

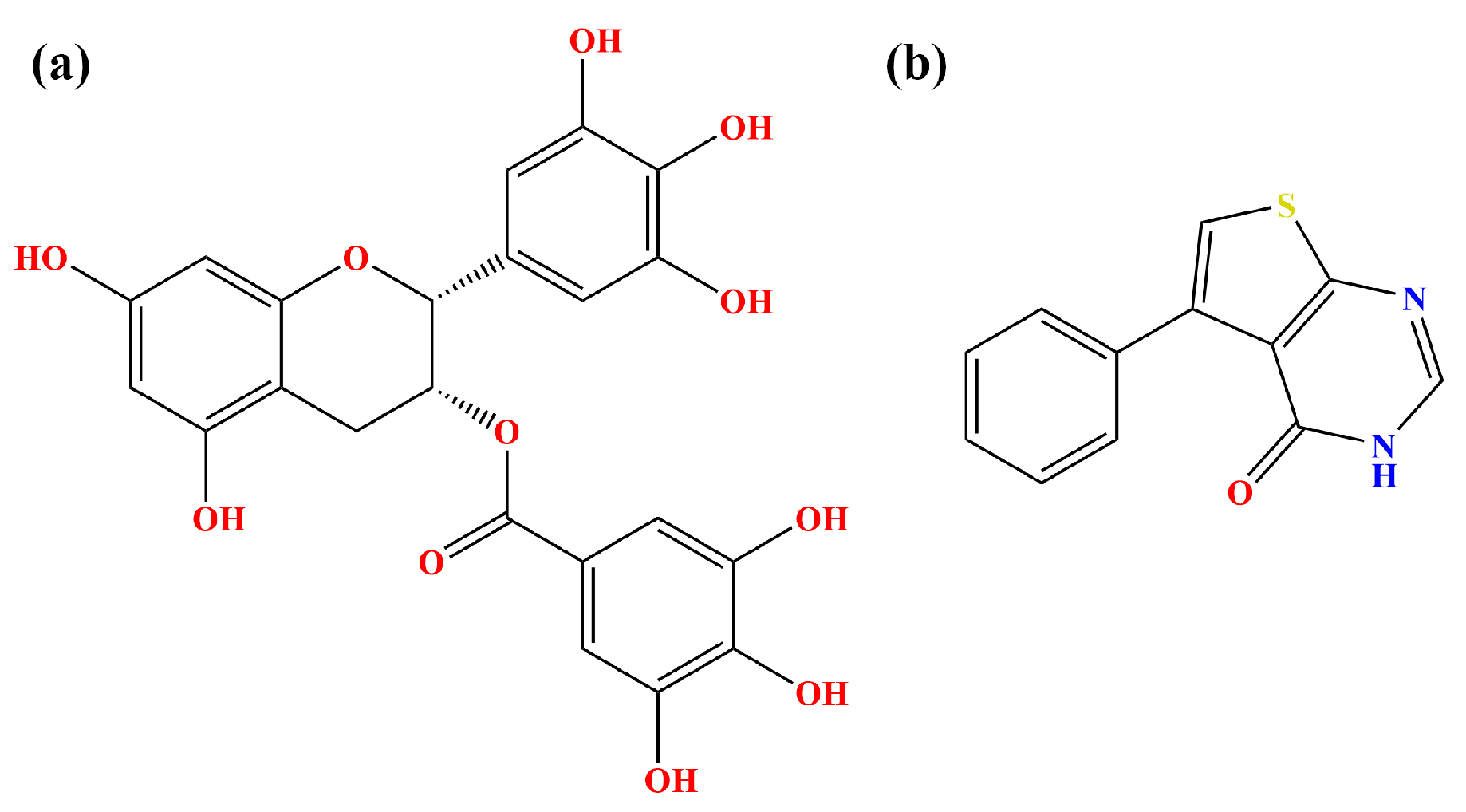
Effect of Fragment 1 on the Binding of Epigallocatechin Gallate to the PD-L1 Dimer Explored by Molecular Dynamics


Abstract
:
1. Introduction
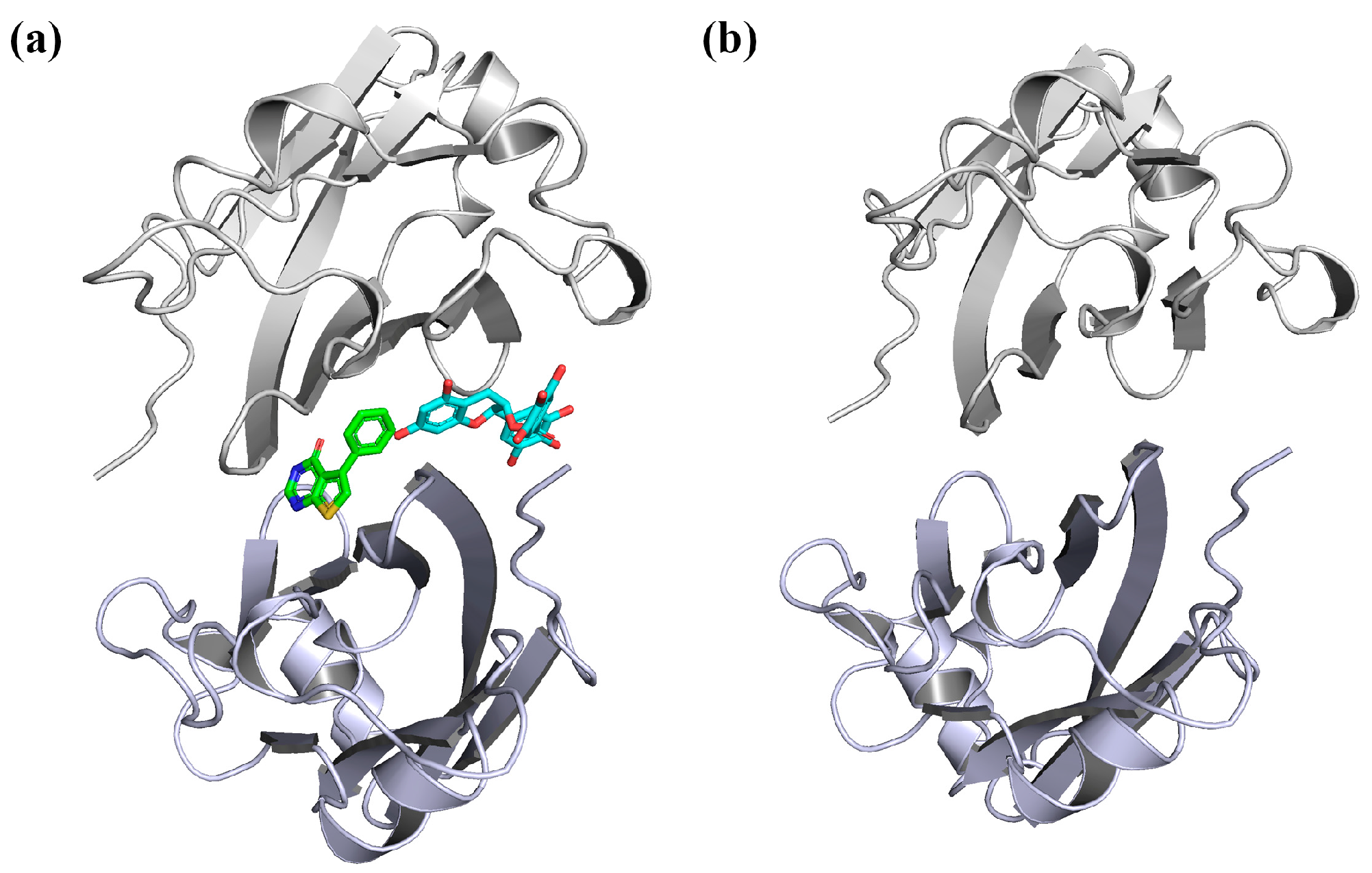
2. Results and Discussion
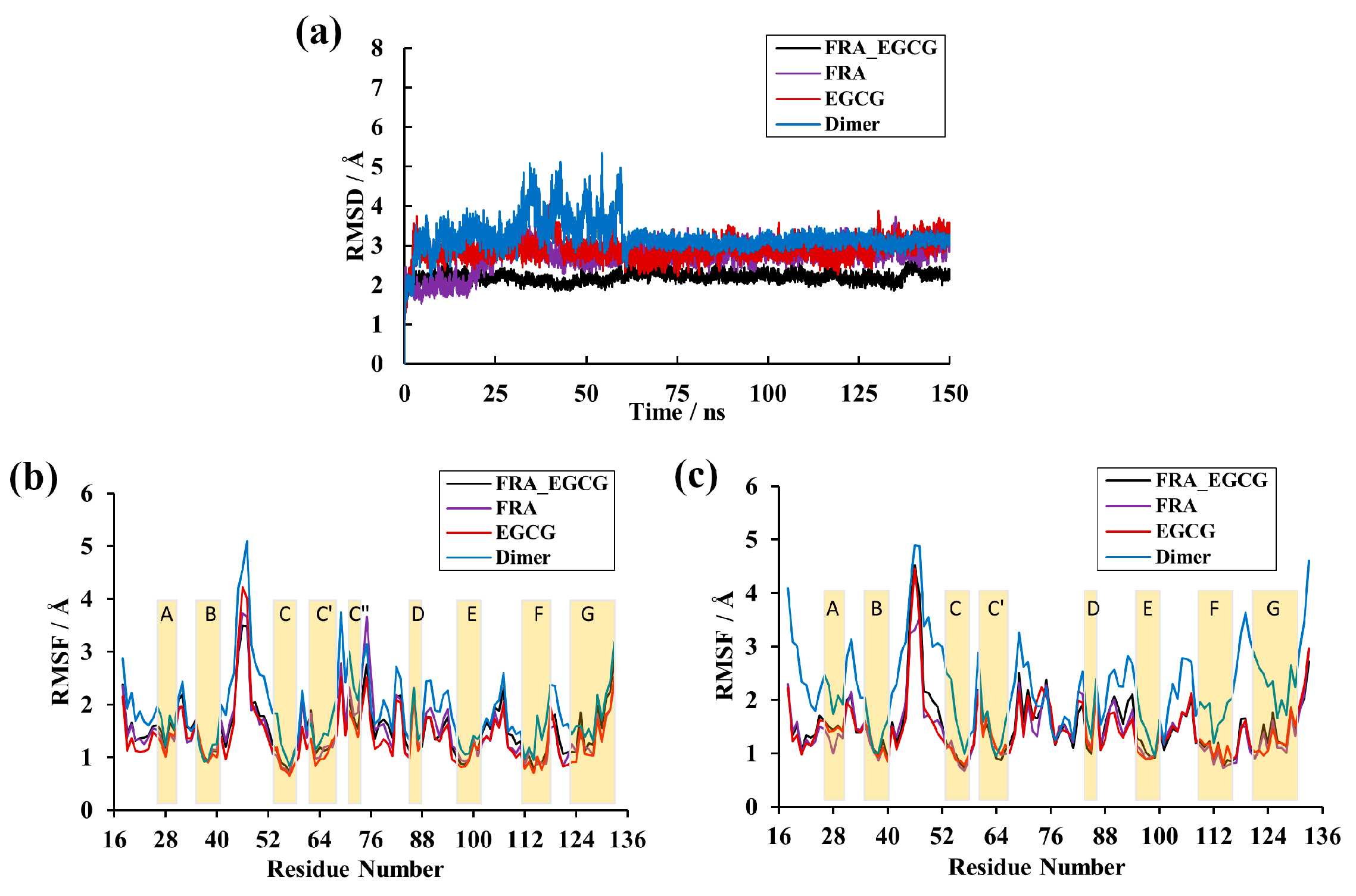
2.1. Root-Mean-Squared Deviation
2.2. Root-Mean-Square Fluctuation
2.3. Binding Free Energy
2.4. Per-Residue Energy Decomposition
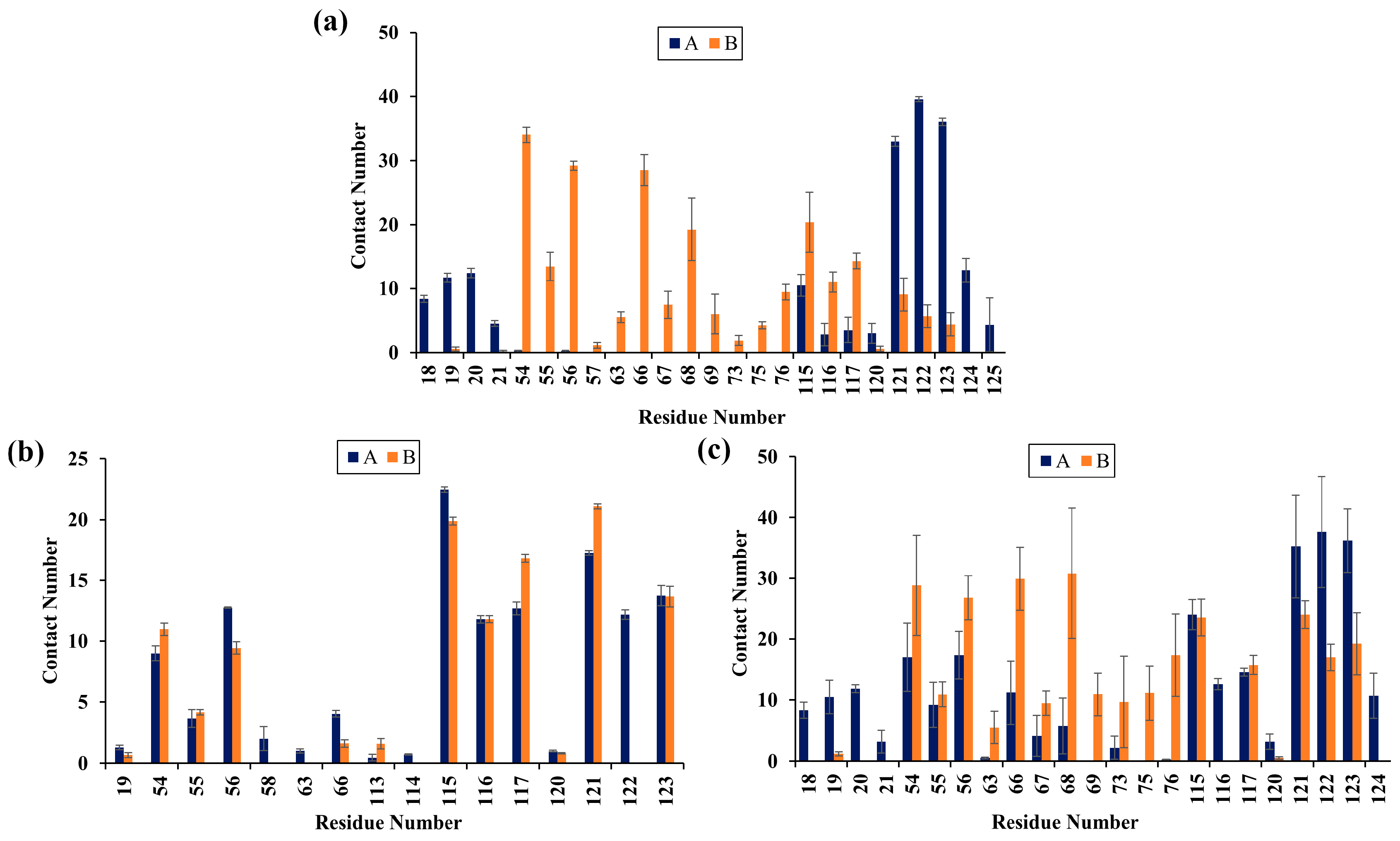
2.5. Contact Numbers
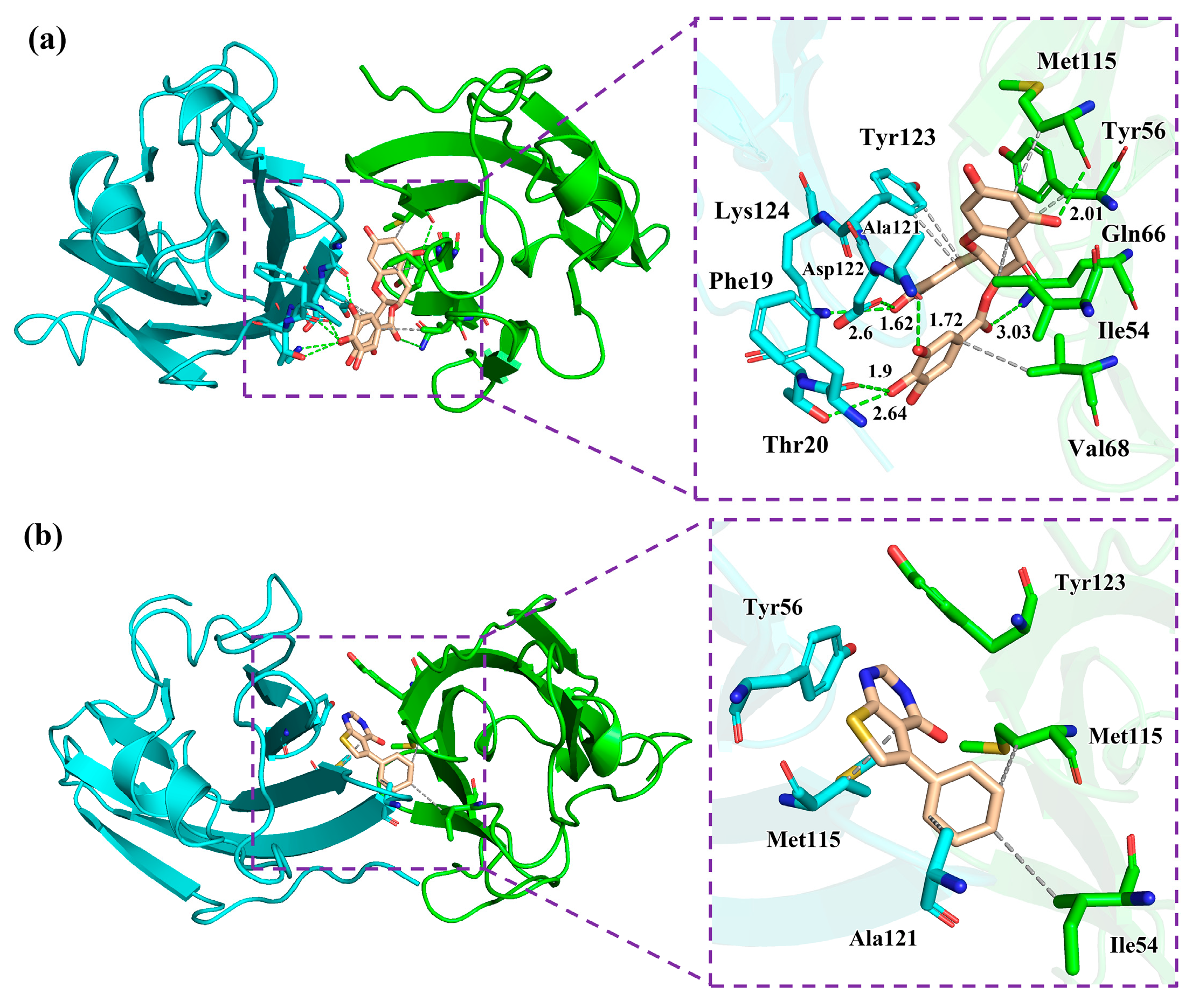
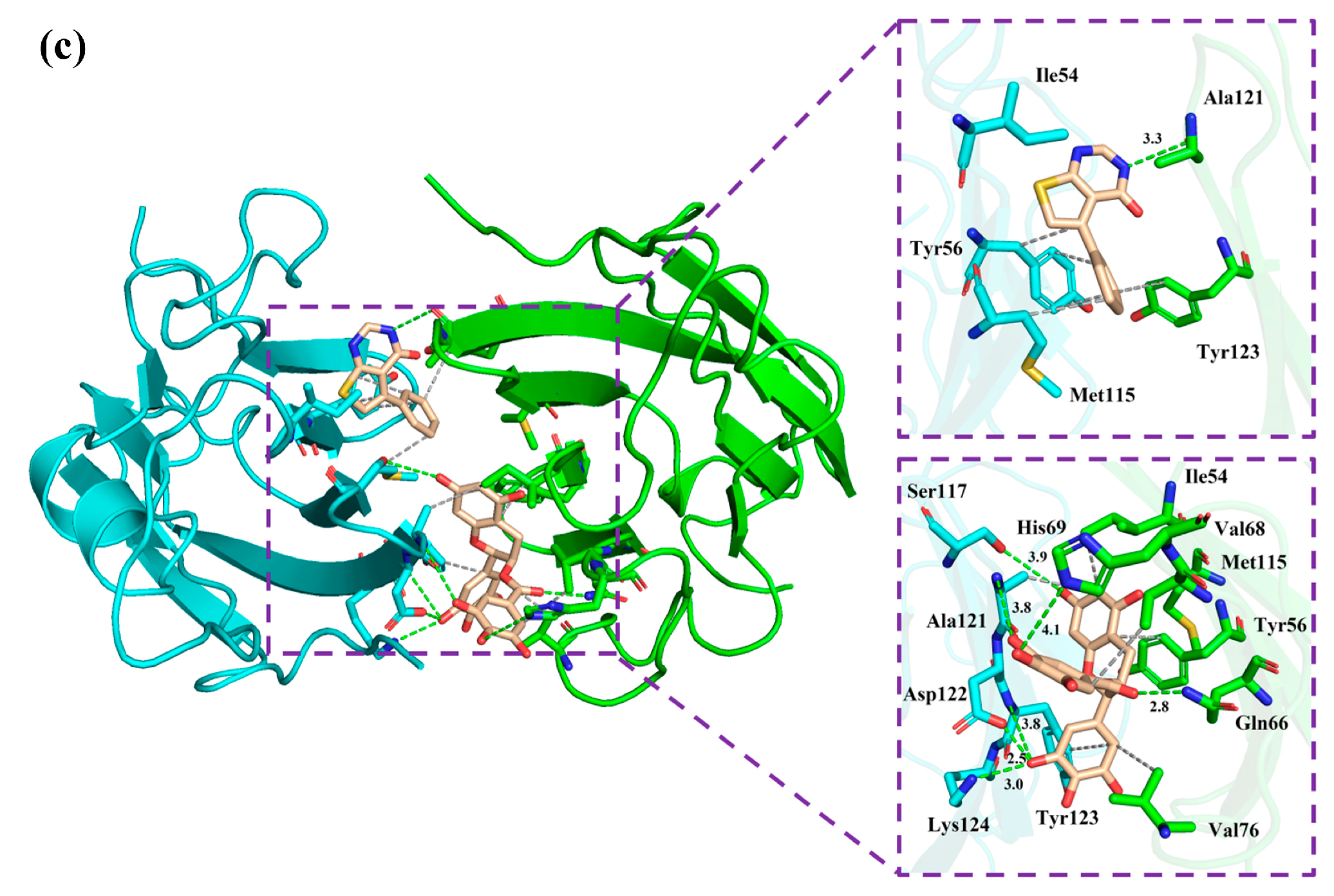
2.6. Non-Bonded Interactions
2.7. Cross-Correlation Matrix Analysis
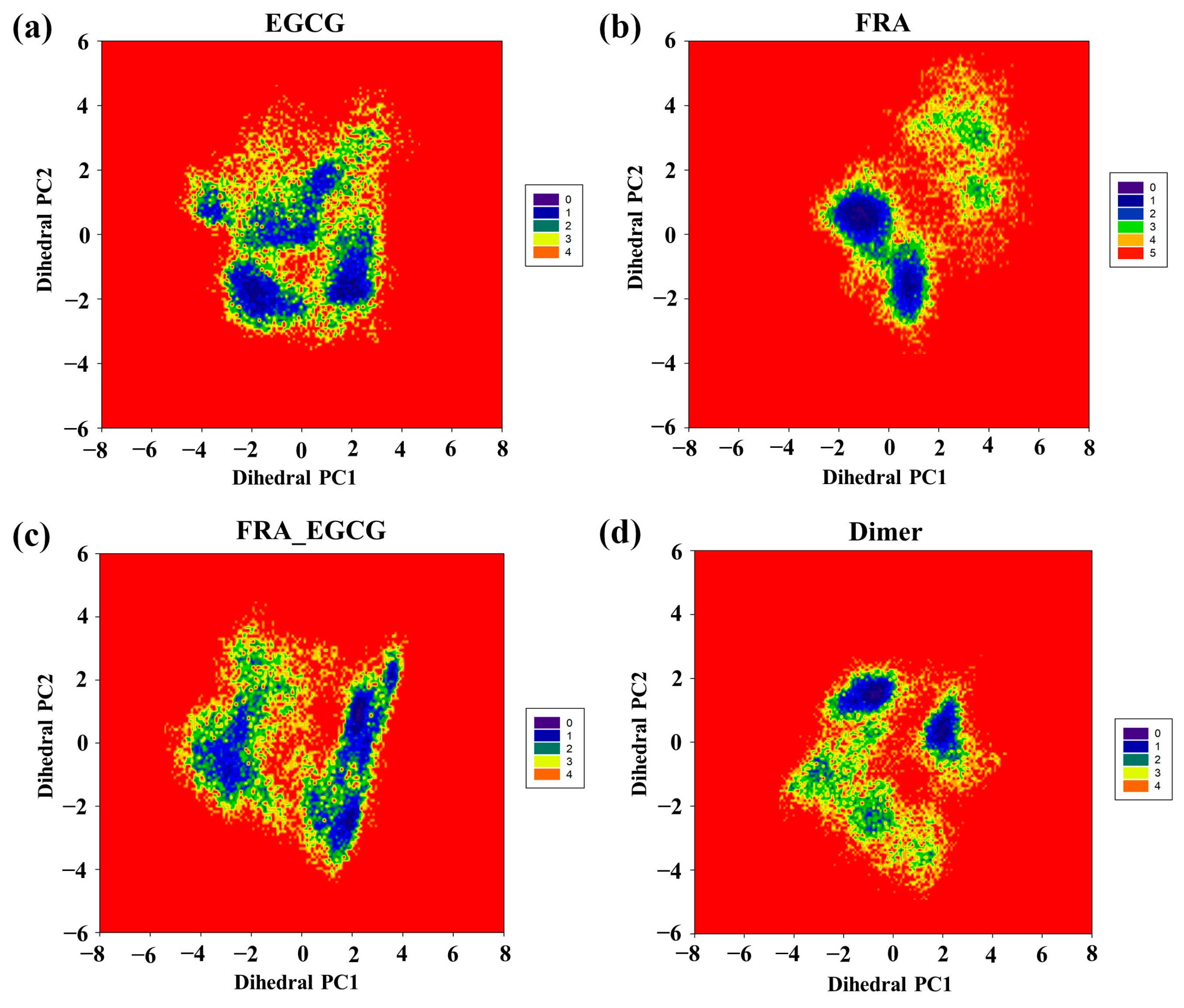
2.8. Free Energy Landscape
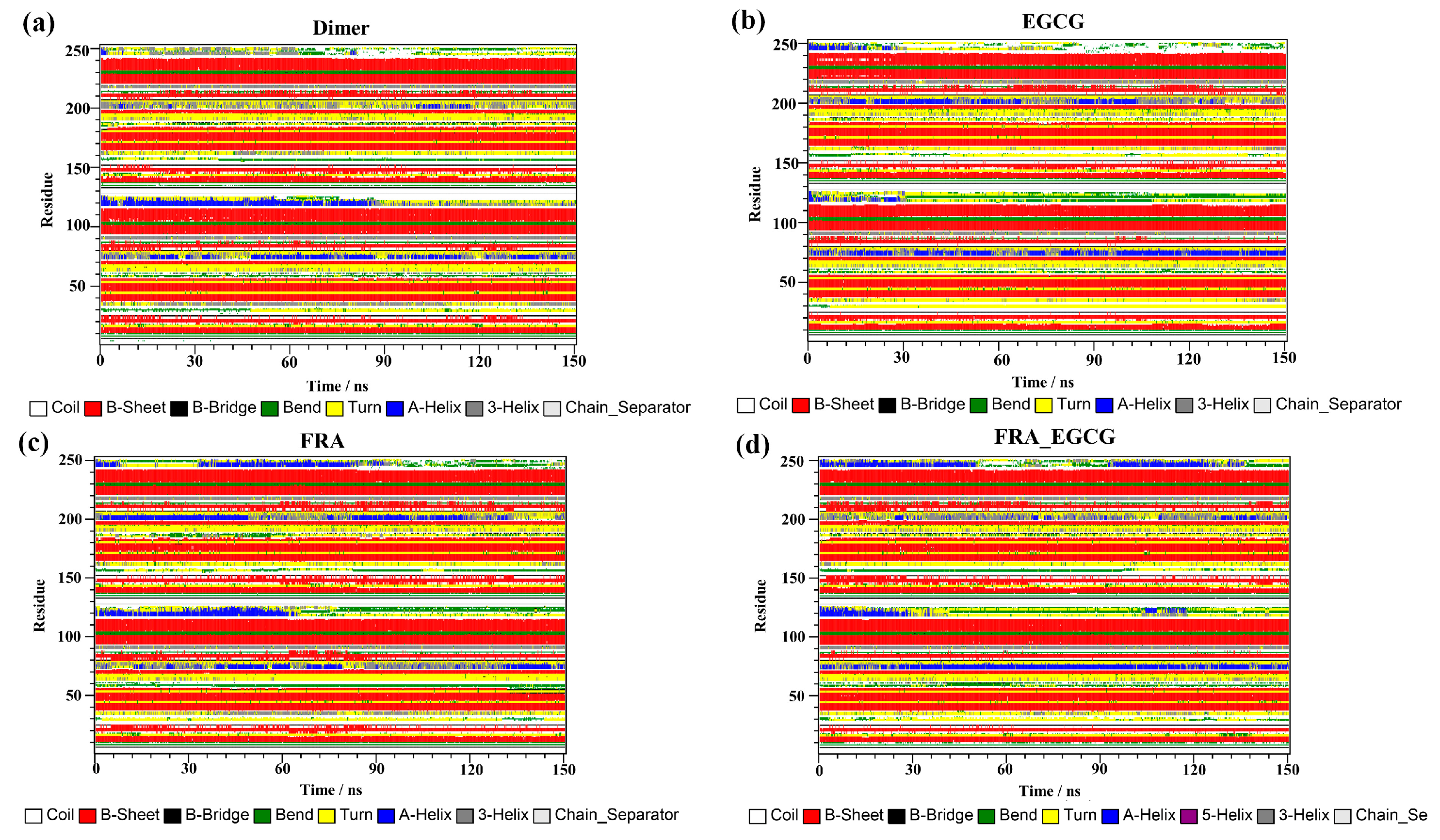
2.9. Secondary Structure
3. Materials and Methods
3.1. Preparation of the Initial Structures and Molecular Docking
3.2. Molecular Dynamics Simulation
3.3. Binding Free Energy Calculation
3.4. Trajectory Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Chen, S.; Song, Z.; Zhang, A. Small-molecule immuno-oncology therapy: Advances, challenges and new directions. Curr. Top. Med. Chem. 2019, 19, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Vanella, V.; Festino, L.; Strudel, M.; Simeone, E.; Grimaldi, A.M.; Ascierto, P.A. PD-L1 inhibitors in the pipeline: Promise and progress. Oncoimmunology 2017, 7, e1365209. [Google Scholar] [CrossRef] [PubMed]
- Kerr, W.G.; Chisholm, J.D. The next generation of immunotherapy for cancer: Small molecules could make big waves. J. Immunol. 2019, 202, 11–19. [Google Scholar] [CrossRef]
- Huck, B.R.; Kotzner, L.; Urbahns, K. Small molecules drive big improvements in immuno-oncology therapies. Angew. Chem. Int. Edit. 2018, 57, 4412–4428. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Yuan, W.E.; Su, J.; Liu, Y.; Chen, J. Recent advances in small molecule based cancer immunotherapy. Eur. J. Med. Chem. 2018, 157, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Torner, R.; Skalniak, L.; Domling, A.; Dubin, G.; Holak, T.A. Small-molecule inhibitors of the programmed cell death-1/programmed death-ligand 1 (PD-1/PD-L1) interaction via transiently induced protein states and dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef]
- Wu, Q.; Jiang, L.; Li, S.C.; He, Q.J.; Yang, B.; Cao, J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol. Sin. 2021, 42, 1–9. [Google Scholar] [CrossRef]
- Hossain, M.; Liu, G.; Dai, B.; Si, Y.; Yang, Q.; Wazir, J.; Birnbaumer, L.; Yang, Y. Reinvigorating exhausted CD8+ cytotoxic T lymphocytes in the tumor microenvironment and current strategies in cancer immunotherapy. Med. Res. Rev. 2021, 41, 156–201. [Google Scholar] [CrossRef]
- Wu, M.; Huang, Q.; Xie, Y.; Wu, X.; Ma, H.; Zhang, Y.; Xia, Y. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J. Hematol. Oncol. 2022, 15, 24. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, L.; Zuo, Y.; Qian, H.; Liu, C. Immune checkpoint blockade in cancer immunotherapy: Mechanisms, clinical outcomes, and safety profiles of PD-1/PD-L1 inhibitors. Arch. Immunol. Ther. Exp. 2020, 68, 1–15. [Google Scholar] [CrossRef]
- Guzik, K.; Tomala, M.; Muszak, D.; Konieczny, M.; Hec, A.; Blaszkiewicz, U.; Pustula, M.; Butera, R.; Domling, A.; Holak, T.A. Development of the inhibitors that target the PD-1/PD-L1 interaction-a brief look at progresson small molecules, peptides and macrocycles. Molecules 2019, 24, 2071. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, Q.; Liu, Z.; Chen, Y.; Feng, F.; Sun, H. Peptide-based and small synthetic molecule inhibitors on PD-1/PD-L1 pathway: A new choice for immunotherapy? Eur. J. Med. Chem. 2019, 161, 378–398. [Google Scholar] [CrossRef] [PubMed]
- Shaabani, S.; Huizinga, H.P.; Butera, R.; Kouchi, A.; Guzik, K.; Magiera-Mularz, K.; Holak, T.A.; Dömling, A. A patent review on PD-1/PD-L1 antagonists: Small molecules, peptides, and macrocycles (2015–2018). Expert Opin. Ther. Pat. 2018, 28, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Ahmed, M.; Okoye, I.; Arutyunova, E.; Babu, D.; Turnbull, W.L.; Kundu, J.K.; Shields, J.; Agopsowicz, K.C.; Xu, L.; et al. Comprehensive in vitro characterization of PD-L1 small molecule inhibitors. Sci. Rep. 2019, 9, 12392. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Baidoo, J.N.E.; Fried, A.; Banerjee, P. Using curcumin to turn the innate immune system against cancer. Biochem. Pharmacol. 2020, 176, 113824. [Google Scholar] [CrossRef] [PubMed]
- Shafabakhsh, R.; Pourhanifeh, M.H.; Mirzaei, H.R.; Sahebkar, A.; Asemi, Z.; Mirzaei, H. Targeting regulatory T cells by curcumin: A potential for cancer immunotherapy. Pharmacol. Res. 2019, 147, 104353. [Google Scholar] [CrossRef]
- Trung, L.Q.; An, D.T.T. Is Resveratrol a Cancer Immunomodulatory Molecule? Front. Pharmacol. 2018, 9, 1255. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Resveratrol targets PD-L1 glycosylation and dimerization to enhance antitumor T-cell immunity. Aging 2020, 12, 8. [Google Scholar] [CrossRef]
- Deng, L.J.; Qi, M.; Li, N.; Lei, Y.H.; Zhang, D.M.; Chen, J.X. Natural products and their derivatives: Promising modulators of tumor immunotherapy. J. Leukoc. Biol. 2020, 108, 493–508. [Google Scholar] [CrossRef]
- Ri, M.H.; Ma, J.; Jin, X. Development of natural products for anti-PD-1/PD-L1 immunotherapy against cancer. J. Ethnopharmacol. 2021, 281, 114370. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, Y.S.; Choi, J.G.; Li, W.; Lee, E.J.; Park, J.W.; Song, J.; Chung, H.S. Kaempferol and its glycoside, kaempferol 7-O-rhamnoside, inhibit PD-1/PD-L1 interaction in vitro. Int. J. Mol. Sci. 2020, 21, 3239. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.G.; Kim, Y.S.; Kim, J.H.; Kim, T.I.; Li, W.; Oh, T.W.; Jeon, C.H.; Kim, S.J.; Chung, H.S. Anticancer effect of Salvia plebeia and its active compound by improving T-cell activity via blockade of PD-1/PD-L1 interaction in humanized PD-1 mouse model. Front. Immunol. 2020, 11, 598556. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kim, T.I.; Kim, J.H.; Chung, H.S. Immune checkpoint PD-1/PD-L1 CTLA-4/CD80 are blocked by Rhus verniciflua stokes and its active compounds. Molecules 2019, 24, 4062. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Gao, Y.; He, T.; Wang, D.; Guo, N.; Zhang, X.; Chen, S.; Wang, H. PD-1/PD-L1 inhibitor screening of caffeoylquinic acid compounds using surface plasmon resonance spectroscopy. Anal. Biochem. 2018, 547, 52–56. [Google Scholar] [CrossRef]
- Guo, Y.; Liang, J.; Liu, B.; Jin, Y. Molecular mechanism of food-derived polyphenols on PD-L1 dimerization: A molecular dynamics simulation study. Int. J. Mol. Sci. 2021, 22, 10924. [Google Scholar] [CrossRef]
- Perry, E.; Mills, J.J.; Zhao, B.; Wang, F.; Sun, Q.; Christov, P.P.; Tarr, J.C.; Rietz, T.A.; Olejniczak, E.T.; Lee, T.; et al. Fragment-based screening of programmed death ligand 1 (PD-L1). Bioorg. Med. Chem. Lett. 2019, 29, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.W.; Li, J.; Zhao, Z.; Zhang, L.; Peng, G.; Liang, L. Molecular dynamics study on the mechanism of polynucleotide encapsulation by chitosan. Sci. Rep. 2017, 7, 5050. [Google Scholar] [CrossRef]
- Niu, Y.; Shi, D.; Li, L.; Guo, J.; Liu, H.; Yao, X. Revealing inhibition difference between PFI-2 enantiomers against SETD7 by molecular dynamics simulations, binding free energy calculations and unbinding pathway analysis. Sci. Rep. 2017, 7, 46547. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep. 2020, 10, 17716. [Google Scholar] [CrossRef]
- Cao, X.; Zao, X.; Xue, B.; Chen, H.; Zhang, J.; Li, S.; Li, X.; Zhu, S.; Guo, R.; Li, X.; et al. The mechanism of TiaoGanYiPi formula for treating chronic hepatitis B by network pharmacology and molecular docking verification. Sci. Rep. 2021, 1, 8402. [Google Scholar] [CrossRef]
- Zhu, J.; Lv, Y.; Han, X.; Xu, D.; Han, W. Understanding the differences of the ligand binding/unbinding pathways between phosphorylated and non-phosphorylated ARH1 using molecular dynamics simulations. Sci. Rep. 2017, 7, 12439. [Google Scholar] [CrossRef]
- Aarthy, M.; Panwar, U.; Singh, S.K. Structural dynamic studies on identification of EGCG analogues for the inhibition of Human Papillomavirus E7. Sci. Rep. 2020, 10, 8661. [Google Scholar] [CrossRef]
- Guo, Y.; Jin, Y.; Wang, B.; Liu, B. Molecular Mechanism of Small-Molecule Inhibitors in Blocking the PD-1/PD-L1 Pathway through PD-L1 Dimerization. Int. J. Mol. Sci. 2021, 22, 4766. [Google Scholar] [CrossRef]
- Jung, S.W.; Cho, A.E.; Yu, W. Exploring the ligand efficacy of cannabinoid receptor 1 (CB1) using molecular dynamics simulations. Sci. Rep. 2018, 8, 13787. [Google Scholar] [CrossRef]
- Skalniak, L.; Zak, K.M.; Guzik, K.; Magiera, K.; Musielak, B.; Pachota, M.; Szelazek, B.; Kocik, J.; Grudnik, P.; Tomala, M.; et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 2017, 8, 72167. [Google Scholar] [CrossRef]
- Wang, T.; Cai, S.; Cheng, Y.; Zhang, W.; Wang, M.; Sun, H.; Guo, B.; Li, Z.; Xiao, Y.; Jiang, S. Discovery of small-molecule inhibitors of the PD-1/PD-L1 axis that promote PD-L1 internalization and degradation. J. Med. Chem. 2022, 65, 3879–3893. [Google Scholar] [CrossRef]
- Kitel, R.; Rodríguez, I.; del Corte, X.; Atmaj, J.; Żarnik, M.; Surmiak, E.; Muszak, D.; Magiera-Mularz, K.; Popowicz, G.M.; Holak, T.A.; et al. Exploring the surface of the ectodomain of the PD-L1 immune checkpoint with small-molecule fragments. ACS Chem. Biol. 2022, 17, 2655–2663. [Google Scholar] [CrossRef]
- Valente, R.; Souza, R.C.; de Medeiros Muniz, G.; Ferreira, J.E.V.; de Miranda, R.M.; AHL, E.L.; Vianez Junior, J. Using accelerated molecular dynamics simulation to elucidate the effects of the T198F mutation on the molecular flexibility of the west nile virus envelope protein. Sci. Rep. 2020, 10, 9625. [Google Scholar] [CrossRef]
- Guan, S.; Wang, T.; Kuai, Z.; Qian, M.; Tian, X.; Zhang, X.; Yu, Y.; Wang, S.; Zhang, H.; Li, H.; et al. Exploration of binding and inhibition mechanism of a small molecule inhibitor of influenza virus H1N1 hemagglutinin by molecular dynamics simulation. Sci. Rep. 2017, 7, 3786. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhou, S.; Chen, Q.; Wang, L.; Liang, Z.; Wang, J. Understanding the molecular mechanism for the differential inhibitory activities of compounds against MTH1. Sci. Rep. 2017, 7, 40557. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 6, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Dis. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 16, 119. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 2018, 10, 1039. [Google Scholar] [CrossRef] [PubMed]
- Spivak, M.; Stone, J.E.; Ribeiro, J.; Saam, J.; Freddolino, P.L.; Bernardi, R.C.; Tajkhorshid, E. VMD as a platform for interactive small molecule preparation and visualization in quantum and classical simulations. J. Chem. Inf. Model. 2023, 63, 4664–4678. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.S.; Paidesetty, S.K.; Dehury, B.; Sahoo, J.; Vedithi, S.C.; Mahapatra, N.; Hussain, T.; Padhy, R.N. Molecular docking and simulation study for synthesis of alternative dapsone derivative as a newer antileprosy drug in multidrug therapy. J. Cell. Biochem. 2018, 119, 9838–9852. [Google Scholar] [CrossRef]
- Dehury, B.; Behera, S.K.; Mahapatra, N. Structural dynamics of casein kinase I (CKI) from malarial parasite plasmodium falciparum (isolate 3D7): Insights from theoretical modelling and molecular simulations. J. Mol. Graph. Model. 2017, 71, 154–166. [Google Scholar] [CrossRef]
- Wang, R.; Liu, W.; Zhou, L.; Ma, Y.; Wang, R. Probing the acting mode and advantages of RMC-4550 as a src-homology 2 domain-containing protein tyrosine phosphatase (SHP2) inhibitor at molecular level through molecular docking and molecular dynamics. J. Biomol. Struct. Dyn. 2020, 38, 1525–1538. [Google Scholar] [CrossRef]
- Al-Khafaji, K.; Tok, T.T. Molecular dynamics simulation, free energy landscape and binding free energy computations in exploration the anti-invasive activity of amygdalin against metastasis. Comput. Methods Programs Biomed. 2020, 195, 105660. [Google Scholar] [CrossRef]
- Oshima, H.; Re, S.; Sugita, Y. Replica-exchange umbrella sampling combined with Gaussian accelerated molecular dynamics for free-energy calculation of biomolecules. J. Chem. Theory Comput. 2019, 15, 5199–5208. [Google Scholar] [CrossRef] [PubMed]
- Capelli, R.; Bochicchio, A.; Piccini, G.; Casasnovas, R.; Carloni, P.; Parrinello, M. Chasing the full free energy landscape of neuroreceptor/ligand unbinding by metadynamics simulations. J. Chem. Theory Comput. 2019, 15, 3354–3361. [Google Scholar] [CrossRef]
- Shahzaib, A.; Dinesh, G.; Vijay, K. Targeting SARS-CoV-2 nucleocapsid oligomerization: Insights from molecular docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2022, 40, 2430–2443. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contribution | EGCG | FRA | FRA_EGCG | Dimer |
|---|---|---|---|---|
| ΔEvdw a | −42.88 ± 1.70 | −37.14 ± 0.51 | −74.75 ± 2.19 | −44.59 ± 9.85 |
| ΔEele b | −23.14 ± 3.14 | −3.43 ± 0.13 | −32.17 ± 2.29 | −124.35 ± 23.36 |
| ΔEPB c | 50.43 ± 3.76 | 19.43 ± 0.83 | 76.89 ± 2.94 | 211.28 ± 17.07 |
| ΔESA d | −4.73 ± 0.16 | −3.14 ± 0.02 | −7.48 ± 0.28 | −6.23 ± 0.37 |
| ΔEpolar,total e | 27.29 ± 1.42 | 16.00 ± 0.97 | 44.72 ± 2.31 | 86.94 ± 10.00 |
| ΔEnonpolar,total f | −47.61 ± 1.75 | −40.28 ± 0.53 | −82.23 ± 2.35 | −50.82 ± 10.06 |
| ΔG g | −20.31 ± 0.35 | −24.27 ± 0.27 | −37.51 ± 0.43 | 36.11 ± 0.89 |
| Donor | Donor H | Acceptor | Occupancy (%) |
|---|---|---|---|
| EGCG@O50 | H50 | AAla121@O | 80.40 |
| EGCG@O10 | H10 | AAsp122@N | 59.80 |
| EGCG@O47 | H47 | APhe19@O | 59.47 |
| EGCG@O10 | H10 | AAsp122@OD1 | 39.53 |
| EGCG@O03 | H03 | BMet115@O | 43.52 |
| Donor | Donor H | Acceptor | Occupancy (%) |
|---|---|---|---|
| EGCG@O10 | H10 | AAsp122@OD2 | 100.00 |
| EGCG@O50 | H50 | AAsp122@OD2 | 93.38 |
| EGCG@O47 | H47 | AThr20@OG1 | 23.84 |
| ALys124@NZ | HZ1 | EGCG@O10 | 17.55 |
| EGCG@O03 | H03 | BGln66@OE1 | 80.64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Guo, Y.; Guo, Z.; Liu, B.; Xu, J. Effect of Fragment 1 on the Binding of Epigallocatechin Gallate to the PD-L1 Dimer Explored by Molecular Dynamics. Molecules 2023, 28, 7881. https://doi.org/10.3390/molecules28237881
Guo Y, Guo Y, Guo Z, Liu B, Xu J. Effect of Fragment 1 on the Binding of Epigallocatechin Gallate to the PD-L1 Dimer Explored by Molecular Dynamics. Molecules. 2023; 28(23):7881. https://doi.org/10.3390/molecules28237881
Chicago/Turabian StyleGuo, Yan, Yilin Guo, Zichao Guo, Boping Liu, and Jianguo Xu. 2023. "Effect of Fragment 1 on the Binding of Epigallocatechin Gallate to the PD-L1 Dimer Explored by Molecular Dynamics" Molecules 28, no. 23: 7881. https://doi.org/10.3390/molecules28237881
APA StyleGuo, Y., Guo, Y., Guo, Z., Liu, B., & Xu, J. (2023). Effect of Fragment 1 on the Binding of Epigallocatechin Gallate to the PD-L1 Dimer Explored by Molecular Dynamics. Molecules, 28(23), 7881. https://doi.org/10.3390/molecules28237881