
Dipeptides Containing Pyrene and Modified Photochemically Reactive Tyrosine: Noncovalent and Covalent Binding to Polynucleotides

 , ,
, ,  and
and
Abstract
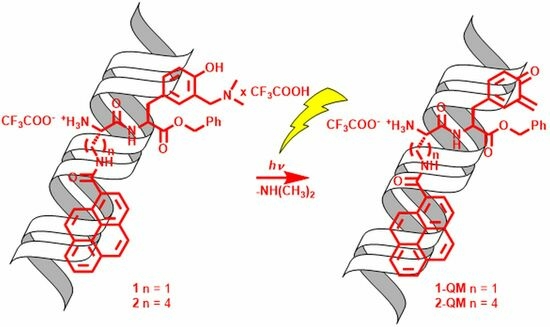
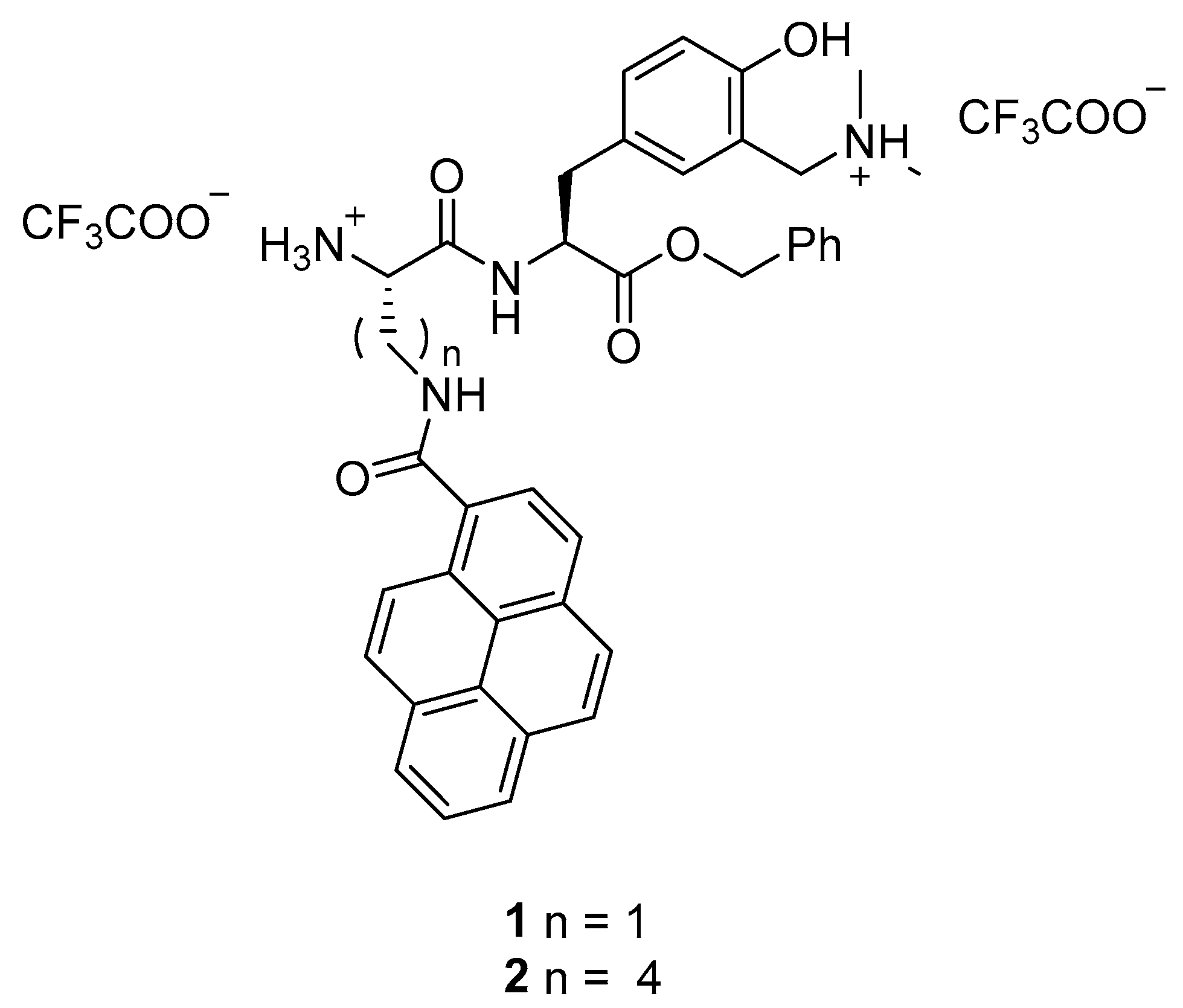
:
1. Introduction
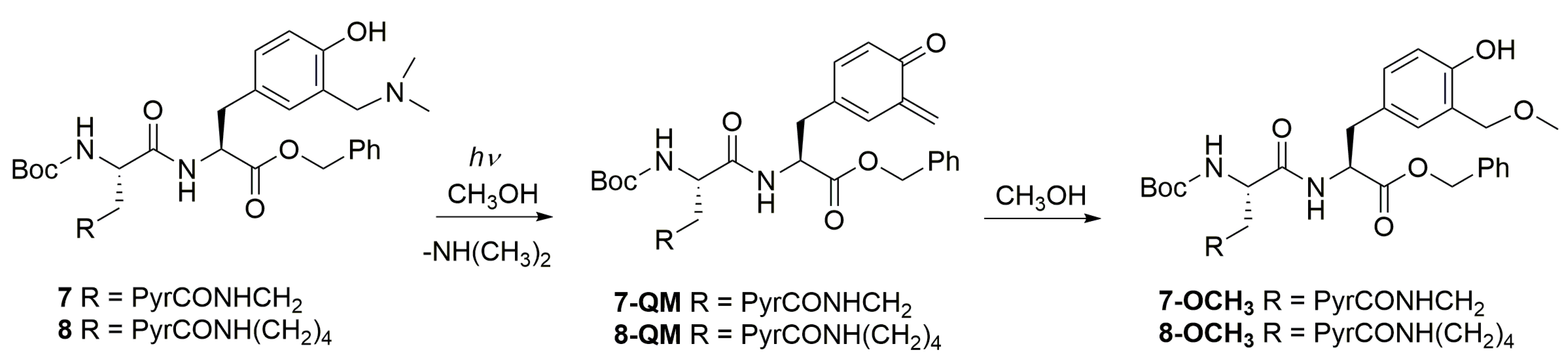
2. Results and Discussion
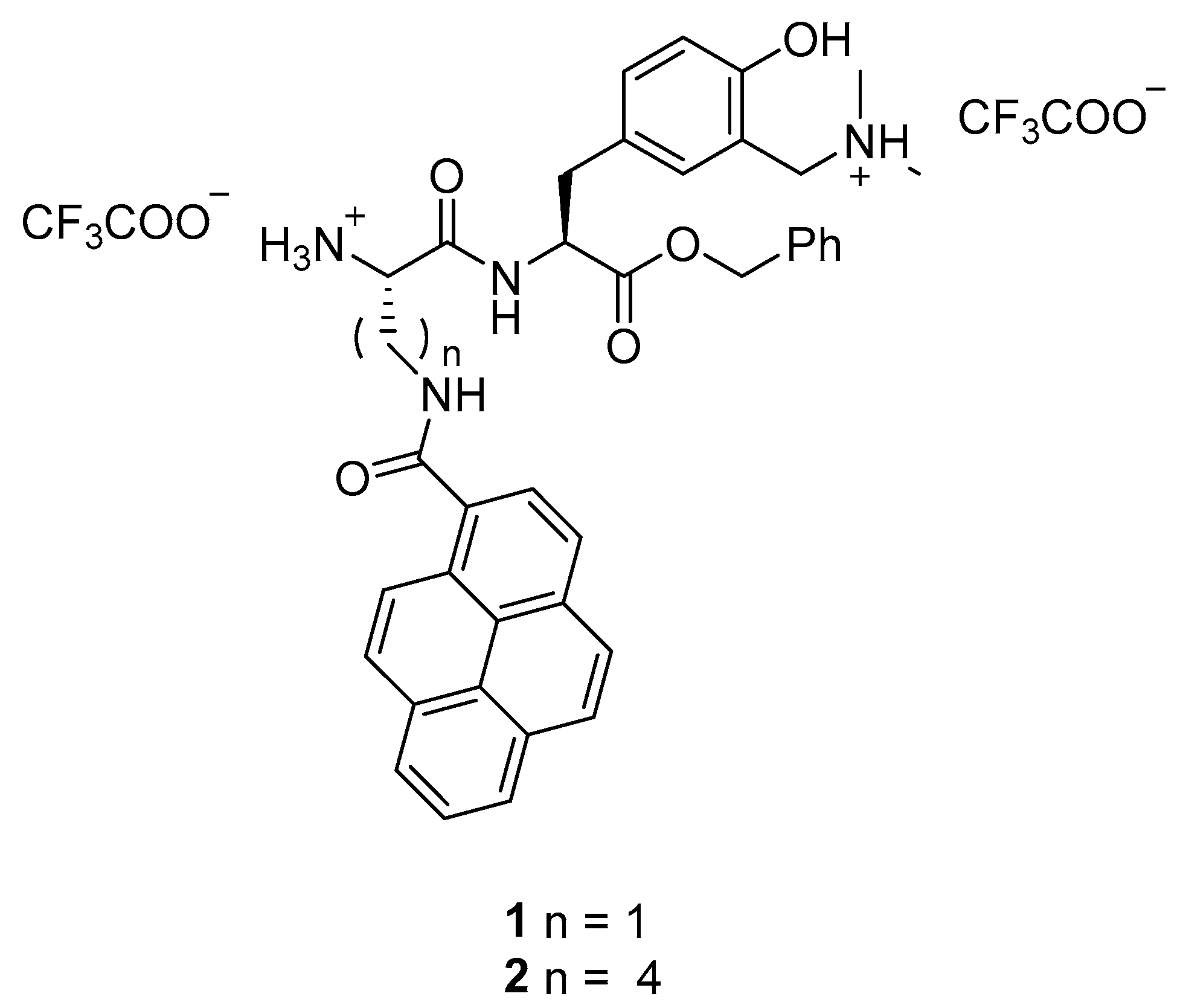
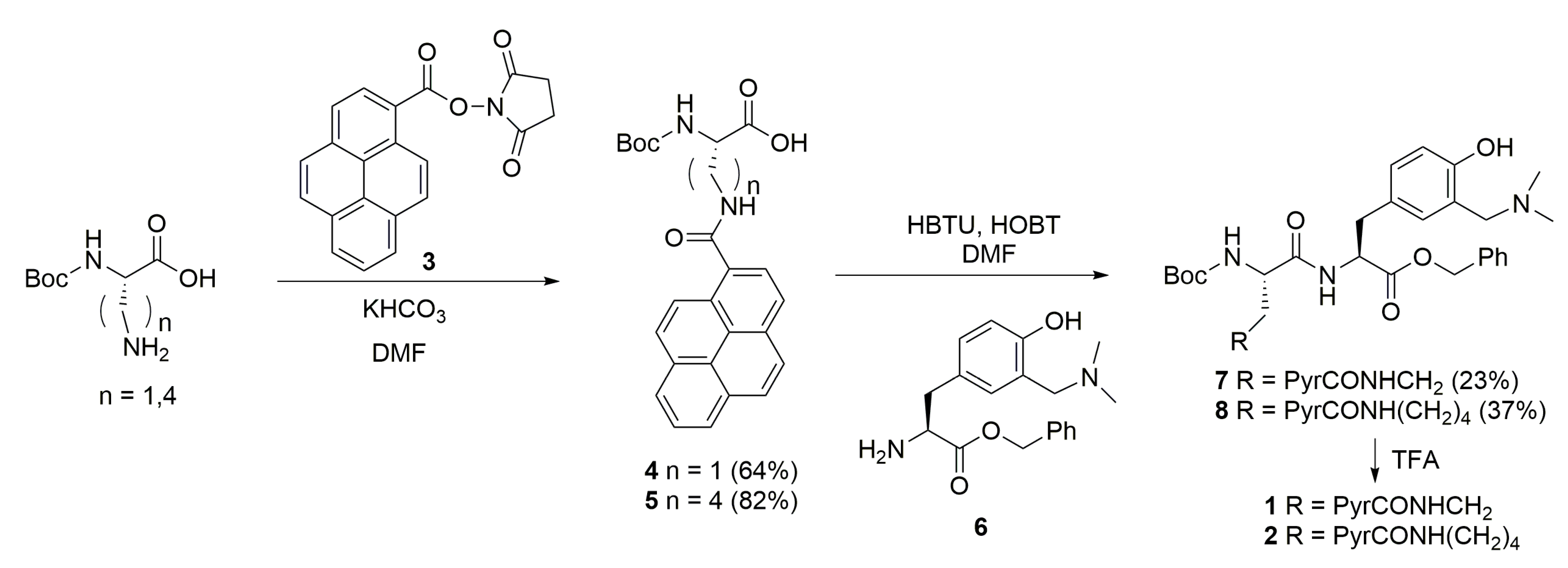
2.1. Synthesis
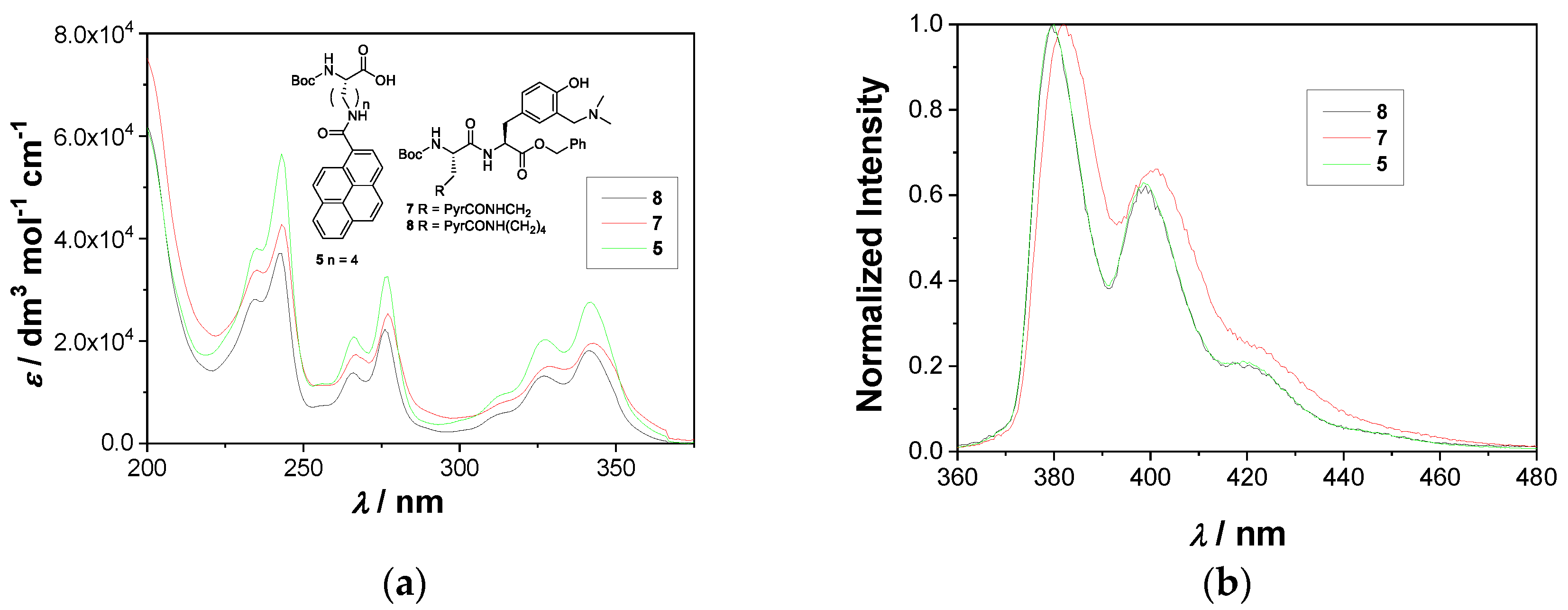
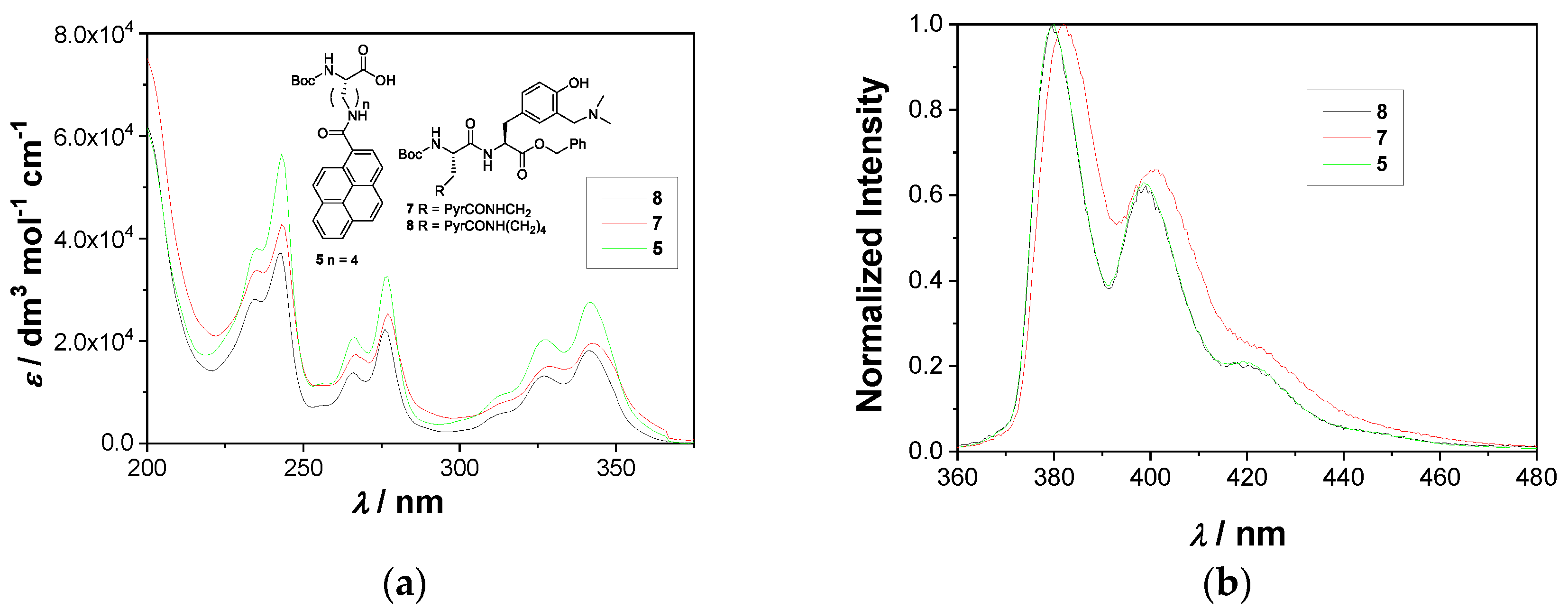
2.2. Photophysical Properties
2.3. Photochemical Reactivity
2.4. Noncovalent Binding to Polynucleotides
2.4.1. Thermal Denaturation Experiments
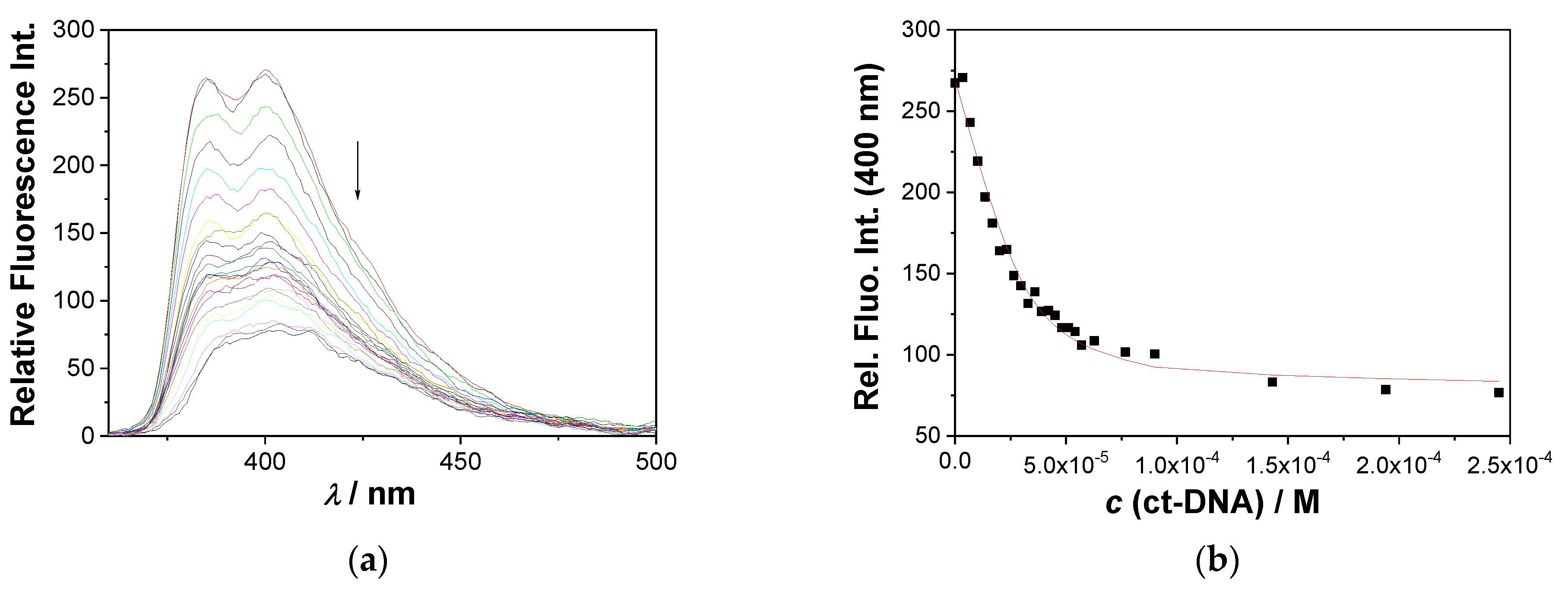
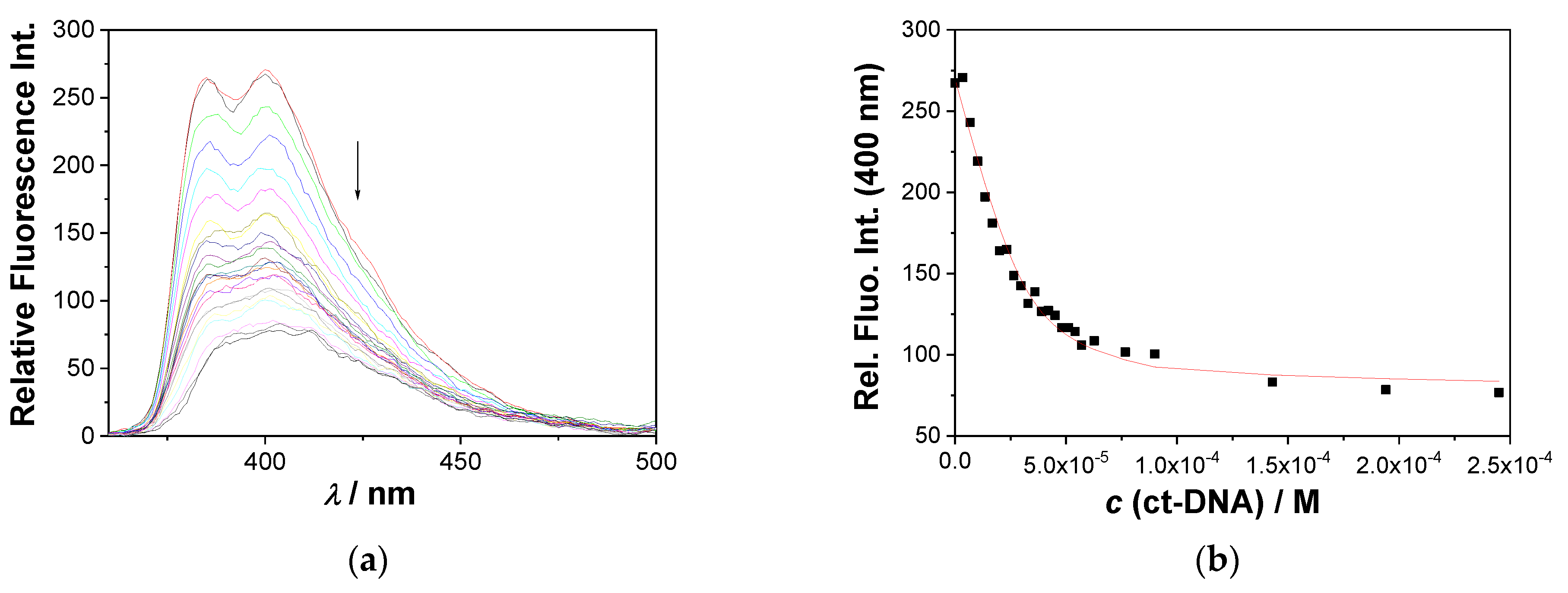
2.4.2. Fluorescence Titrations
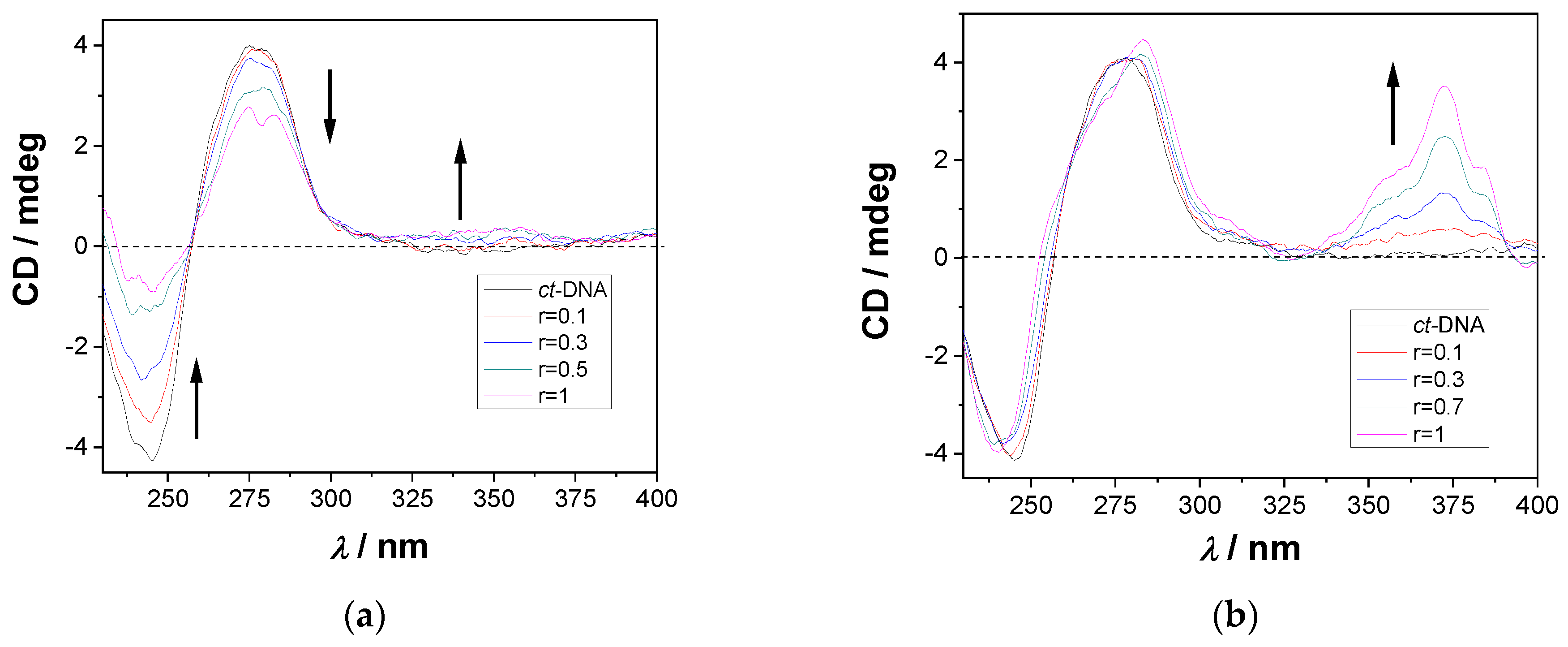
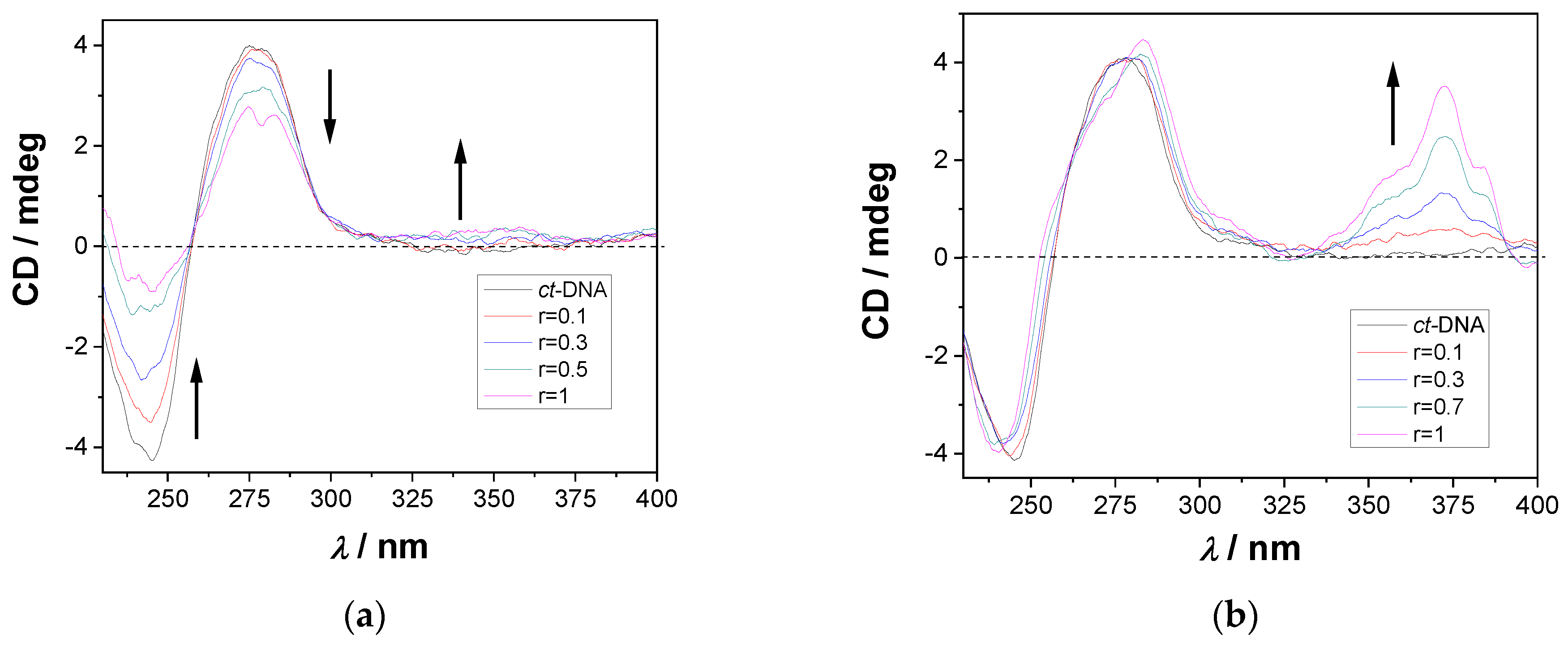
2.4.3. CD Titrations
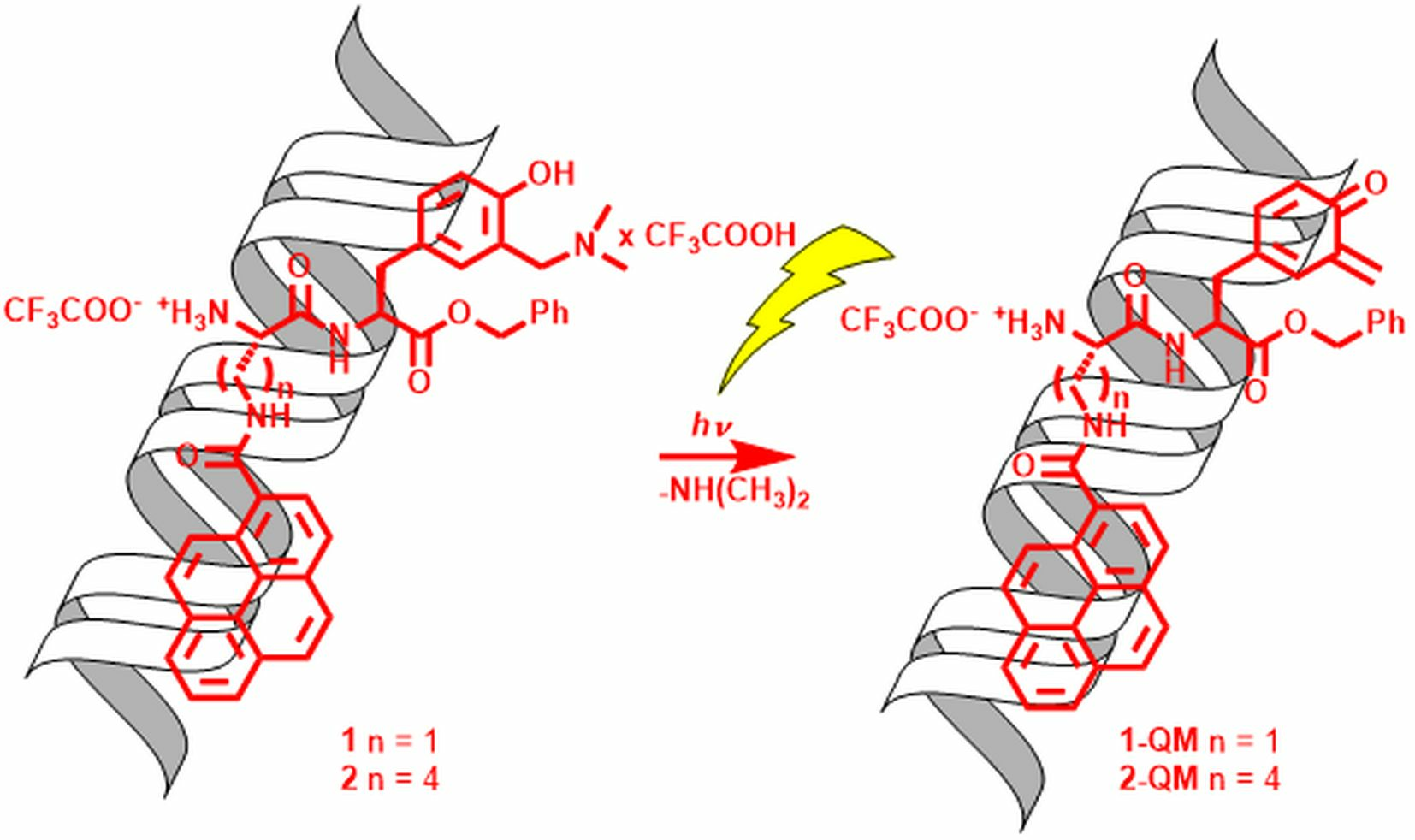
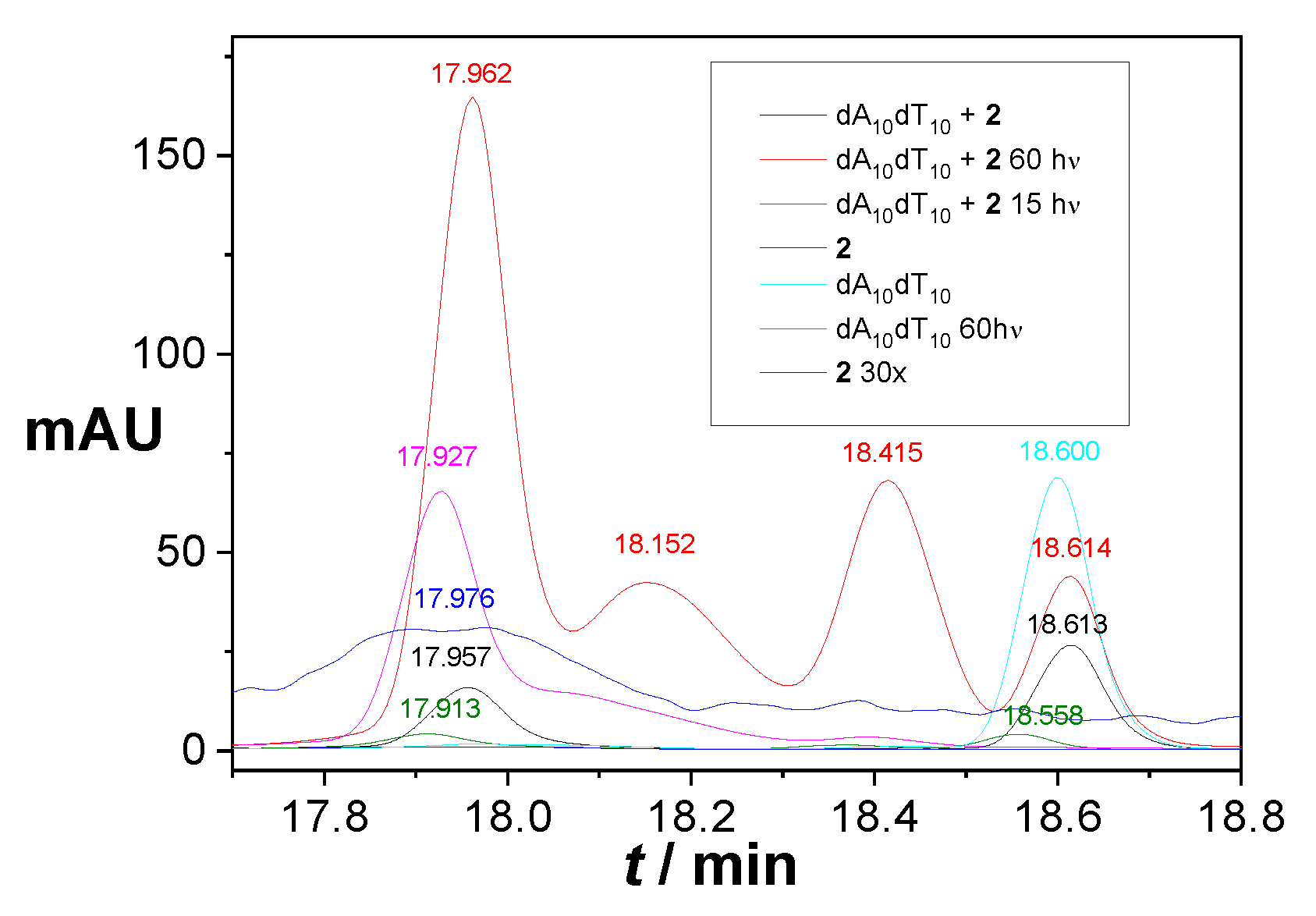
2.5. Covalent Binding to Polynucleotides
3. Materials and Methods
3.1. General Procedure for the Preparation of Pyrene Amino Acids 4 and 5
3.1.1. (S)-2-((Tert-butoxycarbonyl)amino)-3-(pyrene-1-carboxamido)propanoic Acid (4)
3.1.2. (S)-2-((Tert-butoxycarbonyl)amino)-6-(pyrene-1-carboxamido)hexanoic Acid (5)
3.2. General Procedure for the Preparation of Dipeptides 7 and 8
3.2.1. (S)-Benzyl 2-((S)-2-((tert-butoxycarbonyl)amino)-3-(pyrene-1-carboxamido)propanamido)-3-(3-((dimethylamino)methyl)-4-hydroxyphenyl)propanoate (7)
3.2.2. (S)-Benzyl 2-((S)-2-((tert-butoxycarbonyl)amino)-6-(pyrene-1-carboxamido)hexanamido)-3-(3-((dimethylamino)methyl)-4-hydroxyphenyl)propanoate (8)
3.3. General Procedure for the Removal of the BOC Protective Group from 7 and 8
3.4. Preparative Irradiations in CH3OH
3.4.1. (S)-Benzyl 2-((S)-2-((tert-butoxycarbonyl)amino)-3-(pyrene-1-carboxamido)propanamido)-3-(4-hydroxy-3-(methoxymethyl)phenyl)propanoate (7-OCH3)
3.4.2. (S)-Benzyl 2-((S)-2-((tert-butoxycarbonyl)amino)-6-(pyrene-1-carboxamido)hexanamido)-3-(4-hydroxy-3-(methoxymethyl)phenyl)propanoate (8-OCH3)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kastin, A.J. (Ed.) Handbook of Biologically Active Peptides; Elsevier: San Diego, CA, USA, 2013. [Google Scholar]
- Hamley, I.W. Small Bioactive Peptides for Biomaterials Design and Therapeutics. Chem. Rev. 2017, 117, 14015–14041. [Google Scholar] [CrossRef] [PubMed]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. 2021, 20, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Kaminker, R.; Anastasaki, A.; Gutekunst, W.R.; Luo, Y.; Lee, S.-H.; Hawker, C.J. Tuning of protease resistance in oligopeptides through N-alkylation. Chem. Commun. 2018, 54, 9631–9634. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.E.; Lee, K.H. Synthesis of Novel Unnatural Amino Acid as a Building Block and its Incorporation into an Antimicrobial Peptide. Bioorg. Med. Chem. 1999, 7, 2985–2990. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zambaldo, C.; Liu, T.; Zhang, Y.; Xuan, W.; Wang, C.; Reed, S.A.; Yang, P.-Y.; Wang, R.E.; Javahishvili, T.; et al. Recombinant thiopeptides containing noncanonical amino acids. Proc. Natl. Acad. Sci. USA 2016, 113, 3615–3620. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T. Unusual Amino Acids in Medicinal Chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef]
- Deber, C.M.; Madison, V.; Blout, E.R. Why cyclic peptides? Complementary approaches to conformations. Acc. Chem. Res. 1976, 9, 106–113. [Google Scholar]
- Kessler, H. Conformation and Biological Activity of Cyclic Peptides. Angew. Chem. Int. Ed. 1982, 21, 512–523. [Google Scholar] [CrossRef]
- Joo, S.H. Cyclic Peptides as Therapeutic Agents and Biochemical Tools. Biomol. Ther. 2012, 20, 19–26. [Google Scholar] [CrossRef]
- Vrettos, E.I.; Mező, G.; Tzakos, A.G. On the design principles of peptide-drug conjugates for targeted drug delivery to the malignant tumor site. Beilstein J. Org. Chem. 2018, 14, 930–954. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Qiao, H.; He, J.; Li, W.; Chen, R.; Wang, J.; Wu, L.; Hu, R.; Duan, J.; Chen, Z. Functional oligopeptide as a novel strategy for drug delivery. J. Drug Target. 2017, 25, 597–607. [Google Scholar] [CrossRef]
- Mitchell, P.J.; Tjian, R. Transcriptional Regulationin Mamnalian Cells by Sequence-Specific DNA Binding Proteins. Science 1989, 245, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Pabo, C.O.; Sauer, R.T. Transcription Factors: Structural Families and Principles of DNA Recognition. Annu. Rev. Biochem. 1992, 61, 1053–1095. [Google Scholar] [CrossRef]
- Boga, S.; Bouzada, D.; García Peña, D.; Vázquez López, M.; Eugenio Vázquez, M. Sequence-Specific DNA Recognition with Designed Peptides. Eur. J. Org. Chem. 2018, 2018, 249–261. [Google Scholar] [CrossRef]
- Vázquez, M.E.; Caamaño, A.M.; Mascareñas, J.L. From Transcription Factors to Designed Sequence-Specific DNA-Binding Peptides. Chem. Soc. Rev. 2003, 32, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Matić, J.; Tumir, L.-M.; Radić Stojković, M.; Piantanida, I. Advances in Peptide-based DNA/RNA-Intercalators. Curr. Protein Pept. Sci. 2016, 17, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Caamaño, A.M.; Vázquez, M.E.; Martínez-Costas, J.; Castedo, L.; Mascareñas, J.L. A Light-Modulated Sequence-Specific DNA-Binding Peptide. Angew. Chem. 2000, 112, 3234–3237. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Zhao, M.; Liu, L.; Zheng, M.; Wang, Y.; Wu, J.; Peng, S. A Class of Trp-Trp-AA-OBzl: Synthesis, in vitro anti-Proliferation/in vivo anti-Tumor Evaluation, Intercalation-Mechanism Investigation and 3D QSAR Analysis. Eur. J. Med. Chem. 2011, 46, 3410–3419. [Google Scholar] [CrossRef]
- Toseland, C.P. Fluorescent Labeling and Modification of Proteins. J. Chem. Biol. 2013, 6, 85–95. [Google Scholar] [CrossRef]
- Ladokh, A.S. Fluorescence Spectroscopy in Peptide and Protein Analysis. In Encyclopedia of Analytical Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Maity, D. Selected Peptide-based Fluorescent Probes for Biological Applications. Beilstein J. Org. Chem. 2020, 16, 2971–2982. [Google Scholar] [CrossRef]
- Wu, J.; Zou, Y.; Li, C.; Sicking, W.; Piantanida, I.; Yi, T.; Schmuck, C. A Molecular Peptide Beacon for the Ratiometric Sensing of Nucleic Acids. J. Am. Chem. Soc. 2012, 134, 1958–1961. [Google Scholar] [CrossRef] [PubMed]
- Maity, D.; Jiang, J.; Ehlers, M.; Wu, J.; Schmuck, C. A FRET-Enabled Molecular Peptide Beacon with a Significant Red Shift for the Ratiometric Detection of Nucleic Acids. Chem. Commun. 2016, 52, 6134–6137. [Google Scholar] [CrossRef] [PubMed]
- Maity, D.; Li, M.; Ehlers, M.; Schmuck, C. A Metal-Free Fluorescence Turn-on Molecular Probe for Detection of Nucleoside Triphosphates. Chem. Commun. 2017, 53, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Matić, J.; Šupljika, F.; Tandarić, T.; Dukši, M.; Piotrowski, P.; Vianello, R.; Brozović, A.; Piantanida, I.; Schmuck, C.; Radić Stojković, M. DNA/RNA recognition controlled by the glycine linker and the guanidine moiety of phenanthridine peptides. Int. J. Biol. Macromol. 2019, 134, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Krošl, I.; Košćak, M.; Ribičić, K.; Žinić, B.; Majhen, D.; Božinović, K.; Piantanida, I. Impact of the Histidine-Triazole and Tryptophan-Pyrene Exchange in the WHW Peptide: Cu(II) Binding, DNA/RNA Interactions and Bioactivity. Int. J. Mol. Sci. 2022, 23, 7006. [Google Scholar] [CrossRef] [PubMed]
- Košćak, M.; Krošl, I.; Žinić, B.; Piantanida, I. Fluorescent Analogues of FRH Peptide: Cu(II) Binding and Interactions with ds-DNA/RNA. Chemosensors 2022, 10, 34. [Google Scholar] [CrossRef]
- Guelev, V.; Lee, J.; Ward, J.; Sorey, S.; Hoffman, D.W.; Iverson, B.L. Peptide bis-intercalator binds DNA via threading mode with sequence specific contacts in the major groove. Chem. Biol. 2001, 8, 415–425. [Google Scholar] [CrossRef]
- Erben, A.; Matić, J.; Basarić, N.; Piantanida, I. The Phenanthridine-modified Tyrosine Dipeptide: Synthesis and Non-covalent Binding to DNA and RNA. Croat. Chem. Acta 2019, 92, 249–258. [Google Scholar] [CrossRef]
- Erben, A.; Sviben, I.; Mihaljević, B.; Piantanida, I.; Basarić, N. Non-Covalent Binding of Tripeptides-Containing Tryptophan to Polynucleotides and Photochemical Deamination of Modified Tyrosine to Quinone Methide Leading to Covalent Attachment. Molecules 2021, 26, 4315. [Google Scholar] [CrossRef]
- Husak, A.; Noichl, B.P.; Šumanovac Ramljak, T.; Sohora, M.; Škalamera, Đ.; Budiša, N.; Basarić, N. Photochemical Formation of Quinone Methides from Peptides Containing Modified Tyrosine. Org. Biomol. Chem. 2016, 14, 10894–10905. [Google Scholar] [CrossRef] [PubMed]
- Rokita, S.E. (Ed.) Quinone Methides; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Singh, M.S.; Nagaraju, A.; Anand, N.; Chowdhury, S. ortho-Quinone Methide (o-QM): A Highly Reactive, Ephemeral and Versatile Intermediate in Organic Synthesis. RSC Adv. 2014, 4, 55924–55959. [Google Scholar] [CrossRef]
- Bai, W.-J.; David, J.G.; Feng, Z.-G.; Weaver, M.G.; Wu, K.-L.; Pettus, T.R.R. The Domestication of ortho-Quinone Methides. Acc. Chem. Res. 2014, 47, 3655–3664. [Google Scholar] [CrossRef] [PubMed]
- Frecero, M. Quinone Methides as Alkylating and Cross-Linking Agents. Mini Rev. Org. Chem. 2004, 1, 403–415. [Google Scholar] [CrossRef]
- Wang, P.; Song, Y.; Zhang, L.; He, H.; Zhou, X. Quinone Methide Derivatives: Important Intermediates to DNA Alkylating and DNA Cross-Linking Actions. Curr. Med. Chem. 2005, 12, 2893–2913. [Google Scholar] [CrossRef] [PubMed]
- McCracken, P.G.; Bolton, J.L.; Thatcher, G.R.J. Covalent Modification of Proteins and Peptides by the Quinone Methide from 2-tert-Butyl-4,6-dimethylphenol: Selectivity and Reactivity with Respect to Competitive Hydration. J. Org. Chem. 1997, 62, 1820–1825. [Google Scholar] [CrossRef]
- Arumugam, S.; Guo, J.; Mbua, N.E.; Friscourt, F.; Lin, N.; Nekongo, E.; Boons, G.-J.; Popik, V.V. Selective and Reversible Photochemical Derivatization of Cysteine Residues in Peptides and Proteins. Chem. Sci. 2014, 5, 1591–1598. [Google Scholar] [CrossRef]
- Pérez-Ruiz, R.; Molins-Molina, O.; Lence, E.; González-Bello, C.; Miranda, M.A.; Consuelo Jiménez, M. Photogeneration of Quinone Methides as Latent Electrophiles for Lysine Targeting. J. Org. Chem. 2018, 83, 13019–13029. [Google Scholar] [CrossRef]
- Pande, P.; Shearer, J.; Yang, J.; Greenberg, W.A.; Rokita, S.E. Alkylation of Nucleic Acids by a Model Quinone Methide. J. Am. Chem. Soc. 1999, 121, 6773–6779. [Google Scholar] [CrossRef]
- Veldhuyzen, W.F.; Shallop, A.J.; Jones, R.A.; Rokita, S.E. Thermodynamic versus Kinetic Products of DNA Alkylation as Modeled by Reaction of Deoxyadenosine. J. Am. Chem. Soc. 2001, 123, 11126–11132. [Google Scholar] [CrossRef]
- Veldhuyzen, W.F.; Pande, P.; Rokita, S.E. A Transient Product of DNA Alkylation Can Be Stabilized by Binding Localization. J. Am. Chem. Soc. 2003, 125, 14005–14013. [Google Scholar] [CrossRef]
- Richter, S.N.; Maggi, S.; Colloredo Mels, S.; Palumbo, M.; Freccero, M. Binol Quinone Methides as Bisalkylating and DNA Cross-Linking Agents. J. Am. Chem. Soc. 2004, 126, 13973–13979. [Google Scholar] [CrossRef] [PubMed]
- Verga, D.; Nadai, M.; Doria, F.; Percivalle, C.; Di Antonio, M.; Palumbo, M.; Richter, S.N.; Freccero, M. Photogeneration and Reactivity of Naphthoquinone Methides as Purine Selective DNA Alkylating Agents. J. Am. Chem. Soc. 2010, 132, 14625–14637. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wahi, M.S.; Rokita, S.E. Immortalizing a Transient Electrophile for DNA Cross-Linking. Angew. Chem. Int. Ed. 2008, 47, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Rokita, S.E. Dynamic Cross-Linking is Retained in Duplex DNA after Multiple Exchange of Strands. Angew. Chem. Int. Ed. 2010, 49, 5957–5960. [Google Scholar] [CrossRef] [PubMed]
- Rossiter, C.S.; Modica, E.; Kumar, D.; Rokita, S.E. Few Constraints Limit the Design of Quinone Methide-Oligonucleotide Self-Adducts for Directing DNA Alkylation. Chem. Commun. 2011, 47, 1476–1478. [Google Scholar] [CrossRef] [PubMed]
- Di Antonio, M.; Doria, F.; Richter, S.N.; Bertipaglia, C.; Mella, M.; Sissi, C.; Palumbo, M.; Freccero, M. Quinone Methides Tethered to Naphthalene Diimides as Selective G-Quadruplex Alkylating Agents. J. Am. Chem. Soc. 2009, 131, 13132–13141. [Google Scholar] [CrossRef]
- Nadai, M.; Doria, F.; Di Antonio, M.; Sattin, G.; Germani, L.; Percivalle, C.; Palumbo, M.; Richter, S.N.; Freccero, M. Naphthalene Diimide Scaffolds with Dual Reversible and Covalent Interaction Properties towards G-Quadruplex. Biochimie 2011, 93, 1328–1340. [Google Scholar] [CrossRef]
- Doria, F.; Nadai, M.; Folini, M.; Di Antonio, M.; Germani, L.; Percivalle, C.; Sissi, C.; Zaffaroni, N.; Alcaro, S.; Artese, A.; et al. Hybrid Ligand-Alkylating Agents Targeting Telomeric G-Quadruplex Structures. Org. Biomol. Chem. 2012, 10, 2798–2806. [Google Scholar] [CrossRef]
- Doria, F.; Nadai, M.; Folini, M.; Scalabrin, M.; Germani, L.; Sattin, G.; Mella, M.; Palumbo, M.; Zaffaroni, N.; Fabris, D.; et al. Targeting Loop Adeninesin G-Quadruplexby a Selective Oxirane. Chem. Eur. J. 2013, 19, 78–81. [Google Scholar] [CrossRef]
- Basarić, N.; Mlinarić-Majerski, K.; Kralj, M. Quinone Methides: Photochemical Generation and its Application in Biomedicine. Curr. Org. Chem. 2014, 18, 3–18. [Google Scholar] [CrossRef]
- Percivalle, C.; Doria, F.; Freccero, M. Quinone Methides as DNA Alkylating Agents: An Overview on Efficient Activation Protocols for Enhanced Target Selectivity. Curr. Org. Chem. 2014, 18, 19–43. [Google Scholar] [CrossRef]
- Lerch, M.M.; Hansen, M.J.; van Dam, G.M.; Szymanski, W.; Feringa, B.L. Emerging Targets in Photopharmacology. Angew. Chem. Int. Ed. 2016, 55, 10978–10999. [Google Scholar] [CrossRef] [PubMed]
- Chass, G.A.; Lovas, S.; Murphy, R.F.; Csizmadia, I.G. The role of enhanced aromatic π-electron donating aptitude of the tyrosyl sidechain with respect to that of phenylalanyl in intramolecular interactions. Eur. Phys. J. D 2002, 20, 481–497. [Google Scholar] [CrossRef]
- Hirata, Y.; Kanda, Y.; Mataga, N. Picosecond Laser Photolysis and Transient Photocurrent Studies of the Ionic Dissociation Mechanism of Heteroexcimers: Pyrene-N,N-Dimethylaniline and Pyrene-p-Dicyanobenzene Systems in Polar Solvents. J. Phys. Chem. 1983, 87, 1659–1662. [Google Scholar] [CrossRef]
- Demeunynck, M.; Bailly, C.; Wilson, W.D. Small Molecule DNA and RNA Binders: From Synthesis to Nucleic Acid Complexes; Wiley-VCH Verlag GmbH & Co.: Weincheim, Germany, 2004. [Google Scholar]
- Siddique, B.; Duhamel, J. Effect of Polypeptide Sequence on Polypeptide Self-Assembly. Langmuir 2011, 27, 6639–6650. [Google Scholar] [CrossRef] [PubMed]
- Dourtoglou, V.; Gross, B.; Lambropoulou, V.; Zioudrou, C. O-Benzotriazolyl-N,N,N′,N′-tetramethyluronium Hexafluorophosphate as Coupling Reagent for the Synthesis of Peptides of Biological Interest. Synthesis 1984, 1984, 572–575. [Google Scholar] [CrossRef]
- Birks, J.B. Photophysics of Aromatic Molecules; Wiley-Interscience: London, UK, 1970. [Google Scholar]
- Montalti, M.; Credi, A.; Prodi, L.; Gandolfi, M.T. Handbook of Photochemistry; CRC Taylor and Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Resch-Genger, U.; Rurack, K. Determination of the photoluminescence quantum yield of dilute dye solutions (IUPAC Technical Report). Pure Appl. Chem. 2013, 85, 2005–2026. [Google Scholar] [CrossRef]
- Sambol, M.; Košćak, M.; Uzelac, L.; Kralj, M.; Piantanida, I.; Basarić, N. Simultaneous staining of endoplasmic reticulum and lipid droplets by naphthol-aminonaphthalimide conjugates and photoinduced antiproliferative effects. Dye Pigment. 2022, 206, 110651. [Google Scholar] [CrossRef]
- Goldstein, S.; Rabani, J. The Ferrioxalate and Iodide-Iodate Actinometers in the UV Region. J. Photochem. Photobiol. 2008, 193, 50–55. [Google Scholar] [CrossRef]
- Chiang, Y.; Kresge, A.J.; Zhu, Y. Kinetics and Mechanisms of Hydration of o-Quinone Methides in Aqueous Solution. J. Am. Chem. Soc. 2000, 122, 9854–9855. [Google Scholar] [CrossRef]
- Chiang, Y.; Kresge, A.J.; Zhu, Y. Flash Photolytic Generation of ortho-Quinone Methide in Aqueous Solution and Study of Its Chemistry in that Medium. J. Am. Chem. Soc. 2001, 123, 8089–8094. [Google Scholar] [CrossRef] [PubMed]
- Toteva, M.M.; Richard, J.P. The Generation and Reactions of Quinone Methides. Adv. Phys. Org. Chem. 2011, 45, 39–91. [Google Scholar] [PubMed]
- Arumugam, S.; Popik, V.V. Photochemical Generation and the Reactivity of o-Naphthoquinone Methides in Aqueous Solutions. J. Am. Chem. Soc. 2009, 131, 11892–11899. [Google Scholar] [CrossRef] [PubMed]
- Mergny, J.L.; Lacroix, L. Analysis of Thermal Melting Curves. Oligonucleotides 2003, 13, 515–537. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.D.; Ratmeyer, L.; Zhao, M.; Strekowski, L.; Boykin, D. The Search for Structure-Specific Nucleic Acid-Interactive Drugs: Effects of Compound Structure on RNA versus DNA Interaction Strength. Biochemistry 1993, 32, 4098–4104. [Google Scholar] [CrossRef] [PubMed]
- McGhee, J.D.; von Hippel, P.H. Theoretical Aspects of DNA-Protein Interactions: Co-operative and Non-co-operative Binding of Large Ligands to a one-Dimensional Homogeneous Lattice. J. Mol. Biol. 1976, 103, 679–681. [Google Scholar] [CrossRef]
- Rodger, A.; Norden, B. Circular Dichroism and Linear Dichroism; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Šmidlehner, T.; Piantanida, I.; Pescitelli, G. Polarization Spectroscopy Methods in the Determination of Interactions of Small Molecules with Nucleic Acids-Tutorial. Beilstein J. Org. Chem. 2018, 14, 84–105. [Google Scholar] [CrossRef]
- Eriksson, M.; Norden, B. Linear and Circular Dichroism of Drug-Nucleic Acid Complexes. Methods Enzymol. 2001, 340, 68–98. [Google Scholar]
- Dendrinos, K.G.; Kalivretenos, A.G. Synthesis of N-hydroxysuccinimide esters using polymer bound HOBt. Tetrahedron Lett. 1998, 39, 1321–1324. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Y. Oligocholate foldamers with ‘prefolded’ macrocycles for enhanced folding in solution and surfactant micelles. Tetrahedron 2013, 69, 6051–6059. [Google Scholar] [CrossRef]
- Saenger, W. Principles of Nucleic Acid Structure; Springer: New York, NY, USA, 1983. [Google Scholar]
- Cantor, C.R.; Schimmel, P.R. Biophysical Chemistry; WH Freeman and Co.: San Francisco, CA, USA, 1980; pp. 1109–1181. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Φf1 | τf (341 nm) 2/ns | τf (281 nm) 2/ns |
|---|---|---|---|
| 7 | 0.4 ± 0.1 | 0.5 ± 0.1 (0.02) 7.02 ± 0.06 (0.71) 27.1 ± 0.5 (0.27) | 0.37 ± 0.05 (0.03) 7.05 ± 0.06 (0.70) 26.4 ± 0.4 (0.27) |
| 8 | 0.3 ± 0.1 | 0.10 ± 0.05 (0.01) 7.7 ± 0.1 (0.85) 15.8 ± 0.9 (0.14) | 0.06 ± 0.04 (0.02) 7.77 ± 0.09 (0.84) 16.6 ± 0.9 (0.14) |
| 5 | 0.5 ± 0.1 | 5 ± 1 (0.01) 25.87 ± 0.07 (0.99) |
| Compound | ΦR/(10−2) |
|---|---|
| 7 | 1.33 ± 0.03 |
| 8 | 2.56 ± 0.05 |
| 1 | 2 | |
|---|---|---|
| ct-DNA | 5.6 | 6.0 |
| pApU | 6.8 | 6.9 |
| p(dAdT)2 | >7 | 6.8 |
| p(dGdC)2 | 6.7 | 6.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sviben, I.; Glavaš, M.; Erben, A.; Bachelart, T.; Pavlović Saftić, D.; Piantanida, I.; Basarić, N. Dipeptides Containing Pyrene and Modified Photochemically Reactive Tyrosine: Noncovalent and Covalent Binding to Polynucleotides. Molecules 2023, 28, 7533. https://doi.org/10.3390/molecules28227533
Sviben I, Glavaš M, Erben A, Bachelart T, Pavlović Saftić D, Piantanida I, Basarić N. Dipeptides Containing Pyrene and Modified Photochemically Reactive Tyrosine: Noncovalent and Covalent Binding to Polynucleotides. Molecules. 2023; 28(22):7533. https://doi.org/10.3390/molecules28227533
Chicago/Turabian StyleSviben, Igor, Mladena Glavaš, Antonija Erben, Thomas Bachelart, Dijana Pavlović Saftić, Ivo Piantanida, and Nikola Basarić. 2023. "Dipeptides Containing Pyrene and Modified Photochemically Reactive Tyrosine: Noncovalent and Covalent Binding to Polynucleotides" Molecules 28, no. 22: 7533. https://doi.org/10.3390/molecules28227533
APA StyleSviben, I., Glavaš, M., Erben, A., Bachelart, T., Pavlović Saftić, D., Piantanida, I., & Basarić, N. (2023). Dipeptides Containing Pyrene and Modified Photochemically Reactive Tyrosine: Noncovalent and Covalent Binding to Polynucleotides. Molecules, 28(22), 7533. https://doi.org/10.3390/molecules28227533