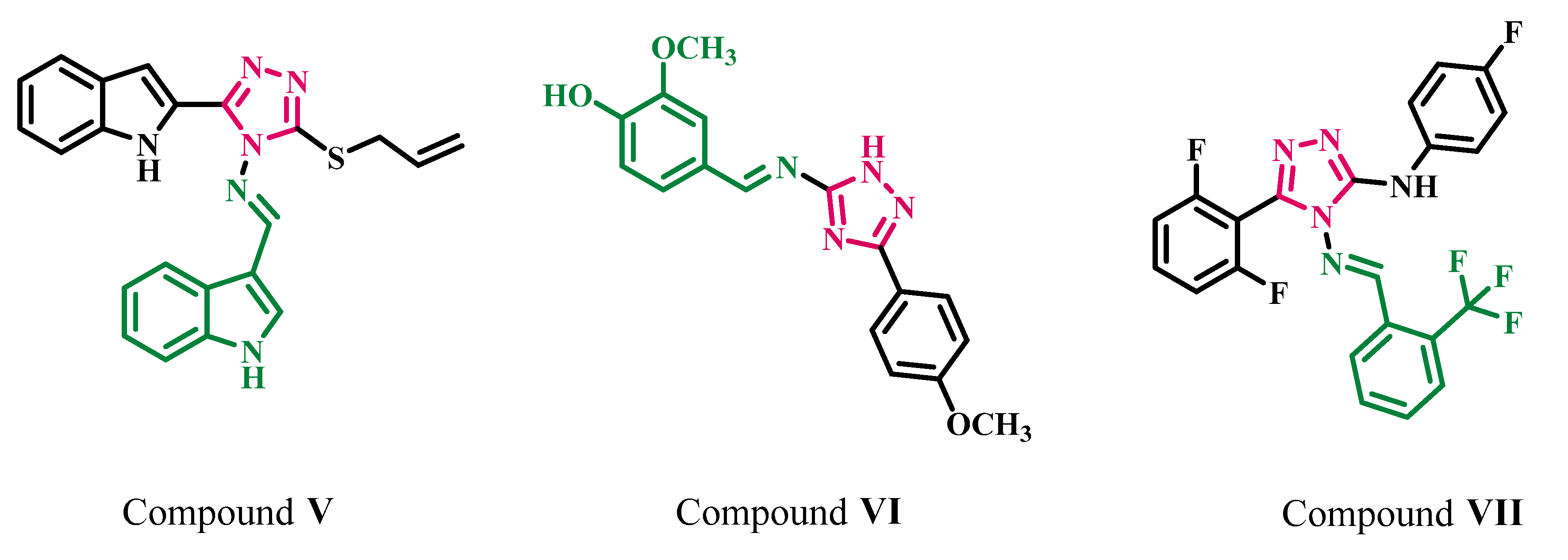
:1. Introduction
2. Results and Discussion
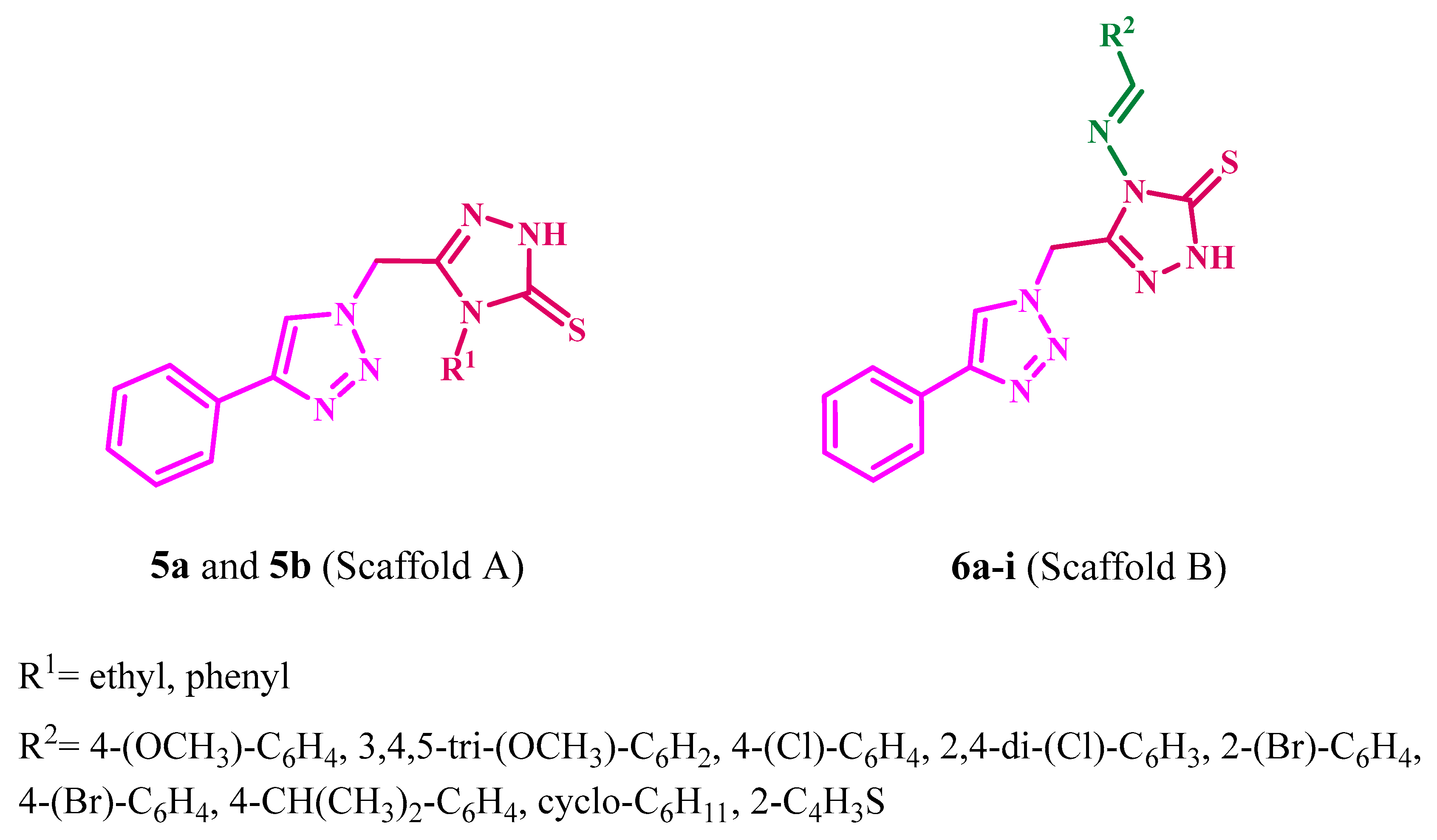
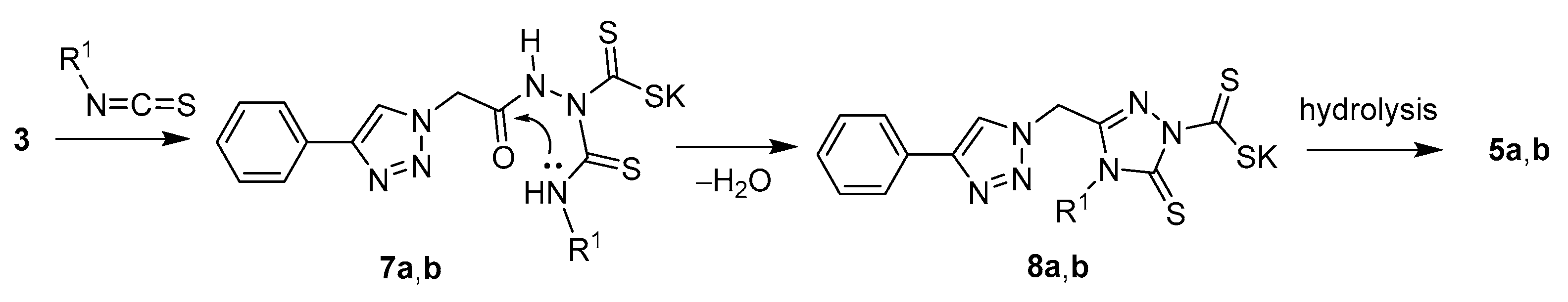
2.1. Chemistry
2.2. Biology
2.2.1. Cell Viability Assay
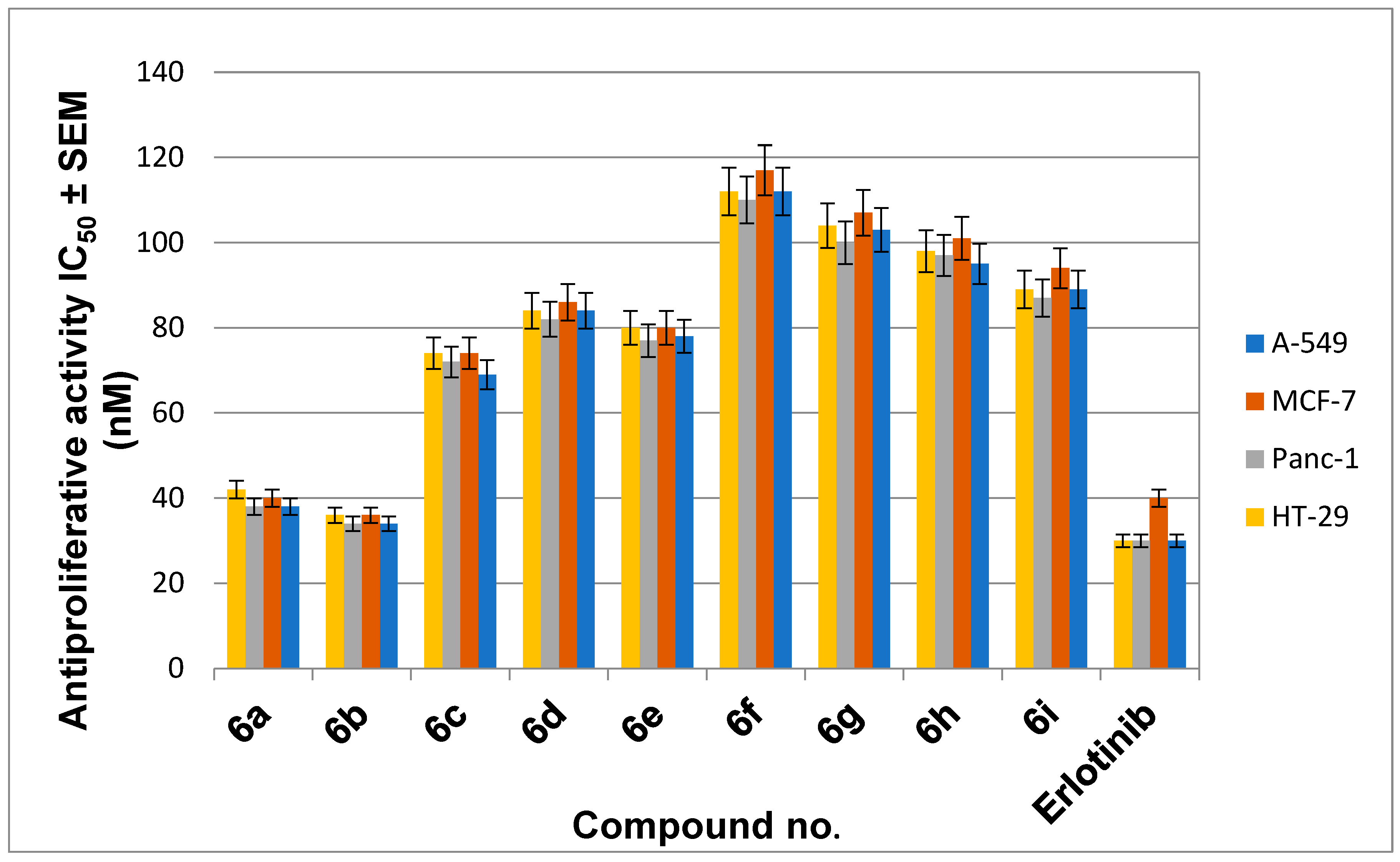
2.2.2. Antiproliferative Assay
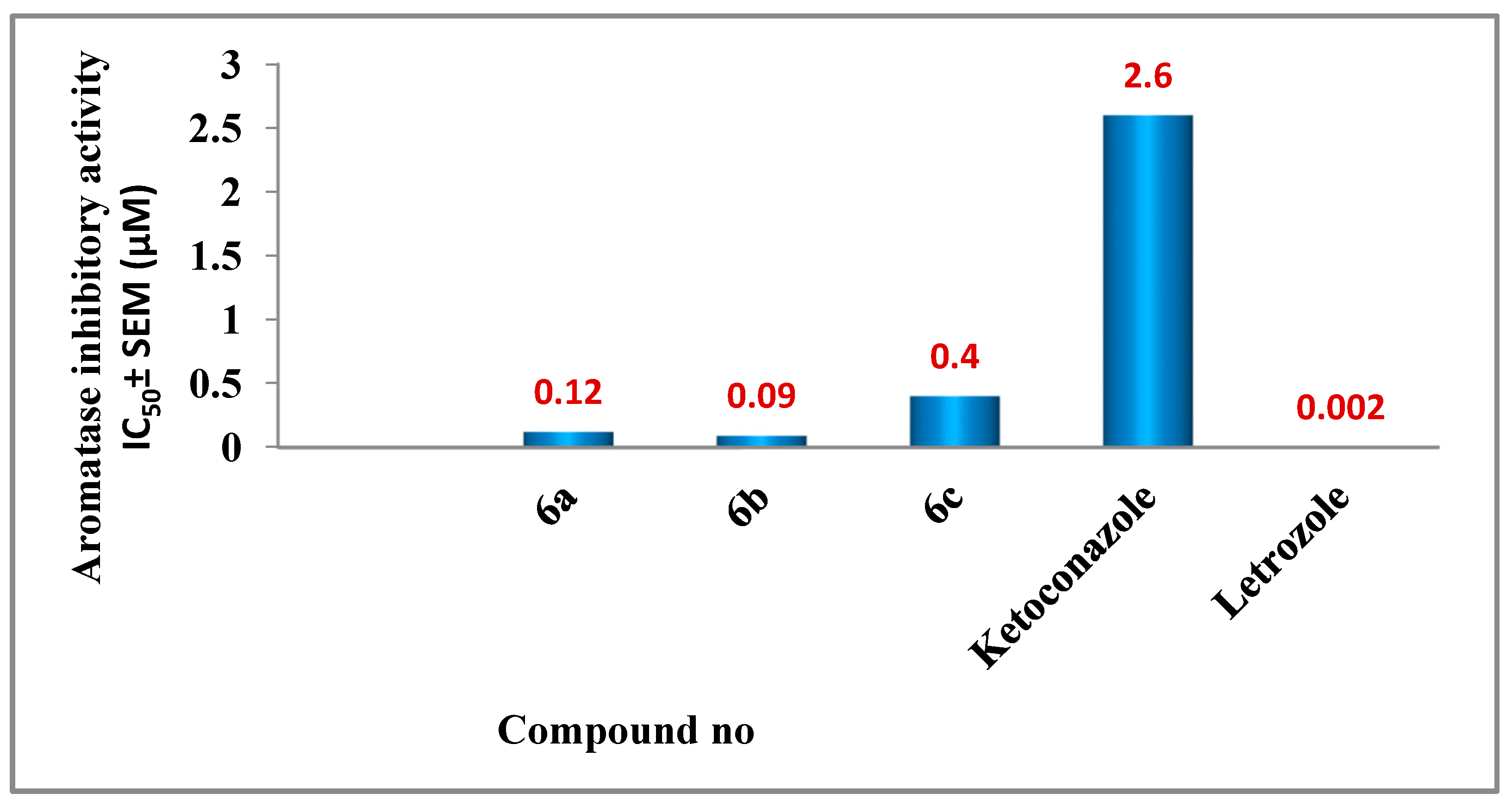
2.2.3. Aromatase Inhibitory Assay
2.2.4. Apoptosis Markers Assays
Caspase-3 Activation Assay
Caspase-8, Bax, Bcl-2 Levels Assays
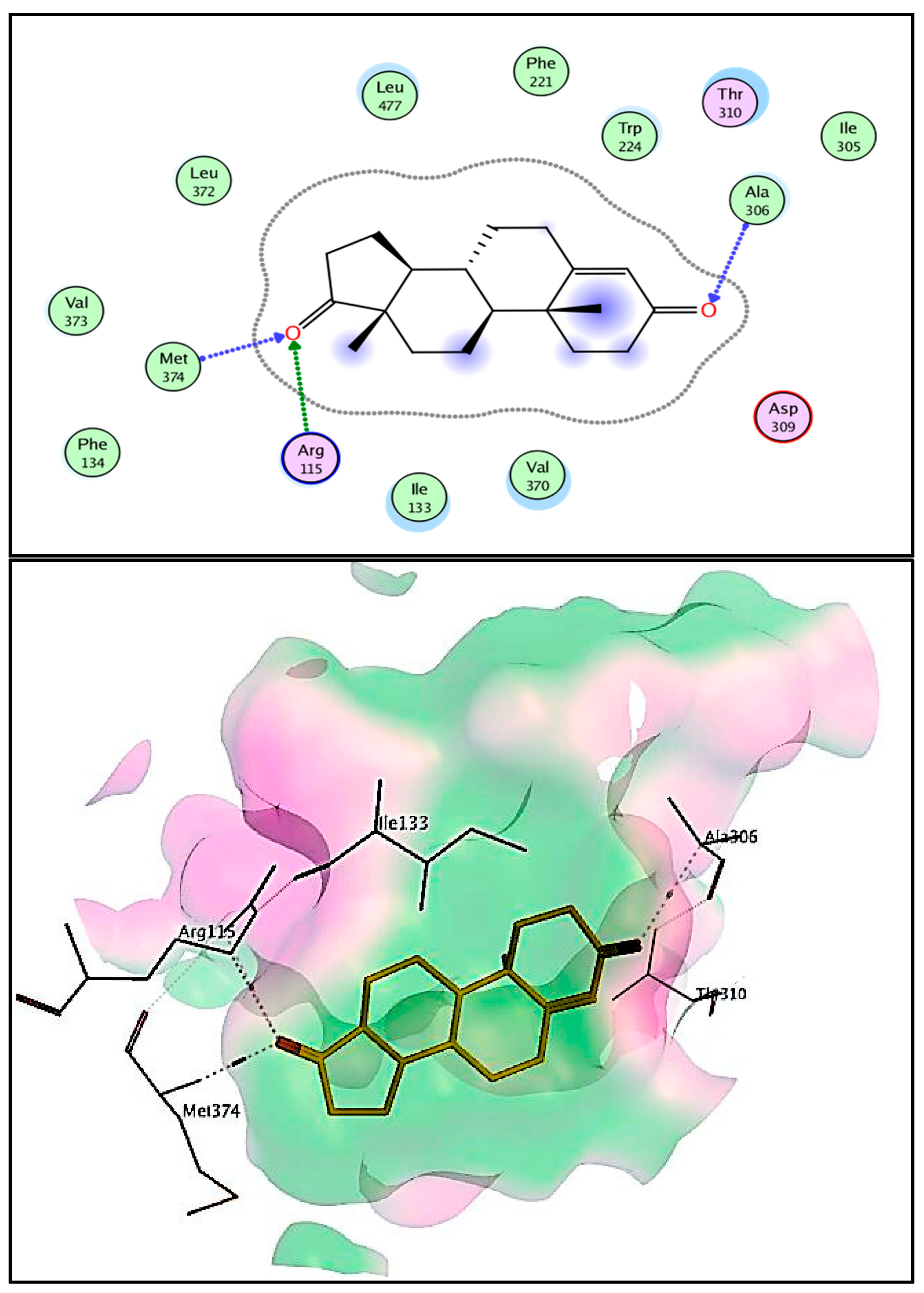
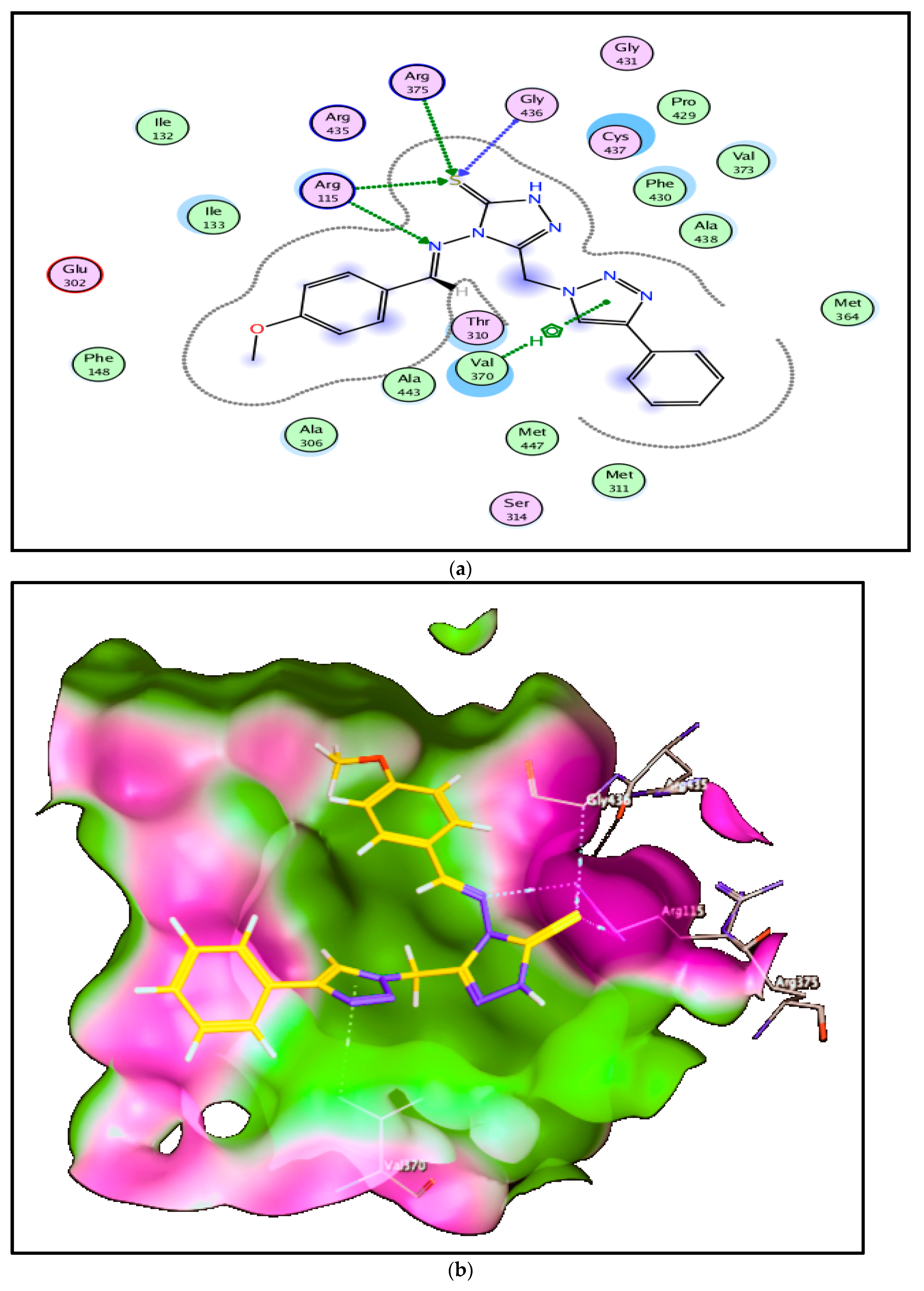
2.3. Docking and Molecular Modeling Studies
2.4. Structure–Activity Relationship (SAR) Analysis of 6a–i

3. Experimental
3.1. Chemistry
3.1.1. General Method for the Synthesis of Compounds 5a and 5b
4-Ethyl-5-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione (5a)
4-Phenyl-5-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)-2,4-dihydro-3H-1,2,4 -triazole-3-thione (5b)
3.1.2. General Method for the Synthesis of Compounds 6a–i
(E)-4-((4-Methoxybenzylidene)amino)-5-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione (6a)
(E)-5-((4-Phenyl-1H-1,2,3-triazol-1-yl)methyl)-4-((3,4,5-trimethoxybenz- ylidene)amino)-2,4-dihydro-3H-1,2,4-triazole-3-thione (6b)
(E)-4-((4-Chlorobenzylidene)amino)-5-((4-phenyl-1H-1,2,3-triazol-1-yl) methyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione (6c)
(E)-4-((2,4-Dichlorobenzylidene)amino)-5-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione (6d)
(E)-4-((2-Bromobenzylidene)amino)-5-((4-phenyl-1H-1,2,3-triazol-1-yl) methyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione (6e)
(E)-4-((4-Bromobenzylidene)amino)-5-((4-phenyl-1H-1,2,3-triazol-1-yl) methyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione (6f)
(E)-4-((4-Isopropylbenzylidene)amino)-5-((4-phenyl-1H-1,2,3-triazol-1-yl) methyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione (6g)
(E)-4-((Cyclohexylmethylene)amino)-5-((4-phenyl-1H-1,2,3-triazol-1-yl) methyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione (6h)
(E)-5-((4-Phenyl-1H-1,2,3-triazol-1-yl)methyl)-4-((thiophen-2-yl-methyl- ene)amino)-2,4-dihydro-3H-1,2,4-triazole-3-thione (6i)
3.2. Biology
3.2.1. Cell Viability Assay
3.2.2. Antiproliferative Assay
3.2.3. Aromatase Inhibitory Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ghosh, D.; Lo, J.; Egbuta, C. Recent progress in the discovery of next generation inhibitors of aromatase from the structure–function perspective. J. Med. Chem. 2016, 59, 5131–5148. [Google Scholar] [CrossRef]
- Ahmad, I. Recent developments in steroidal and nonsteroidal aromatase inhibitors for the chemoprevention of estrogen-dependent breast cancer. Eur. J. Med. Chem. 2015, 102, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, N.; Amin, S.A.; Saha, A.; Jha, T. Combating breast cancer with non-steroidal aromatase inhibitors (NSAIs): Understanding the chemico-biological interactions through comparative SAR/QSAR study. Eur. J. Med. Chem. 2017, 137, 365–438. [Google Scholar] [CrossRef]
- Mohammed Alwan, A.; Tavakol Afshari, J.; Afzaljavan, F. Significance of the Estrogen Hormone and Single Nucleotide Polymorphisms in the Progression of Breast Cancer among Female. Inst. Razi. Arch. 2022, 77, 943–958. [Google Scholar]
- Avvaru, S.P.; Noolvi, M.N.; Aminbhavi, T.M.; Chkraborty, S.; Dash, A.; Shukla, S.S. Aromatase inhibitors evolution as potential class of drugs in the treatment of postmenopausal breast cancer women. Mini Rev. Med. Chem. 2018, 18, 609–621. [Google Scholar] [CrossRef]
- Fabian, C.J. The what, why and how of aromatase inhibitors: Hormonal agents for treatment and prevention of breast cancer. Int. J. Clin. Pract. 2007, 61, 2051–2063. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Chen, S. Aromatase inhibitors: Structural features and biochemical characterization. Ann. N. Y. Acad. Sci. 2006, 1089, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, S.; Rampa, A.; Belluti, F.; Bisi, A. Nonsteroidal aromatase inhibitors for the treatment of breast cancer: An update. Anti-Cancer Agents Med. Chem. 2014, 14, 54–65. [Google Scholar] [CrossRef] [PubMed]
- To, S.Q.; Knower, K.C.; Cheung, V.; Simpson, E.R.; Clyne, C.D. Transcriptional control of local estrogen formation by aromatase in the breast. J. Steroid Biochem. Mol. Biol. 2015, 145, 179–186. [Google Scholar] [CrossRef]
- Karaer, Ö.; Oruç, S.; Koyuncu, F.M. Aromatase inhibitors: Possible future applications. Acta Obstet. Gynecol. Scand. 2004, 83, 699–706. [Google Scholar] [CrossRef]
- Sobral, A.F.; Amaral, C.; Correia-da-Silva, G.; Teixeira, N. Unravelling exemestane: From biology to clinical prospects. J. Steroid Biochem. Mol. Biol. 2016, 163, 1–11. [Google Scholar] [CrossRef]
- Amir, E.; Seruga, B.; Niraula, S.; Carlsson, L.; Ocaña, A. Toxicity of adjuvant endocrine therapy in postmenopausal breast cancer patients: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2011, 103, 1299–1309. [Google Scholar] [CrossRef]
- McAteer, R. Design of New Azole Molecules to Prevent Metal Leaching from Medical Implants. Master’s Thesis, Technological University Dublin, Ireland, Dublin, 2021. [Google Scholar]
- Rafaat, M.S. Evaluation Biological Activity of 1,2,4-Triazole Mannich Base. Master’s Thesis, Firat University, Elazığ, Turkey, 2022. [Google Scholar]
- Terruzzi, S. Multivariate Flexible Metal Organic Frameworks: The Role of Functionalized Linkers, Heterogeneity and Defects in Adsorption Processes. Ph.D. Thesis, Università degli Studi di Milano, Milano, Italy, 2022. [Google Scholar]
- Caporuscio, F.; Rastelli, G.; Imbriano, C.; Del Rio, A. Structure-based design of potent aromatase inhibitors by high-throughput docking. J. Med. Chem. 2011, 54, 4006–4017. [Google Scholar] [CrossRef] [PubMed]
- Caciolla, J.; Martini, S.; Spinello, A.; Belluti, F.; Bisi, A.; Zaffaroni, N.; Magistrato, A.; Gobbi, S. Single-digit nanomolar inhibitors lock the aromatase active site via a dualsteric targeting strategy. Eur. J. Med. Chem. 2022, 244, 114802. [Google Scholar] [CrossRef]
- Osmaniye, D.; Hıdır, A.; Sağlık, B.N.; Levent, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of New Pyrimidine-Triazole Derivatives and Investigation of Their Anticancer Activities. Chem. Biodivers. 2022, 19, e202200216. [Google Scholar] [CrossRef] [PubMed]
- Rani, S.; Raheja, K.; Luxami, V.; Paul, K. A review on diverse heterocyclic compounds as the privileged scaffolds in non-steroidal aromatase inhibitors. Bioorg. Chem. 2021, 113, 105017. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Xiao, X.; Huang, C.; Yuan, Y.; Tang, D.; Dai, X.; Zeng, X. Potent aromatase inhibitors and molecular mechanism of inhibitory action. Eur. J. Med. Chem. 2018, 143, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Liu, Y.; Dai, Z.; Liu, W.; Zhao, K.; Zhang, T.; Hu, Y.; Zhang, X.; Dai, Y. Synthesis and aromatase inhibitory evaluation of 4-N-nitrophenyl substituted amino-4H-1,2,4-triazole derivatives. Bioorg. Med. Chem. 2016, 24, 4723–4730. [Google Scholar] [CrossRef] [PubMed]
- Pingaew, R.; Prachayasittikul, V.; Mandi, P.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Synthesis and molecular docking of 1,2,3-triazole-based sulfonamides as aromatase inhibitors. Bioorg. Med. Chem. 2015, 23, 3472–3480. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, M.; Abd El-All, A.S.; El-Naem, S.I.; Abdalla, M.M.; Rashdan, H.R. New potent 5α-Reductase and aromatase inhibitors derived from 1,2,3-triazole derivative. Molecules 2020, 25, 672. [Google Scholar] [CrossRef]
- Radha, V.P.; Prabakaran, M. Novel thiadiazole-derived Schiff base ligand and its transition metal complexes: Thermal behaviour, theoretical study, chemo-sensor, antimicrobial, antidiabetic and anticancer activity. Appl. Organomet. Chem. 2022, 36, e6872. [Google Scholar] [CrossRef]
- El-Sherief, H.A.; Youssif, B.G.; Abdelazeem, A.H.; Abdel-Aziz, M.; Abdel-Rahman, H.M. Design, synthesis and antiproliferative evaluation of novel 1,2,4-triazole/schiff base hybrids with EGFR and B-RAF inhibitory activities. Anti-Cancer Agents Med. Chem. 2019, 19, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.K.; Vibhute, B.T. Synthesis, characterization, anticancer and DNA photocleavage study of novel quinoline Schiff base and its metal complexes. Arab. J. Chem. 2021, 14, 103285. [Google Scholar] [CrossRef]
- Nafie, M.S.; Boraei, A.T. Exploration of novel VEGFR2 tyrosine kinase inhibitors via design and synthesis of new alkylated indolyl-triazole Schiff bases for targeting breast cancer. Bioorg. Chem. 2022, 122, 105708. [Google Scholar] [CrossRef]
- Kaur, R.; Ranjan Dwivedi, A.; Kumar, B.; Kumar, V. Recent developments on 1,2,4-triazole nucleus in anticancer compounds: A review. Anti-Cancer Agents Med. Chem. 2016, 16, 465–489. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef]
- Kumar, R.; Vats, L.; Bua, S.; Supuran, C.T.; Sharma, P.K. Design and synthesis of novel benzenesulfonamide containing 1,2,3-triazoles as potent human carbonic anhydrase isoforms I, II, IV and IX inhibitors. Eur. J. Med. Chem. 2018, 155, 545–551. [Google Scholar] [CrossRef]
- Ji, D.; Lu, J.; Lu, B.; Xin, C.; Mu, J.; Li, J.; Peng, C.; Bao, X. Efficient synthesis and antimicrobial activity of some novel S-β-d-glucosides of 5-aryl-1,2,4-triazole-3-thiones derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 1997–2000. [Google Scholar] [CrossRef]
- Gomaa, H.A.; Shaker, M.E.; Alzarea, S.I.; Hendawy, O.; Mohamed, F.A.; Gouda, A.M.; Ali, A.T.; Morcoss, M.M.; Abdelrahman, M.H.; Trembleau, L. Optimization and SAR investigation of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as EGFR and BRAFV600E dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg. Chem. 2022, 120, 105616. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, H.A.; El-Sherief, H.A.; Hussein, S.; Gouda, A.M.; Salem, O.I.; Alharbi, K.S.; Hayallah, A.M.; Youssif, B.G. Novel 1,2,4-triazole derivatives as apoptotic inducers targeting p53: Synthesis and antiproliferative activity. Bioorg. Chem. 2020, 105, 104369. [Google Scholar] [CrossRef]
- Marzouk, A.A.; Abdel-Aziz, S.A.; Abdelrahman, K.S.; Wanas, A.S.; Gouda, A.M.; Youssif, B.G.; Abdel-Aziz, M. Design and synthesis of new 1,6-dihydropyrimidin-2-thio derivatives targeting VEGFR-2: Molecular docking and antiproliferative evaluation. Bioorg. Chem. 2020, 102, 104090. [Google Scholar] [CrossRef] [PubMed]
- Youssif, B.G.; Gouda, A.M.; Moustafa, A.H.; Abdelhamid, A.A.; Gomaa, H.A.; Kamal, I.; Marzouk, A.A. Design and synthesis of new triarylimidazole derivatives as dual inhibitors of BRAFV600E/p38α with potential antiproliferative activity. J. Mol. Struct. 2022, 1253, 132218. [Google Scholar] [CrossRef]
- Chamduang, C.; Pingaew, R.; Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Novel triazole-tetrahydroisoquinoline hybrids as human aromatase inhibitors. Bioorg. Chem. 2019, 93, 103327. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Villa-Pulgarin, J.A.; Gajate, C.; Botet, J.; Jimenez, A.; Justies, N.; Varela-M, R.E.; Cuesta-Marbán, Á.; Müller, I.; Modolell, M.; Revuelta, J.L. Mitochondria and lipid raft-located FOF1-ATP synthase as major therapeutic targets in the antileishmanial and anticancer activities of ether lipid edelfosine. PLoS Neglect. Trop. Dis. 2017, 11, e0005805. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Zhang, Q.; Zhu, Z.; Xu, H.; Ding, F.; Wang, M.; Du, S.; Du, Y.; Yan, Z. BHX, a novel pyrazoline derivative, inhibits breast cancer cell invasion by reversing the epithelial-mesenchymal transition and down-regulating Wnt/β-catenin signalling. Sci. Rep. 2017, 7, 9153. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Al-Harbi, S.; Almasan, A. Caspase-3 activation is a critical determinant of genotoxic stress-induced apoptosis. Apoptosis Cancer Methods Protoc. 2015, 1219, 1–9. [Google Scholar]
- Boland, K.; Flanagan, L.; Prehn, J.H. Paracrine control of tissue regeneration and cell proliferation by Caspase-3. Cell Death Dis. 2013, 4, e725. [Google Scholar] [CrossRef]
- Youssif, B.G.; Mohamed, A.M.; Osman, E.E.A.; Abou-Ghadir, O.F.; Elnaggar, D.H.; Abdelrahman, M.H.; Treamblu, L.; Gomaa, H.A. 5-Chlorobenzofuran-2-carboxamides: From allosteric CB1 modulators to potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019, 177, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.P.; Roy, K. Docking and 3D-QSAR studies of diverse classes of human aromatase (CYP19) inhibitors. J. Mol. Model. 2010, 16, 1597–1616. [Google Scholar] [CrossRef] [PubMed]
- Shaykoon, M.S.; Marzouk, A.A.; Soltan, O.M.; Wanas, A.S.; Radwan, M.M.; Gouda, A.M.; Youssif, B.G.; Abdel-Aziz, M. Design, synthesis and antitrypanosomal activity of heteroaryl-based 1,2,4-triazole and 1,3,4-oxadiazole derivatives. Bioorg. Chem. 2020, 100, 103933. [Google Scholar] [CrossRef] [PubMed]

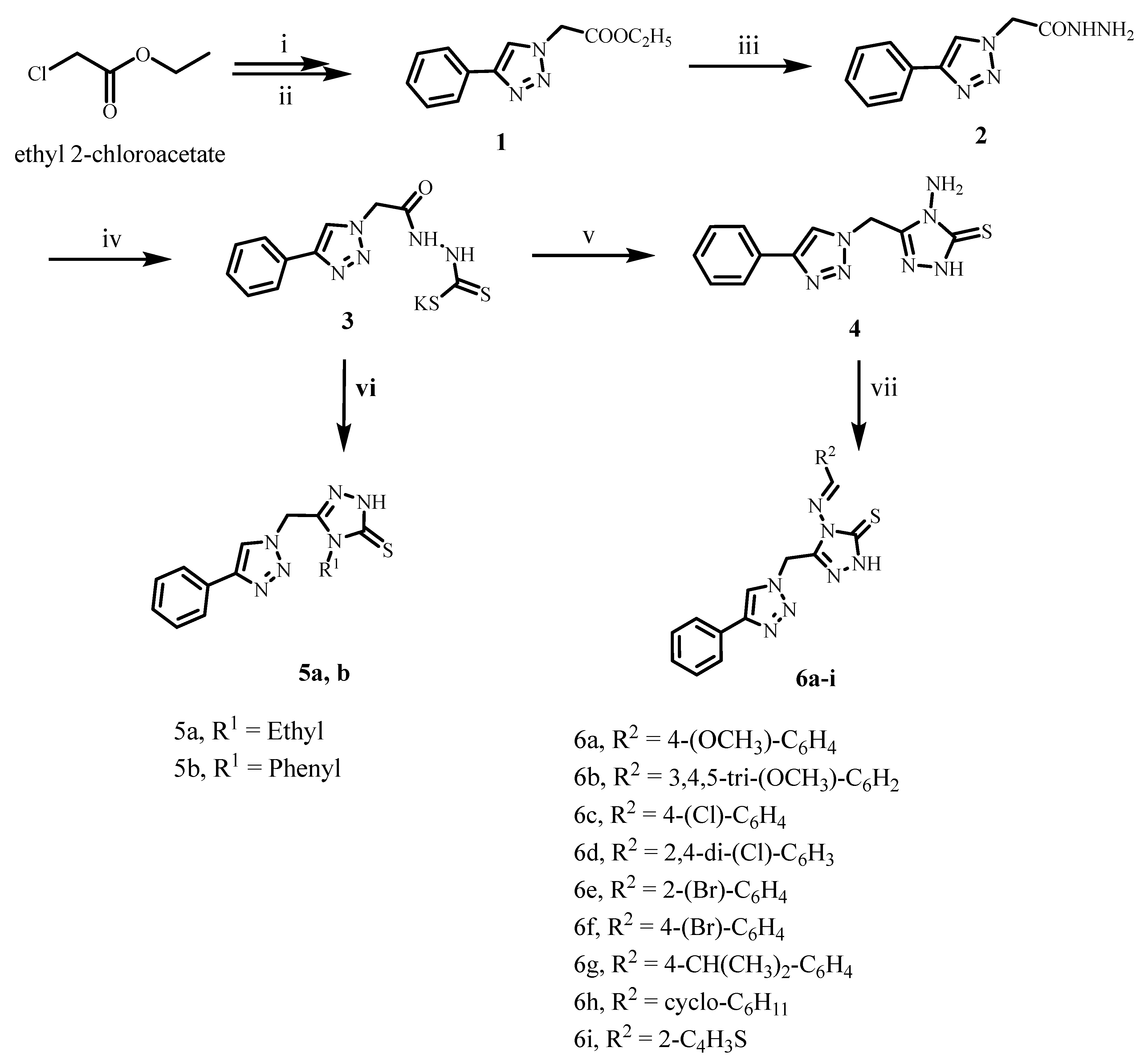


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Cell Viability % | Antiproliferative Activity IC50 ± SEM (nM) | ||||
|---|---|---|---|---|---|---|
| A-549 | MCF-7 | Panc-1 | HT-29 | Average IC50(GI50) | ||
| 5a | 90 | >120 | >120 | >120 | >120 | >120 |
| 5b | 92 | >120 | >120 | >120 | >120 | >120 |
| 6a | 92 | 38 ± 3 | 40 ± 4 | 38 ± 3 | 42 ± 4 | 40 |
| 6b | 91 | 34 ± 3 | 36 ± 3 | 34 ± 3 | 36 ± 3 | 35 |
| 6c | 87 | 69 ± 6 | 74 ± 7 | 72 ± 6 | 74 ± 6 | 72 |
| 6d | 89 | 84 ± 8 | 86 ± 8 | 82 ± 8 | 84 ± 8 | 84 |
| 6e | 90 | 78 ± 7 | 80 ± 8 | 77 ± 7 | 80 ± 7 | 79 |
| 6f | 92 | 112 ± 10 | 117 ± 10 | 110 ± 10 | 112 ± 10 | 113 |
| 6g | 91 | 103 ± 10 | 107 ± 10 | 100 ± 10 | 104 ± 10 | 104 |
| 6h | 87 | 95 ± 9 | 101 ± 9 | 97 ± 9 | 98 ± 9 | 98 |
| 6i | 92 | 89 ± 8 | 94 ± 9 | 87 ± 8 | 89 ± 8 | 90 |
| Erlotinib | ND | 30 ± 3 | 40 ± 3 | 30 ± 3 | 30 ± 3 | 33 |
| Comp. | IC50 ± SEM (µM) |
|---|---|
| 5a | >12.5 |
| 5b | >12.5 |
| 6a | 0.12 ± 0.01 |
| 6b | 0.09 ± 0.01 |
| 6c | 0.40 ± 0.04 |
| 6d | >12.5 |
| 6e | >12.5 |
| 6f | >12.5 |
| 6g | >12.5 |
| Ketoconazole | 2.6 ± 0.20 |
| Letrozole | 0.002 ± 0.0002 |
| Compd. No. | Caspase-3 | Caspase-8 | Bax | Bcl-2 | ||||
|---|---|---|---|---|---|---|---|---|
| Conc (Pg/mL) | Fold Change | Conc (ng/mL) | Fold Change | Conc (Pg/mL) | Fold Change | Conc (ng/mL) | Fold Reduction | |
| 6a | 495 ± 4 | 7.5 | 2.05 | 23 | 295 | 33 | 0.80 | 6 |
| 6b | 525 ± 5 | 8.0 | 2.30 | 25 | 320 | 35 | 0.70 | 7 |
| 6c | 325 ± 3 | 5.0 | ND | ND | ND | ND | ND | ND |
| Staurosporine | 465 ± 4 | 7.0 | 1.85 | 21 | 288 | 32 | 1.00 | 5 |
| Control | 65 | 1.0 | 0.09 | 1 | 9 | 1 | 5.00 | 1 |
| Compd. No. | The Score (kcal/mole) | Types of Interactions | ||
|---|---|---|---|---|
| Hydrogen Bond (Distance Å) | π–π Interactions (Arene-H or Cation) (Distance Å) | Hydrophobic Interactions | ||
| Androstenedione | −8.32 | C=O …Ala306 (3.45 Å) C=O… Arg115 (3.44 Å) C=O… Met374 (2.84 Å) C=O… Ser314 (2.54 Å) C=O… Met311 (3.45 Å) | ---- | Thr310, Leu477, Ile133, Val370, Trp224, Cys437, Arg435, Arg145, Phe430 |
| 6a | −9.46 | S…Arg115, Arg375 & Gly436 (3.79, 4.49 & 3.93 Å) N….Arg115 (3.30 Å) | Val370—Triazole ring (3.74 Å) | Ile133, Cys437, Phe430, Ala306, Ala438, Val373 |
| 6b | −9.94 | S…Arg115, Arg435 & Ile133 (3.19, 4.40 & 3.86 Å) N….Arg115 (3.01 Å) N of triazole ring….Thr310 (3.49 Å) | ---- | Ala306, Leu477, Cys437, Trp224, Val370, Leu152 |
| 6c | −9.18 | S…Thr310 (4.14 Å) N of triazole ring….Arg115 (3.26 Å) | Cys437—Triazole ring (3.81 Å) | Ile133, Phe430, Met311, Ala306, Val373, Ala438, Val370 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maghraby, M.T.-E.; Mazyad Almutairi, T.; Bräse, S.; Salem, O.I.A.; Youssif, B.G.M.; Sheha, M.M. New 1,2,3-Triazole/1,2,4-triazole Hybrids as Aromatase Inhibitors: Design, Synthesis, and Apoptotic Antiproliferative Activity. Molecules 2023, 28, 7092. https://doi.org/10.3390/molecules28207092
Maghraby MT-E, Mazyad Almutairi T, Bräse S, Salem OIA, Youssif BGM, Sheha MM. New 1,2,3-Triazole/1,2,4-triazole Hybrids as Aromatase Inhibitors: Design, Synthesis, and Apoptotic Antiproliferative Activity. Molecules. 2023; 28(20):7092. https://doi.org/10.3390/molecules28207092
Chicago/Turabian StyleMaghraby, Mohamed T-E, Tahani Mazyad Almutairi, Stefan Bräse, Ola I. A. Salem, Bahaa G. M. Youssif, and Mahmoud M. Sheha. 2023. "New 1,2,3-Triazole/1,2,4-triazole Hybrids as Aromatase Inhibitors: Design, Synthesis, and Apoptotic Antiproliferative Activity" Molecules 28, no. 20: 7092. https://doi.org/10.3390/molecules28207092
APA StyleMaghraby, M. T.-E., Mazyad Almutairi, T., Bräse, S., Salem, O. I. A., Youssif, B. G. M., & Sheha, M. M. (2023). New 1,2,3-Triazole/1,2,4-triazole Hybrids as Aromatase Inhibitors: Design, Synthesis, and Apoptotic Antiproliferative Activity. Molecules, 28(20), 7092. https://doi.org/10.3390/molecules28207092