
In Silico and In Vitro Search for Dual Inhibitors of the Trypanosoma brucei and Leishmania major Pteridine Reductase 1 and Dihydrofolate Reductase




Abstract
:1. Introduction
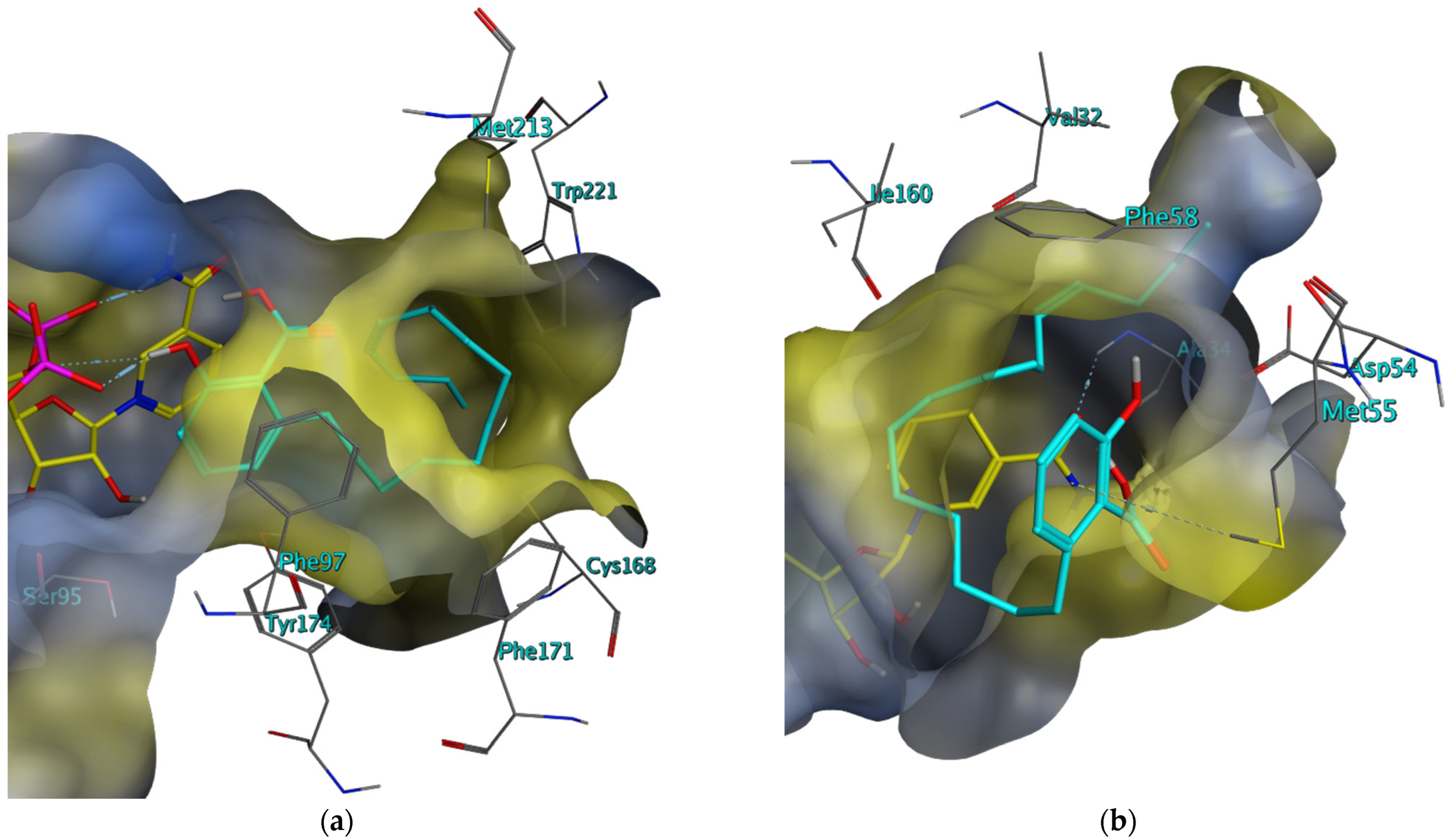
2. Results
2.1. Pharmacophore-Based Virtual Screening for Inhibitors of the T. brucei and L. major PTR1 and DHFR
2.2. In Vitro Evaluation of the In Silico Hits against the T. brucei and L. major PTR1 and DHFR
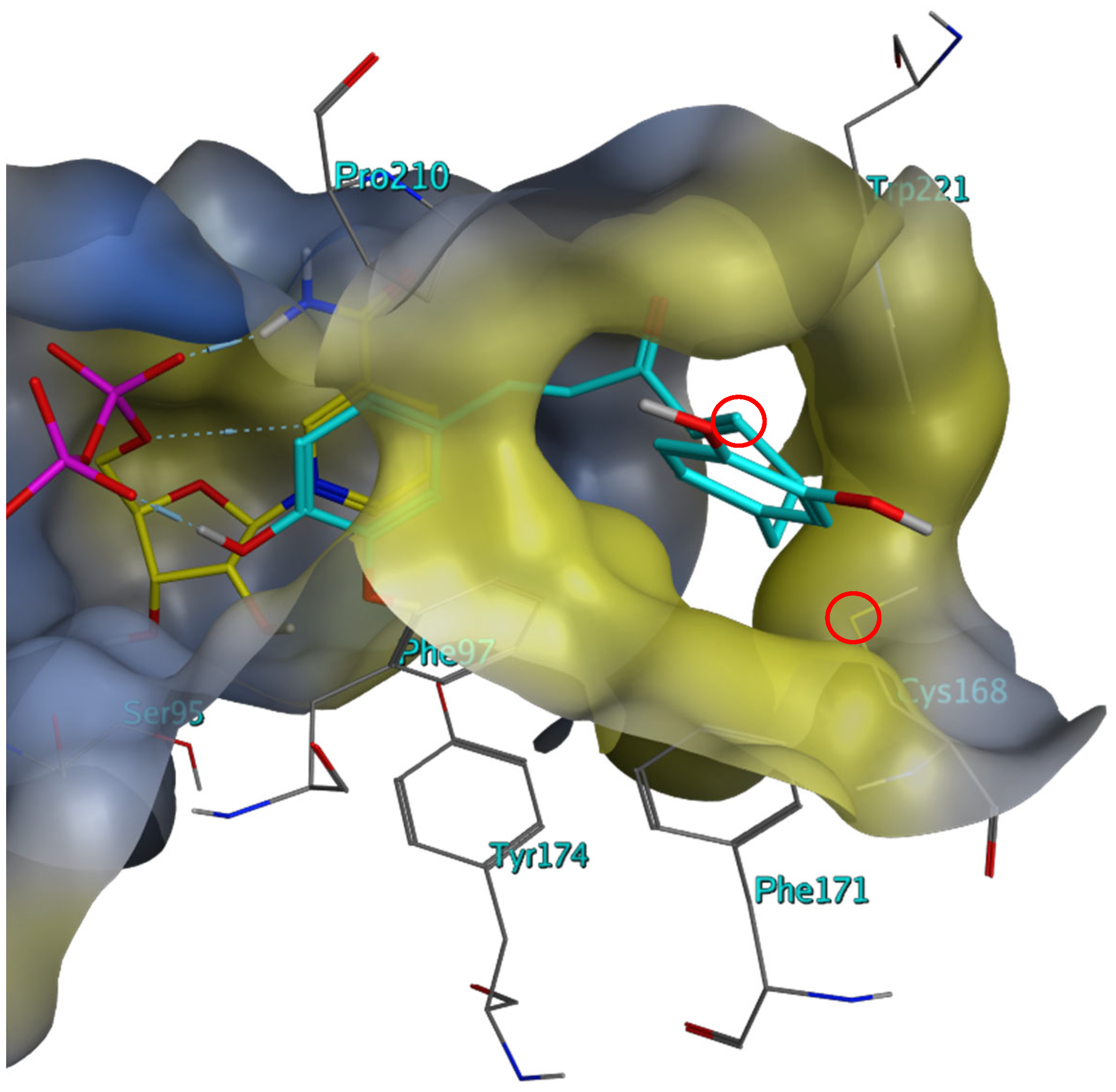
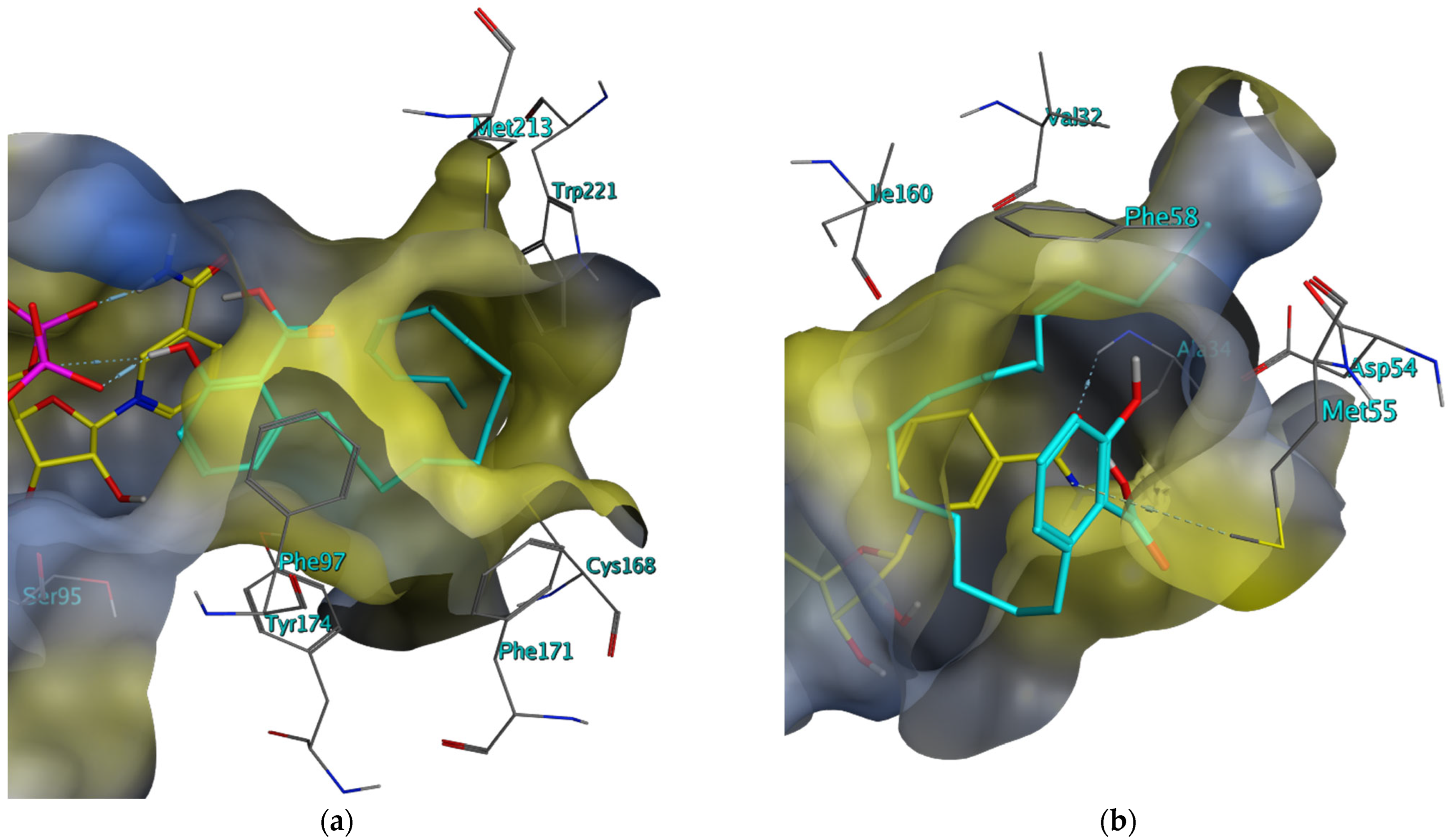
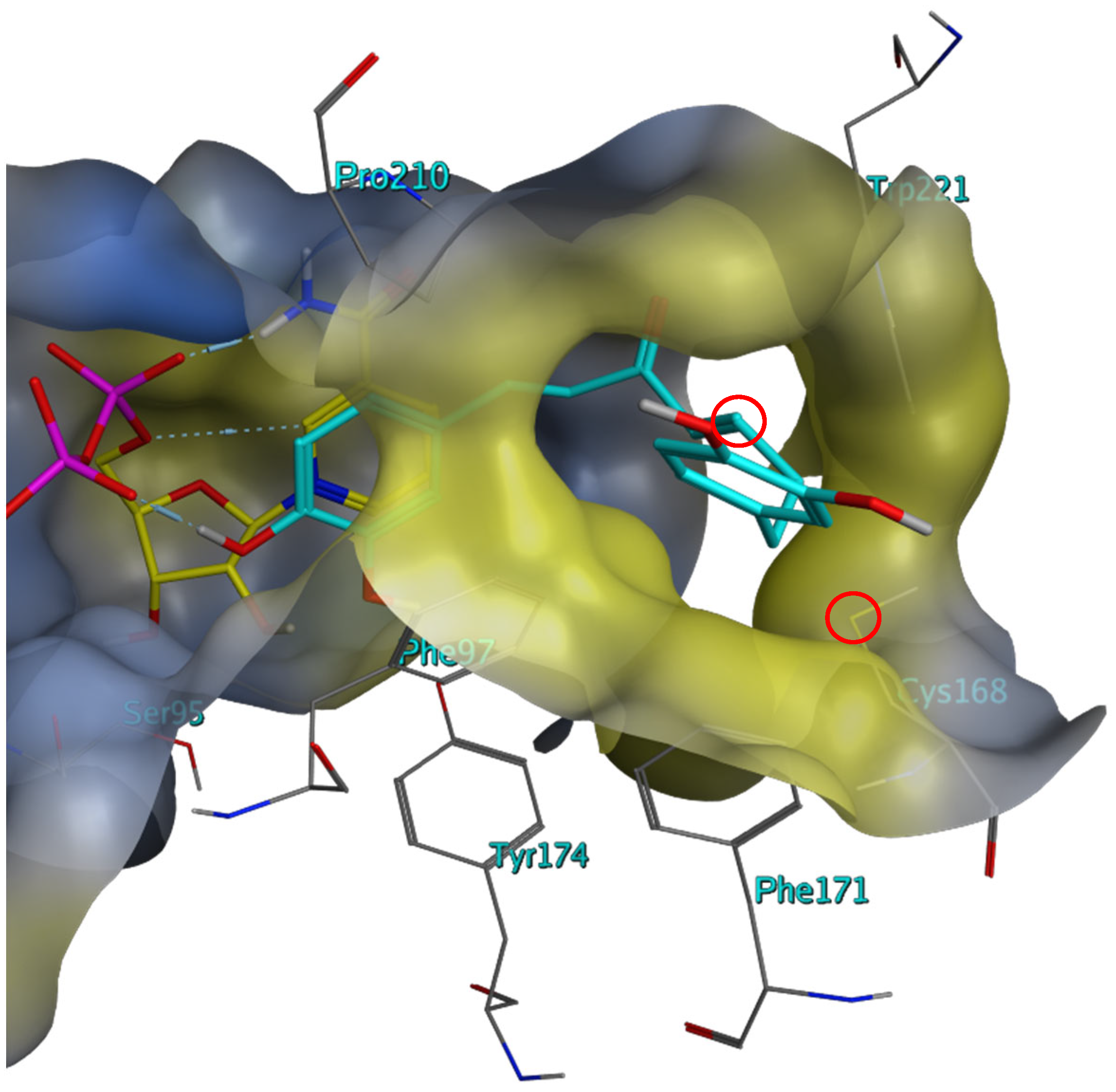
2.3. Investigation of the Mechanism of Inhibition for Selected TbPTR1 Inhibitors
3. Discussion
4. Materials and Methods
4.1. In Silico Procedure
4.1.1. Preparation of the Respective 3D Protein Structures
4.1.2. Homology Modeling
4.1.3. Pharmacophore Design
4.1.4. Virtual Screening of Natural Product Databases
4.1.5. Molecular Docking
4.2. In Vitro Procedure
4.2.1. Cloning of TbDHFR and LmDHFR into the E. coli BL21(DE3) Host Strain
4.2.2. Recombinant Expression and Purification of LmPTR1 and TbPTR1
4.2.3. Recombinant Expression and Purification of LmDHFR and TbDHFR
4.2.4. Recombinant Expression and Purification of hDHFR
4.2.5. Kinetic Characterization
TbPTR1 and LmPTR1
TbDHFR and LmDHFR
hDHFR
4.2.6. Test Compounds
4.2.7. Single-Concentration Enzyme Inhibition Assays
4.2.8. Determination of the IC50 Values
4.2.9. Dilution Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neglected Tropical Diseases—GLOBAL. Available online: https://www.who.int/health-topics/neglected-tropical-diseases#tab=tab_1 (accessed on 22 June 2023).
- Herrmann, F.C. In Silico-Identifikation und In Vitro-Evaluation Natürlicher Inhibitoren Diverser Therapierelevanter Zielenzyme von Humanpathogenen Eukaryoten der Gattungen Trypanosoma, Leishmania und Plasmodium. Ph.D. Thesis, University of Münster, Münster, Germany, 2016. [Google Scholar]
- Herrmann, F.C.; Sivakumar, N.; Jose, J.; Costi, M.P.; Pozzi, C.; Schmidt, T.J. In silico Identification and In vitro Evaluation of Natural Inhibitors of Leishmania major Pteridine Reductase I. Molecules 2017, 22, 2166. [Google Scholar] [CrossRef] [PubMed]
- Possart, K.; Herrmann, F.C.; Jose, J.; Costi, M.P.; Schmidt, T.J. Sesquiterpene Lactones with Dual Inhibitory Activity against the Trypanosoma brucei Pteridine Reductase 1 and Dihydrofolate Reductase. Molecules 2021, 27, 149. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, F.C.; Lenz, M.; Jose, J.; Kaiser, M.; Brun, R.; Schmidt, T.J. In silico Identification and in vitro Activity of Novel Natural Inhibitors of Trypanosoma brucei Glyceraldehyde-3-phosphate-dehydrogenase. Molecules 2015, 20, 16154–16169. [Google Scholar] [CrossRef] [PubMed]
- Trypanosomiasis, Human African (Sleeping Sickness). Available online: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed on 22 June 2023).
- Lucius, R.; Loos-Frank, B.; Lane, R.P. Biologie von Parasiten, 3rd ed.; Springer Spektrum: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Adegboye, O.; Field, M.A.; Kupz, A.; Pai, S.; Sharma, D.; Smout, M.J.; Wangchuk, P.; Wong, Y.; Loiseau, C. Natural-Product-Based Solutions for Tropical Infectious Diseases. Clin. Microbiol. Rev. 2021, 34, e0034820. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Fexinidazole: First Global Approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Leishmaniasis. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 22 June 2023).
- Garza-Tovar, T.F.; Sacriste-Hernández, M.I.; Juárez-Durán, E.R.; Arenas, R. An overview of the treatment of cutaneous leishmaniasis. Fac. Rev. 2020, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, I.H. Inhibitors of dihydrofolate reductase in leishmania and trypanosomes. Biochim. Biophys. Acta Mol. Basis Dis. 2002, 1587, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.W.; Dewar, S.; Ong, H.B.; Sienkiewicz, N.; Fairlamb, A.H. Trypanosoma brucei DHFR-TS Revisited: Characterisation of a Bifunctional and Highly Unstable Recombinant Dihydrofolate Reductase-Thymidylate Synthase. PLoS Neglect. Trop. Dis. 2016, 10, e0004714. [Google Scholar] [CrossRef]
- Nare, B.; Hardy, L.W.; Beverley, S.M. The roles of pteridine reductase 1 and dihydrofolate reductase-thymidylate synthase in pteridine metabolism in the protozoan parasite Leishmania major. J. Biol. Chem. 1997, 272, 13883–13891. [Google Scholar] [CrossRef]
- Sienkiewicz, N.; Ong, H.B.; Fairlamb, A.H. Trypanosoma brucei pteridine reductase 1 is essential for survival in vitro and for virulence in mice. Mol. Microbiol. 2010, 77, 658–671. [Google Scholar] [CrossRef]
- Bisswanger, H. Enzymkinetik: Theorie und Methoden, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Schüttelkopf, A.W.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Structures of Leishmania major pteridine reductase complexes reveal the active site features important for ligand binding and to guide inhibitor design. J. Mol. Biol. 2005, 352, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Gourley, D.G.; Schüttelkopf, A.W.; Leonard, G.A.; Luba, J.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat. Struct. Biol. 2001, 8, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Changtam, C.; de Koning, H.P.; Ibrahim, H.; Sajid, M.S.; Gould, M.K.; Suksamrarn, A. Curcuminoid analogs with potent activity against Trypanosoma and Leishmania species. Eur. J. Med. Chem. 2010, 45, 941–956. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef]
- Pereira, J.M.; Severino, R.P.; Vieira, P.C.; Fernandes, J.B.; Da Silva, M.F.G.F.; Zottis, A.; Andricopulo, A.D.; Oliva, G.; Corrêa, A.G. Anacardic acid derivatives as inhibitors of glyceraldehyde-3-phosphate dehydrogenase from Trypanosoma cruzi. Bioorg. Med. Chem. 2008, 16, 8889–8895. [Google Scholar] [CrossRef] [PubMed]
- Borsari, C.; Luciani, R.; Pozzi, C.; Poehner, I.; Henrich, S.; Trande, M.; Cordeiro-da-Silva, A.; Santarem, N.; Baptista, C.; Tait, A.; et al. Profiling of Flavonol Derivatives for the Development of Antitrypanosomatidic Drugs. J. Med. Chem. 2016, 59, 7598–7616. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.N.; No, J.H.; Lee, G.Y.; Li, W.; Yang, S.Y.; Yang, G.; Schmidt, T.J.; Kang, J.S.; Kim, Y.H. Phenolic Constituents of Medicinal Plants with Activity against Trypanosoma brucei. Molecules 2016, 21, 480. [Google Scholar] [CrossRef]
- Kelly, J.X.; Ignatushchenko, M.V.; Bouwer, H.G.; Peyton, D.H.; Hinrichs, D.J.; Winter, R.W.; Riscoe, M. Antileishmanial drug development: Exploitation of parasite heme dependency. Mol. Biochem. Parasitol. 2003, 126, 43–49. [Google Scholar] [CrossRef]
- Laphookhieo, S.; Maneerat, W.; Koysomboon, S. Antimalarial and cytotoxic phenolic compounds from Cratoxylum maingayi and Cratoxylum cochinchinense. Molecules 2009, 14, 1389–1395. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Bank, R.P.D. RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 22 September 2021).
- Dawson, A.; Tulloch, L.B.; Barrack, K.L.; Hunter, W.N. High resolution structure of TbPTR1 in complex with Pemetrexed. Acta Crystallogr. Sect. D Struct. Biol. 2010, 66, 1334. [Google Scholar]
- Dawson, A.; Tulloch, L.B.; Barrack, K.L.; Hunter, W.N. Structure of PTR1 from Trypanosoma brucei in ternary complex with 2,4-diamino-5-[2-(2,5-dimethoxyphenyl)ethyl]thieno[2,3-d]-pyrimidine and NADP+. Acta Crystallogr. Sect. D Struct. Biol. 2010, 66, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, A.I.; Huggan, J.K.; Suckling, C.J.; Gibson, C.L.; Stewart, K.; Giordani, F.; Barrett, M.P.; Wong, P.E.; Barrack, K.L.; Hunter, W.N. Crystal structure of pteridine reductase 1 (PTR1) from Trypanosoma brucei in ternary complex with cofactor and inhibitor. J. Med. Chem. 2014, 57, 6479. [Google Scholar] [CrossRef] [PubMed]
- Vanichtanankul, J.; Taweechai, S.; Yuvaniyama, J.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Yuthavong, Y. Trypanosoma brucei dihydrofolate reductase pyrimethamine complex. ACS Chem. Biol. 2011, 6. [Google Scholar] [CrossRef]
- Vanichtanankul, J.; Yuvaniyama, J.; Yuthavong, Y. Trypanosoma brucei dihydrofolate reductase (TbDHFR) in complex with WR99210. ACS Chem. Biol. 2011, 6. [Google Scholar] [CrossRef]
- Gourley, D.G.; Hunter, W.N. One active site, two modes of reduction correlate the mechanism of leishmania pteridine reductase with pterin metabolism and antifolate drug resistance in trypanosomes. Nat. Struct. Biol. 2001, 8, 521–525. [Google Scholar] [CrossRef] [PubMed]
- McLuskey, K.; Gibellini, F.; Carvalho, P.; Avery, M.A.; Hunter, W.N. Inhibition of Leishmania major pteridine reductase by 2,4,6-triaminoquinazoline: Structure of the NADPH ternary complex. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1780–1785. [Google Scholar] [CrossRef]
- Cavazzuti, A.; Paglietti, G.; Hunter, W.N.; Gamarro, F.; Piras, S.; Loriga, M.; Allecca, S.; Corona, P.; McLuskey, K.; Tulloch, L.; et al. Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc. Natl. Acad. Sci. USA 2008, 105, 1448–1453. [Google Scholar] [CrossRef]
- Schormann, N.; Velu, S.E.; Murugesan, S.; Senkovich, O.; Walker, K.; Chenna, B.C.; Shinkre, B.; Desai, A.; Chattopadhyay, D. Synthesis and characterization of potent inhibitors of Trypanosoma cruzi dihydrofolate reductase. Bioorg. Med. Chem. 2010, 18, 4056–4066. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Vordenbäumen, S.; Saenger, T.; Braukmann, A.; Tahan, T.; Bleck, E.; Jose, J.; Schneider, M. Human casein alpha s1 induces proinflammatory cytokine expression in monocytic cells by TLR4 signaling. Mol. Nutr. Food Res. 2016, 60, 1079–1089. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 2001. [Google Scholar]
- Perez Brandan, C.; Padilla, A.M.; Xu, D.; Tarleton, R.L.; Basombrio, M.A. Knockout of the dhfr-ts gene in Trypanosoma cruzi generates attenuated parasites able to confer protection against a virulent challenge. PLoS Neglect. Trop. Dis. 2011, 5, e1418. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
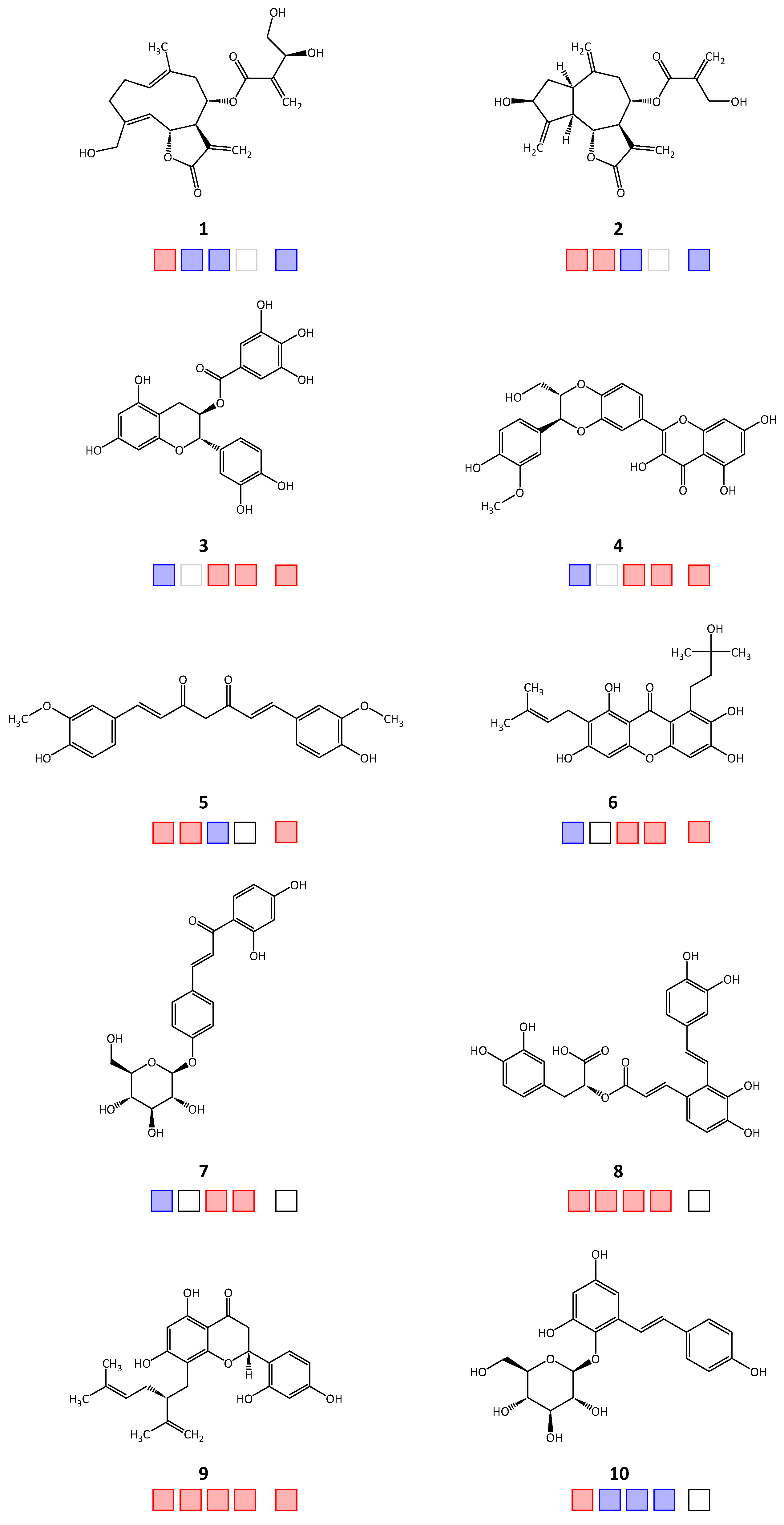
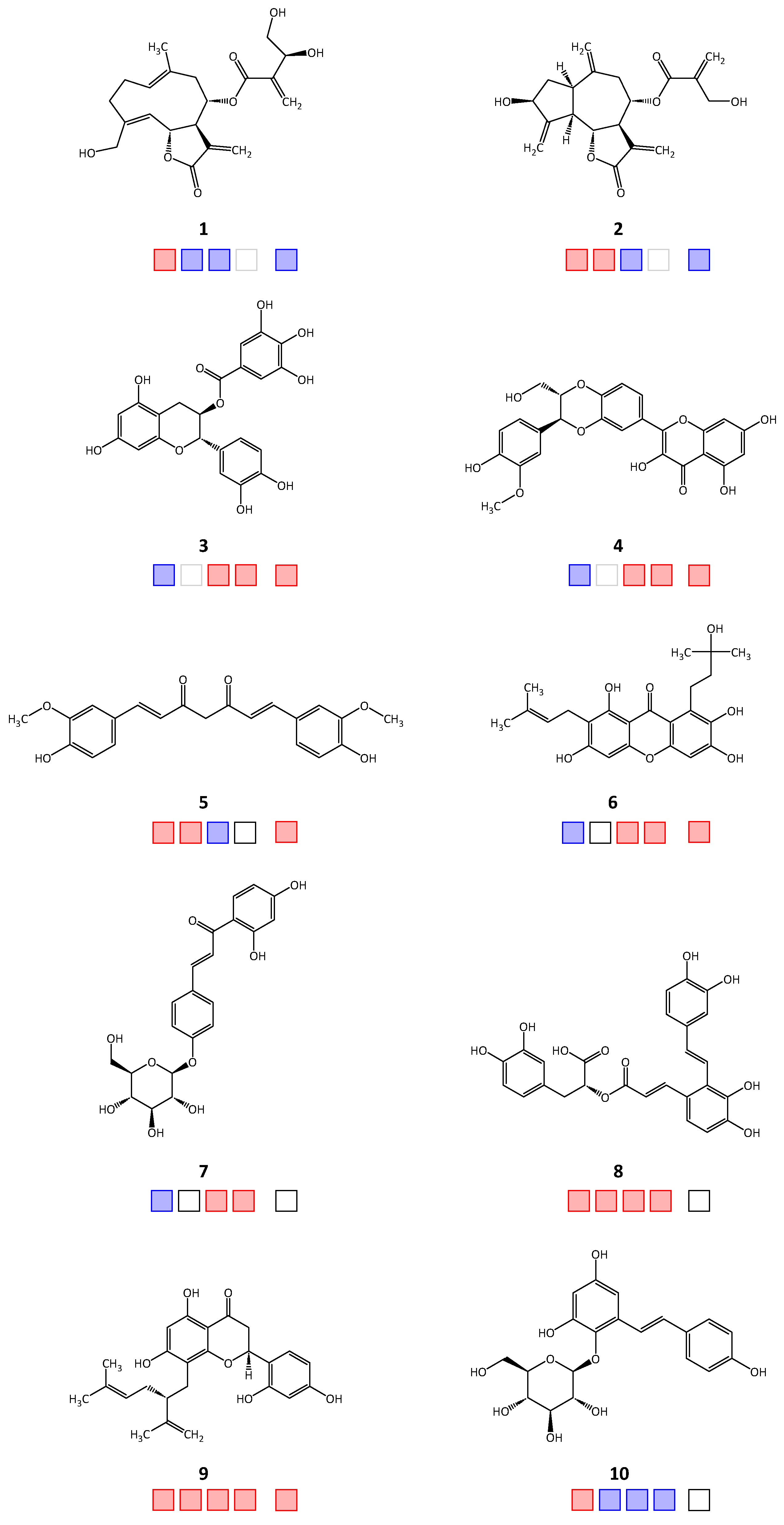
| Compound | IC50 [μM] | ||||
|---|---|---|---|---|---|
| TbPTR1 | TbDHFR | LmPTR1 | LmDHFR | hDHFR | |
| 1 | 12.2 a [4] | >50 | >50 | >50 | |
| 2 | 12.4 [4] | 7.12 | >50 | >50 | |
| 3 | >100 [2] | 75.3 [3] | 0.6 | 4.2 | |
| 4 | >100 | 42.9 [3] | 2.6 | 3.8 a | |
| 5 | 18.5 | 10.1 | >50 | 33.9 | |
| 6 | >100 [2] | 26.3 [3] | 3.0 | 1.7 a | |
| 7 | >100 | 84.5 [3] | 46.2 | >50 | |
| 8 | 85.1 | 2.5 | 42.2 [3] | 18.7 a | >50 |
| 9 | 31.8 | 9.4 | 19.2 [3] | 14 | 31.6 a |
| 10 | 83.6 | >50 | >50 | >50 | n.t. |
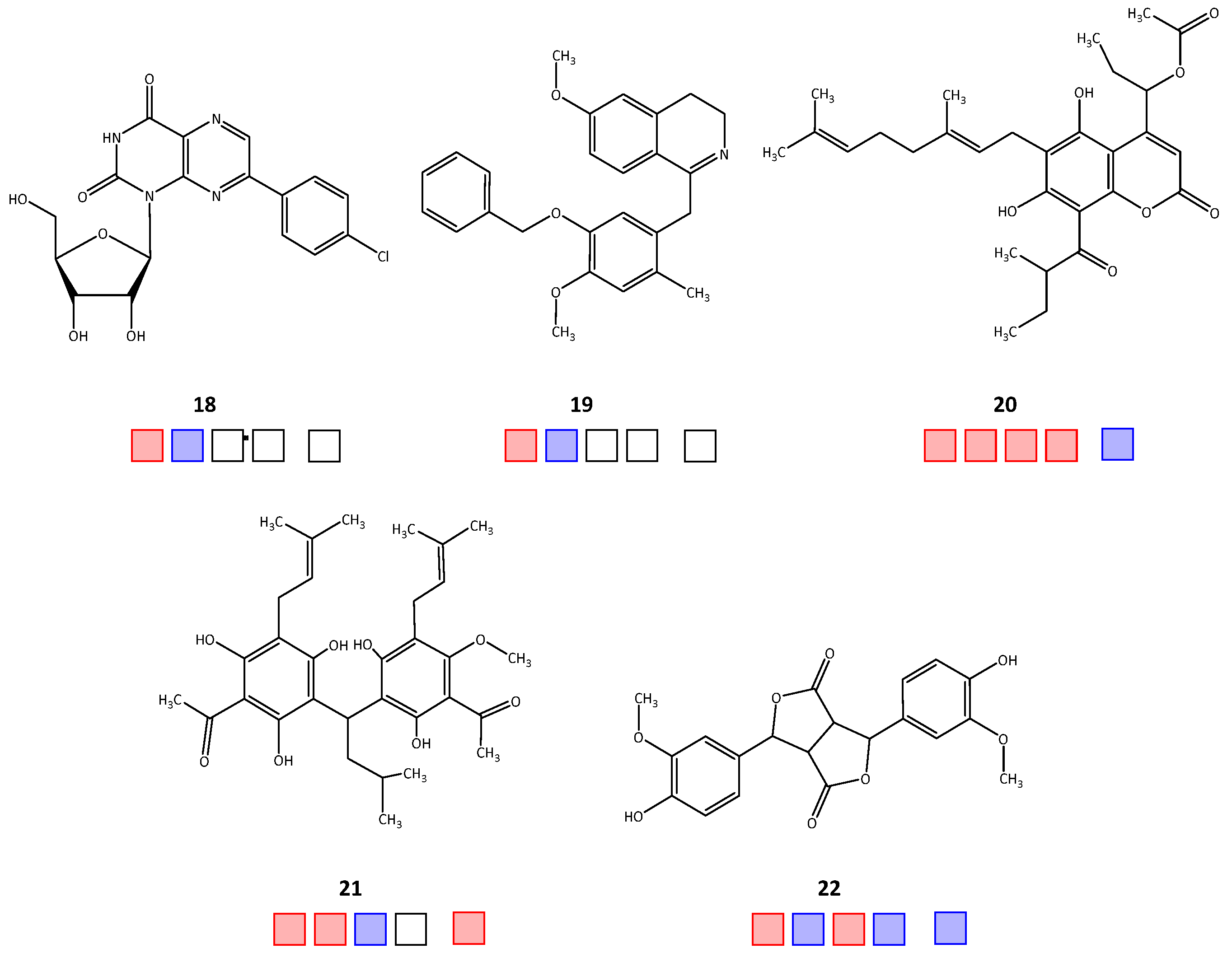
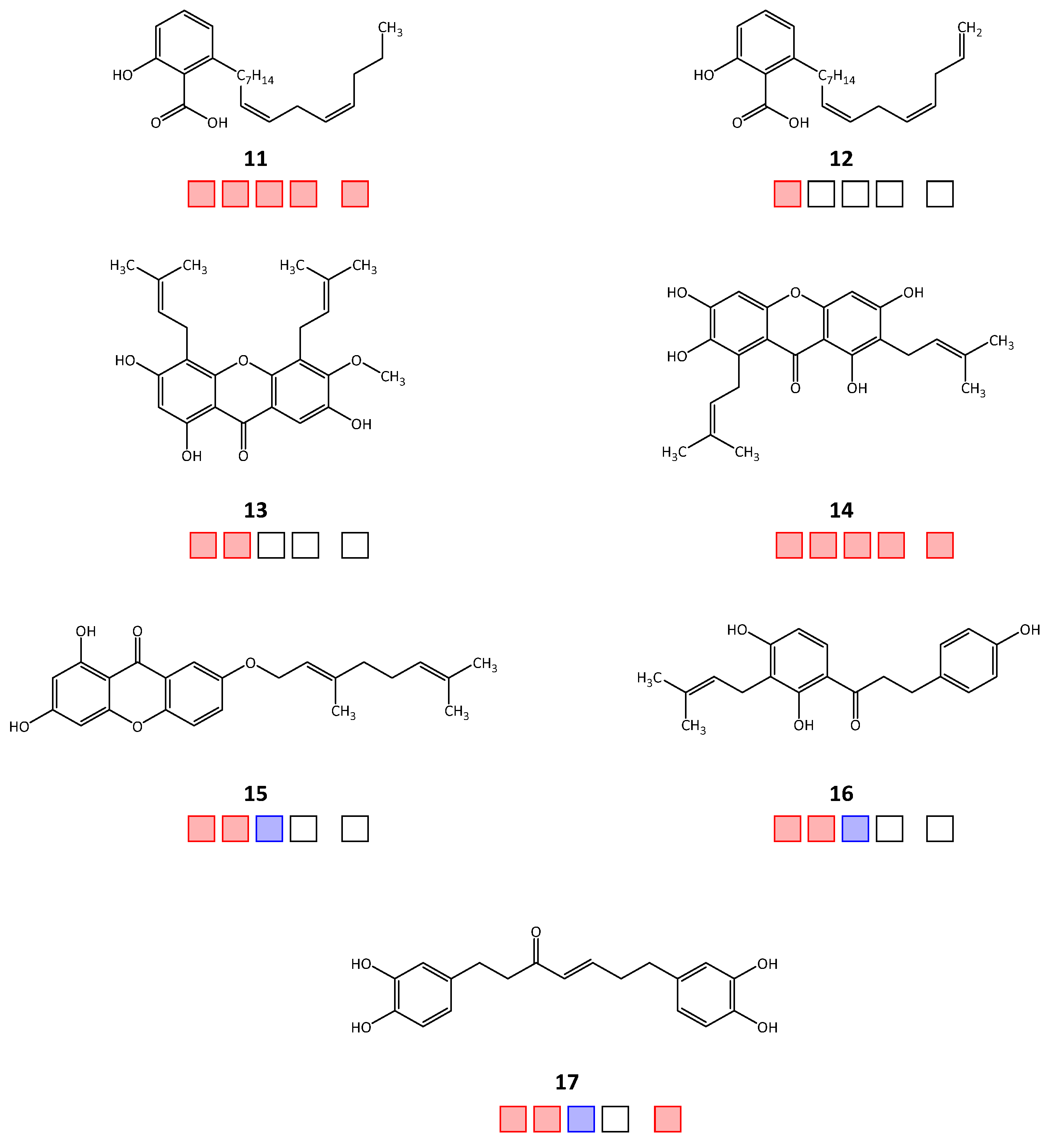
| 11 | 20.1 | 0.2 | 10.2 | 2.6 | 2.4 |
| 12 | 21.7 | n.t. | n.t. | n.t. | n.t. |
| 13 | 74.4 | 0.6 | n.t. | n.t. | n.t. |
| 14 | 71.0 | 0.2 | 10.0 | 1.5 | 2.7 |
| 15 | 32.4 a | 2.9 | >50 | n.t. | |
| 16 | 64.6 | 11.7 | >50 | n.t. | |
| 17 | 8.3 | 17.7 | >50 | 40.7 | |
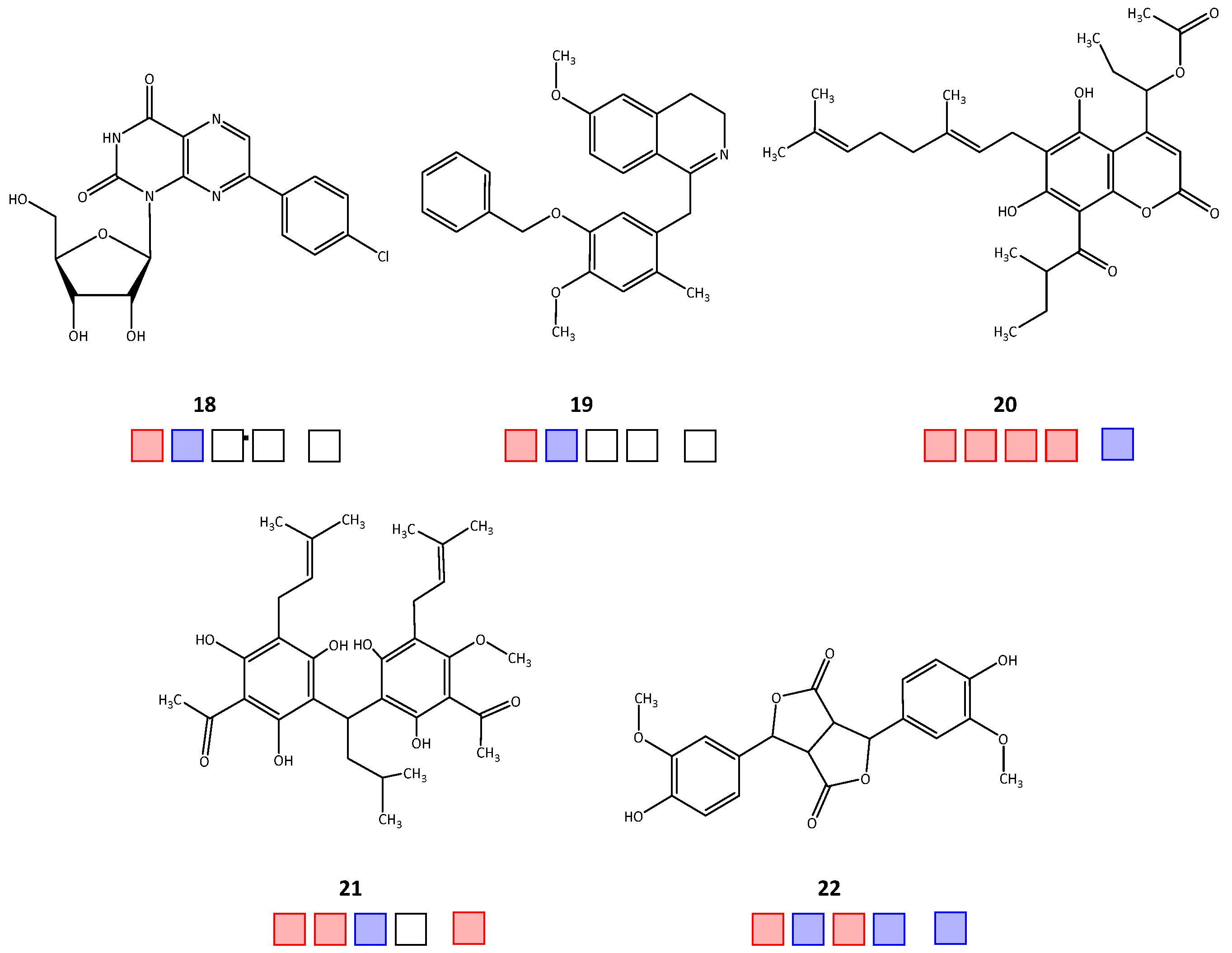
| 18 | 58.6 | >50 | n.t. | n.t. | n.t. |
| 19 | 75.1 | >50 | n.t. | n.t. | n.t. |
| 20 | 28.3 | 2.9 | 35.1 a | 4.7 a | >50 |
| 21 | 36.8 a | 2.1 | >50 | 19.8 | |
| 22 | 37.8 a | >50 | 30.2 | >50 | >50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Possart, K.; Herrmann, F.C.; Jose, J.; Schmidt, T.J. In Silico and In Vitro Search for Dual Inhibitors of the Trypanosoma brucei and Leishmania major Pteridine Reductase 1 and Dihydrofolate Reductase. Molecules 2023, 28, 7526. https://doi.org/10.3390/molecules28227526
Possart K, Herrmann FC, Jose J, Schmidt TJ. In Silico and In Vitro Search for Dual Inhibitors of the Trypanosoma brucei and Leishmania major Pteridine Reductase 1 and Dihydrofolate Reductase. Molecules. 2023; 28(22):7526. https://doi.org/10.3390/molecules28227526
Chicago/Turabian StylePossart, Katharina, Fabian C. Herrmann, Joachim Jose, and Thomas J. Schmidt. 2023. "In Silico and In Vitro Search for Dual Inhibitors of the Trypanosoma brucei and Leishmania major Pteridine Reductase 1 and Dihydrofolate Reductase" Molecules 28, no. 22: 7526. https://doi.org/10.3390/molecules28227526
APA StylePossart, K., Herrmann, F. C., Jose, J., & Schmidt, T. J. (2023). In Silico and In Vitro Search for Dual Inhibitors of the Trypanosoma brucei and Leishmania major Pteridine Reductase 1 and Dihydrofolate Reductase. Molecules, 28(22), 7526. https://doi.org/10.3390/molecules28227526