
Reusable Glucose-Based Crown Ethers Anchored to PVC
 ,
,
Abstract
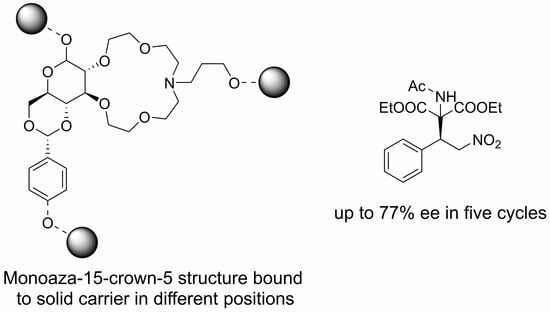
:
1. Introduction
2. Results and Discussion
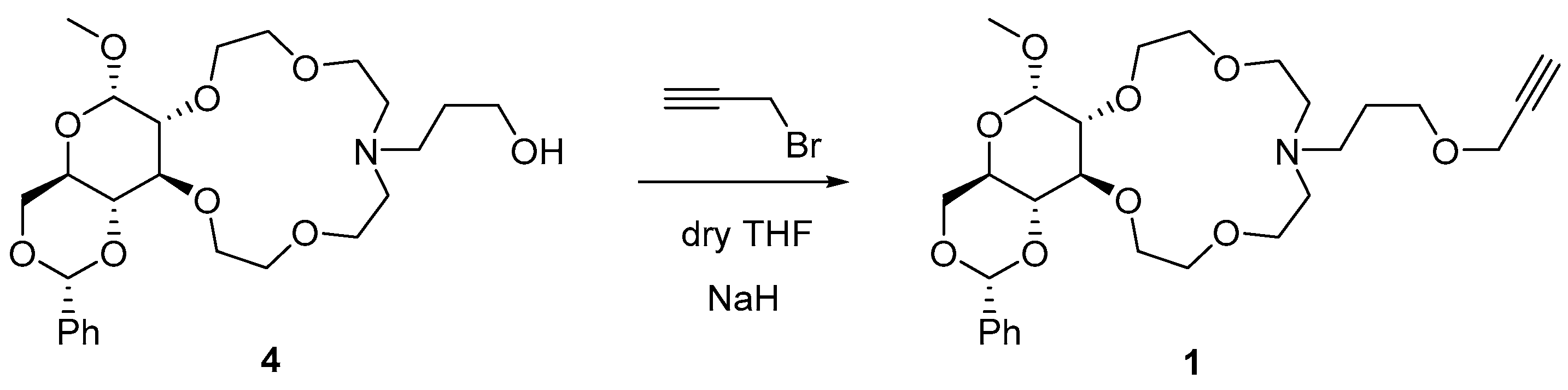
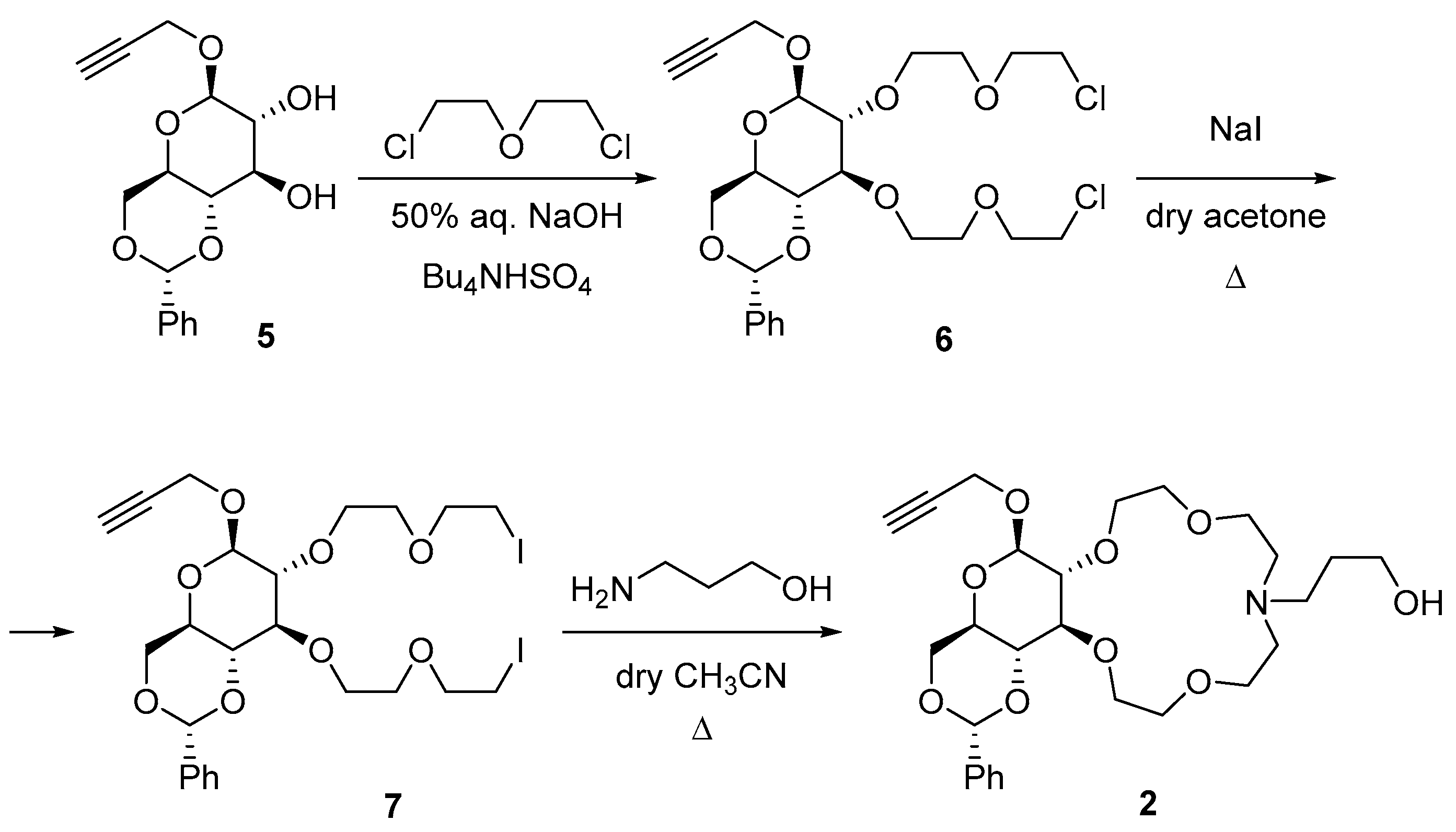
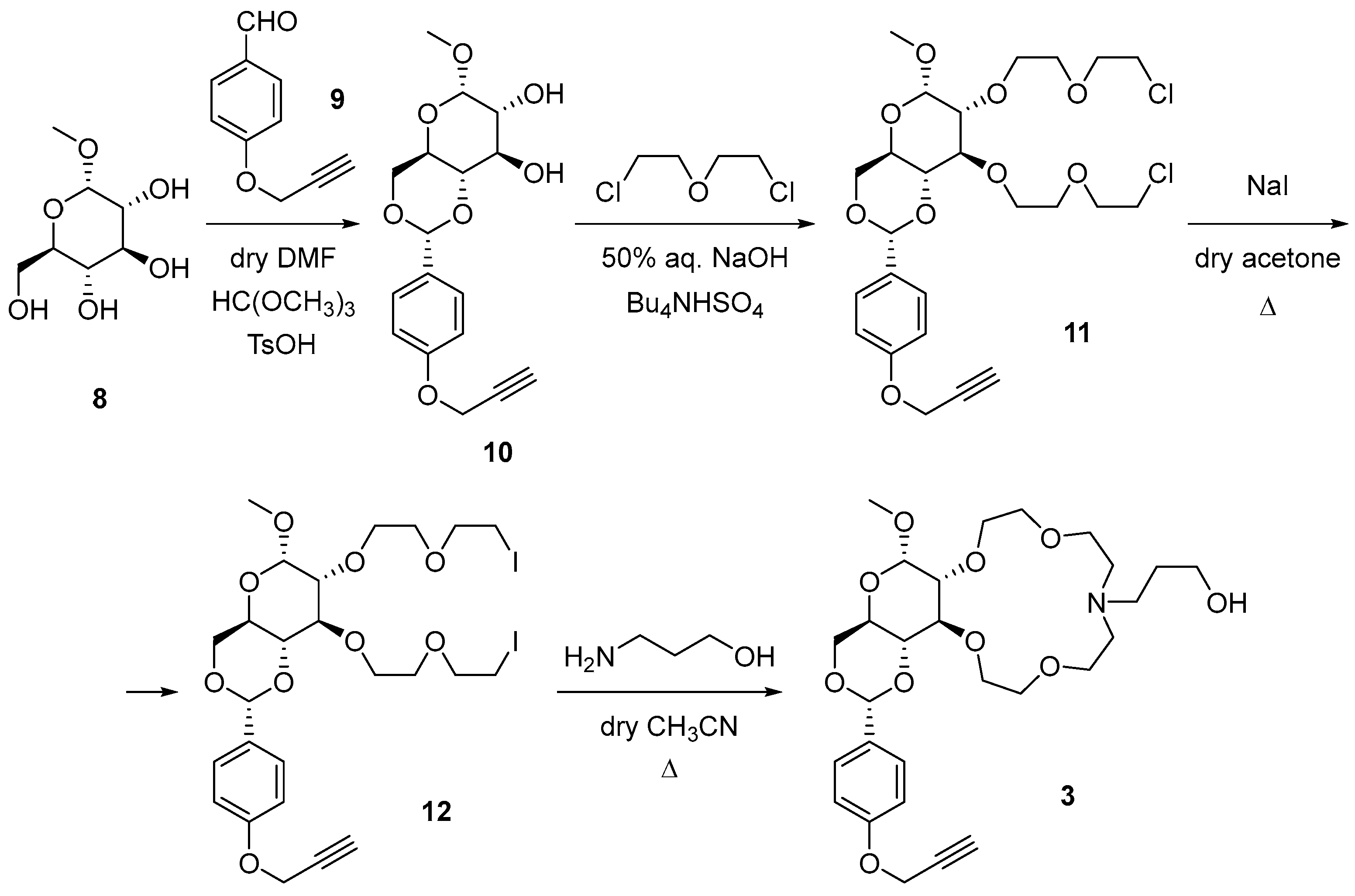
2.1. Synthesis of Glucose-Based Crown Ethers Bearing a Propargyl Group
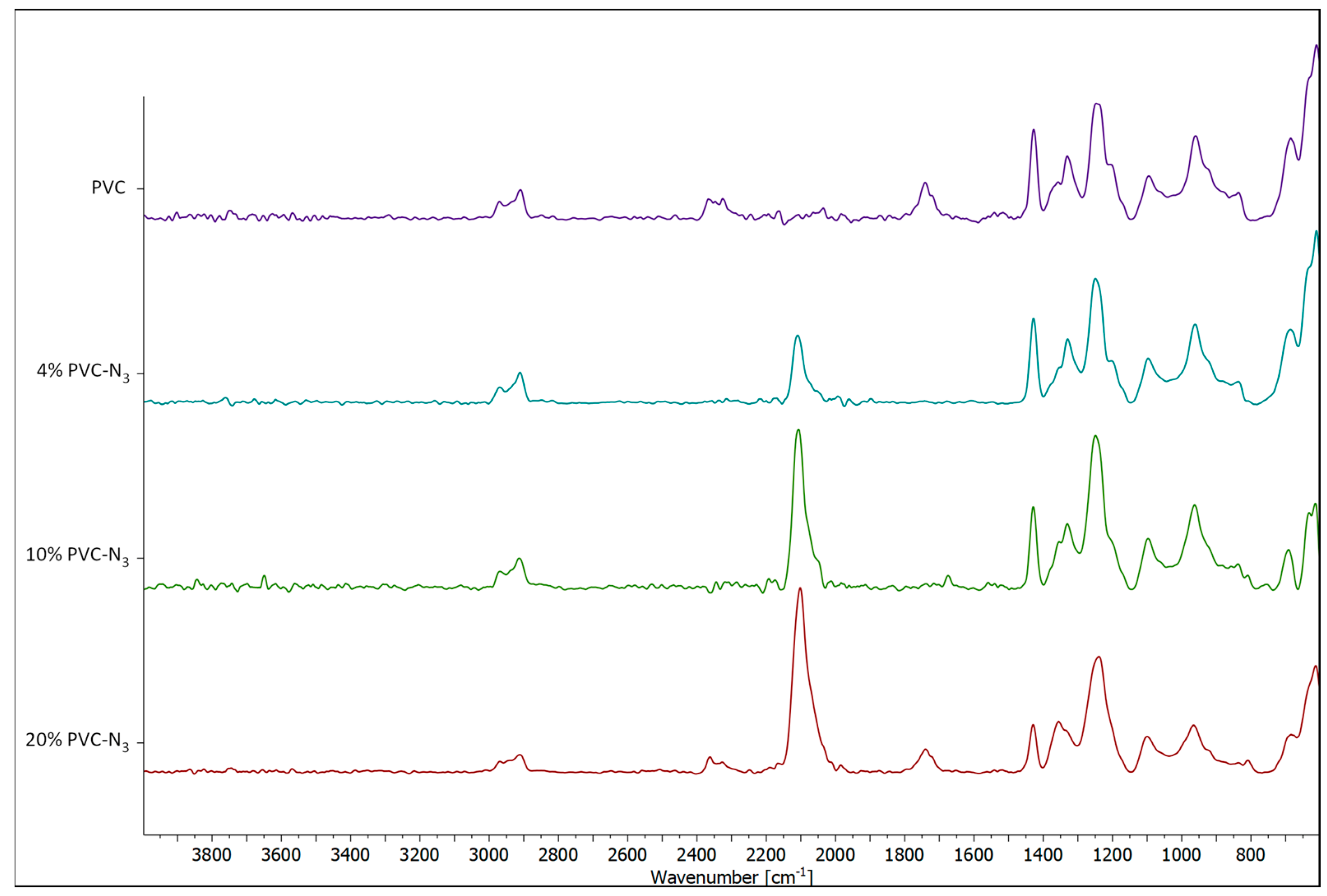
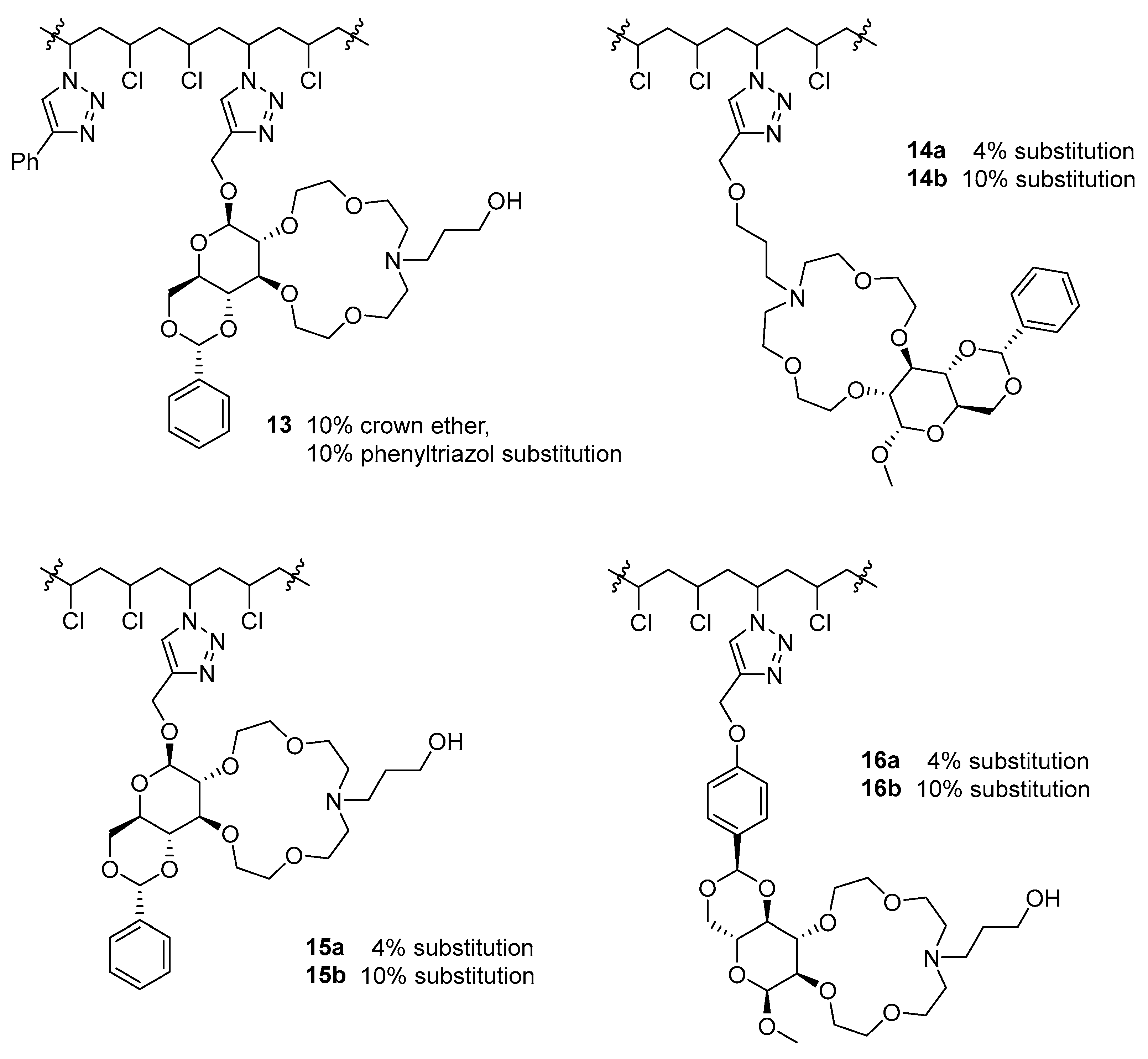
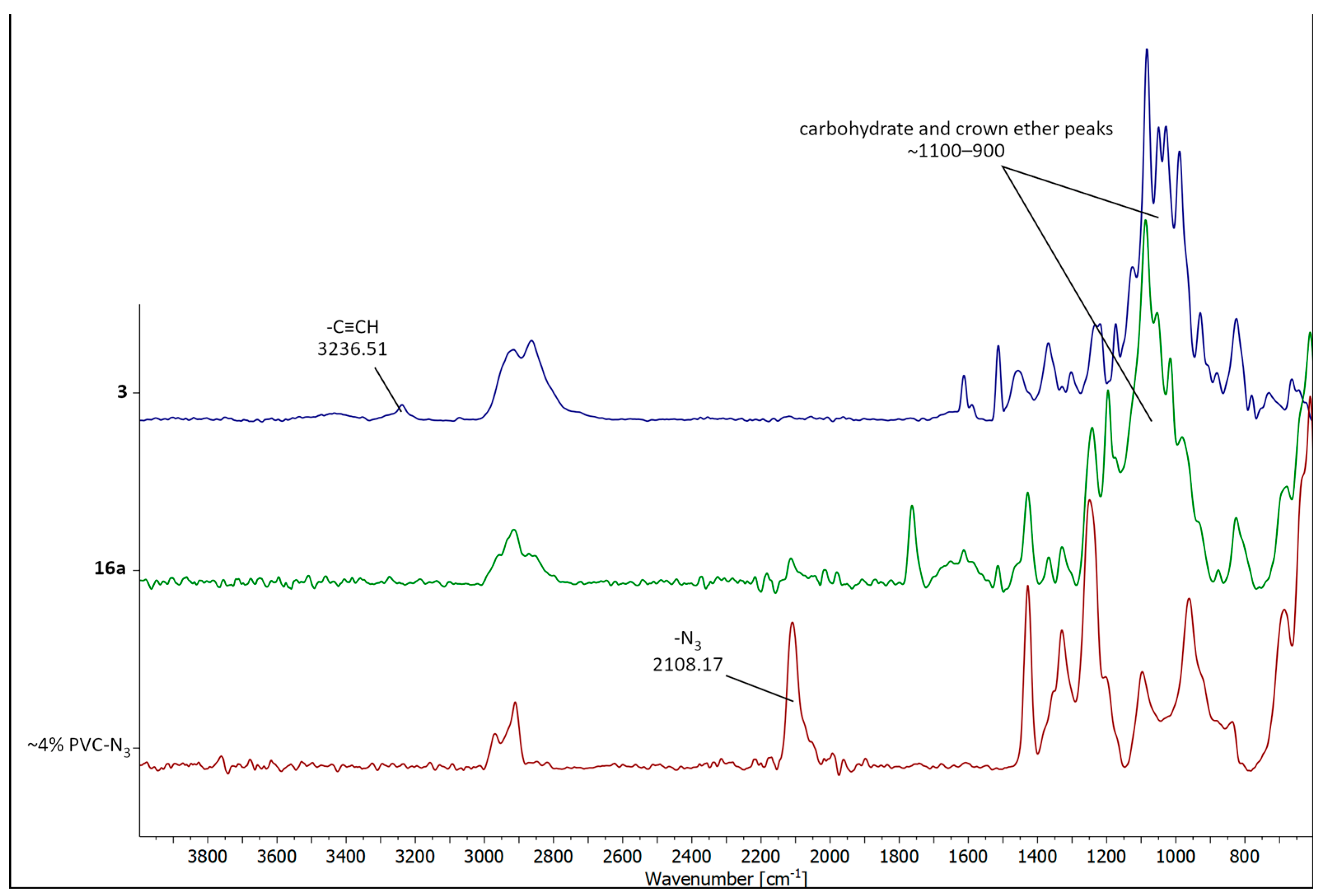
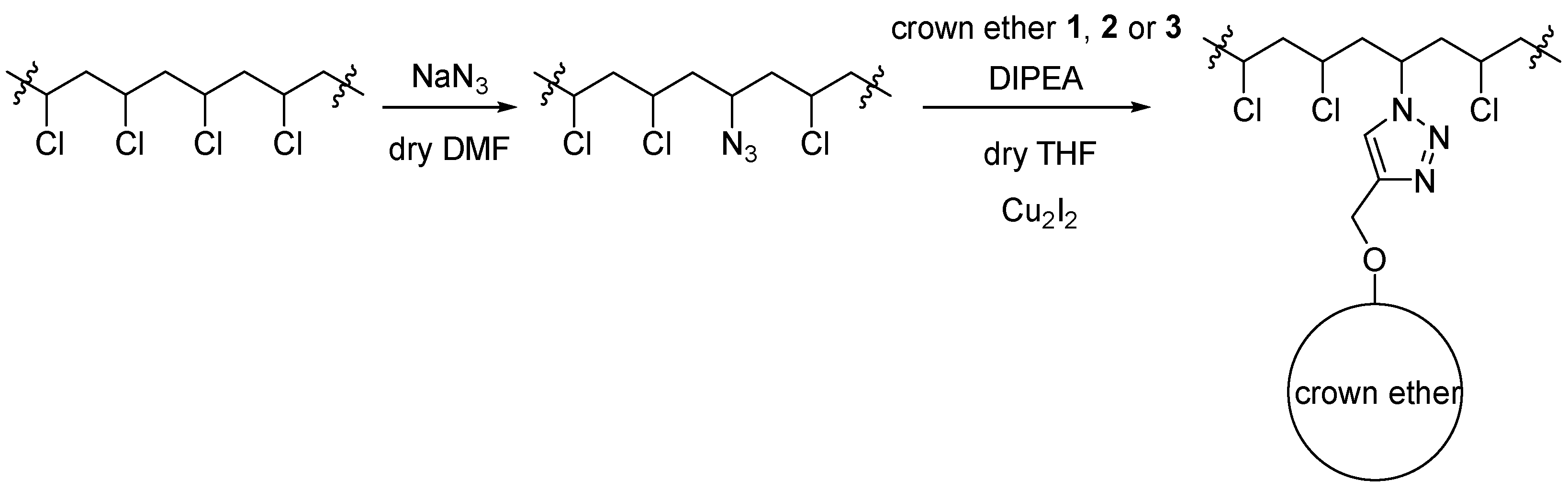
2.2. Immobilizing the Chiral Macrocycles on PVC
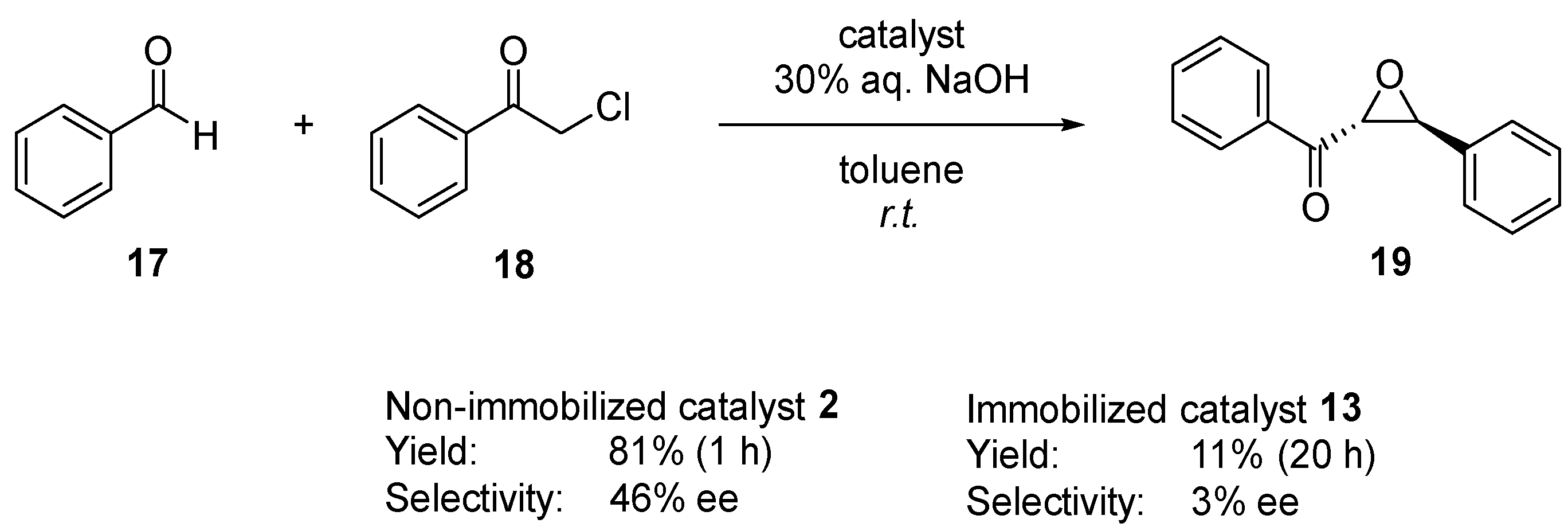
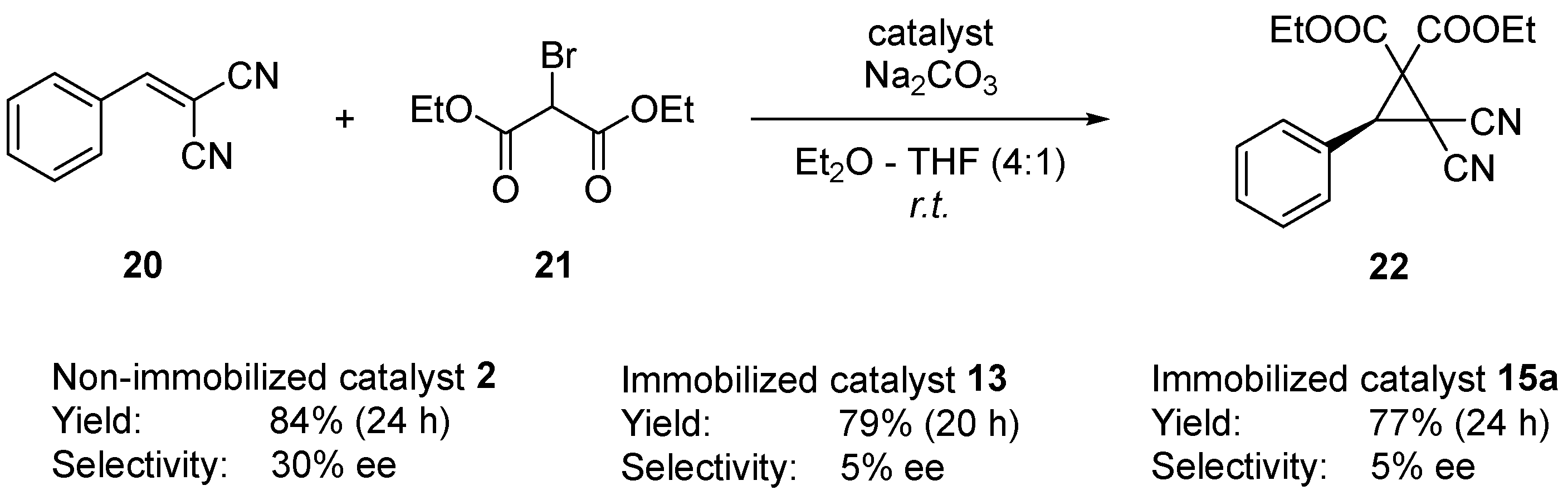
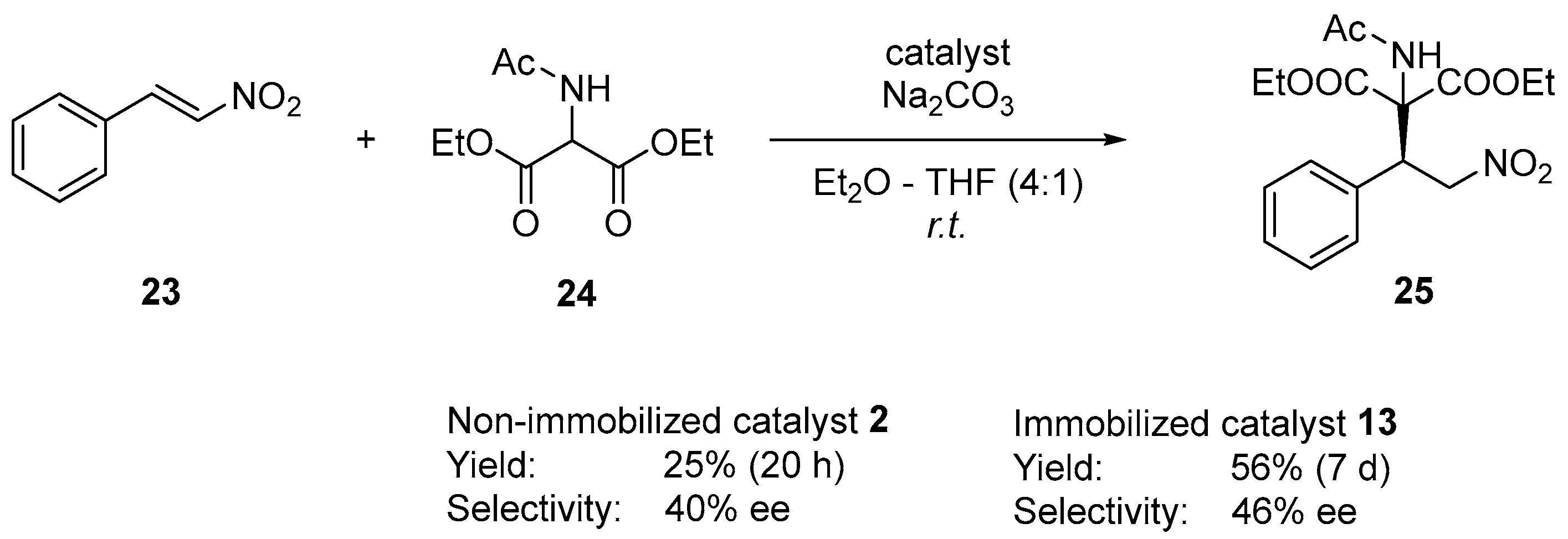
2.3. Examination of Different Phase Transfer Catalytic Systems
2.4. Catalyst Evaluation
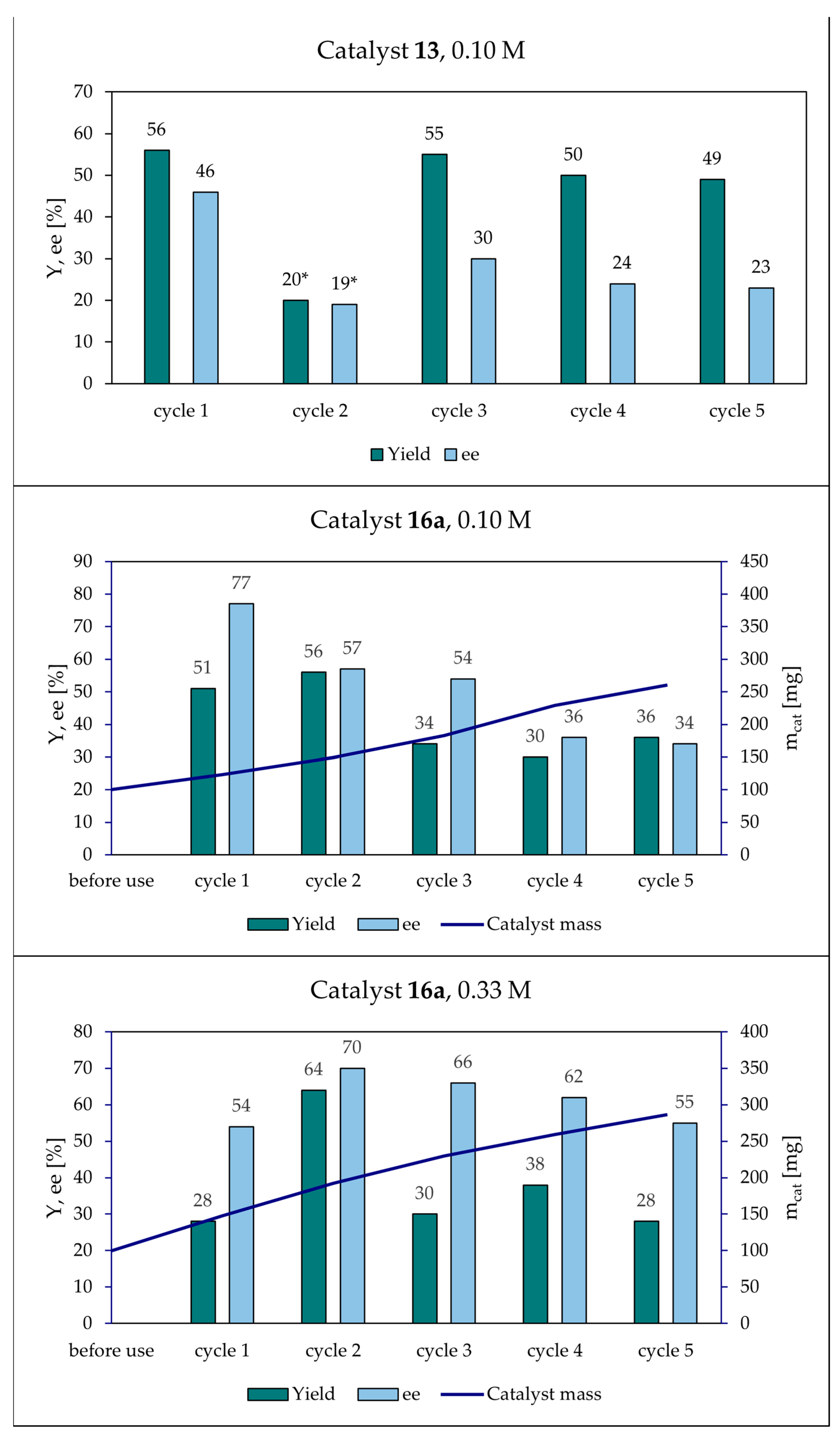
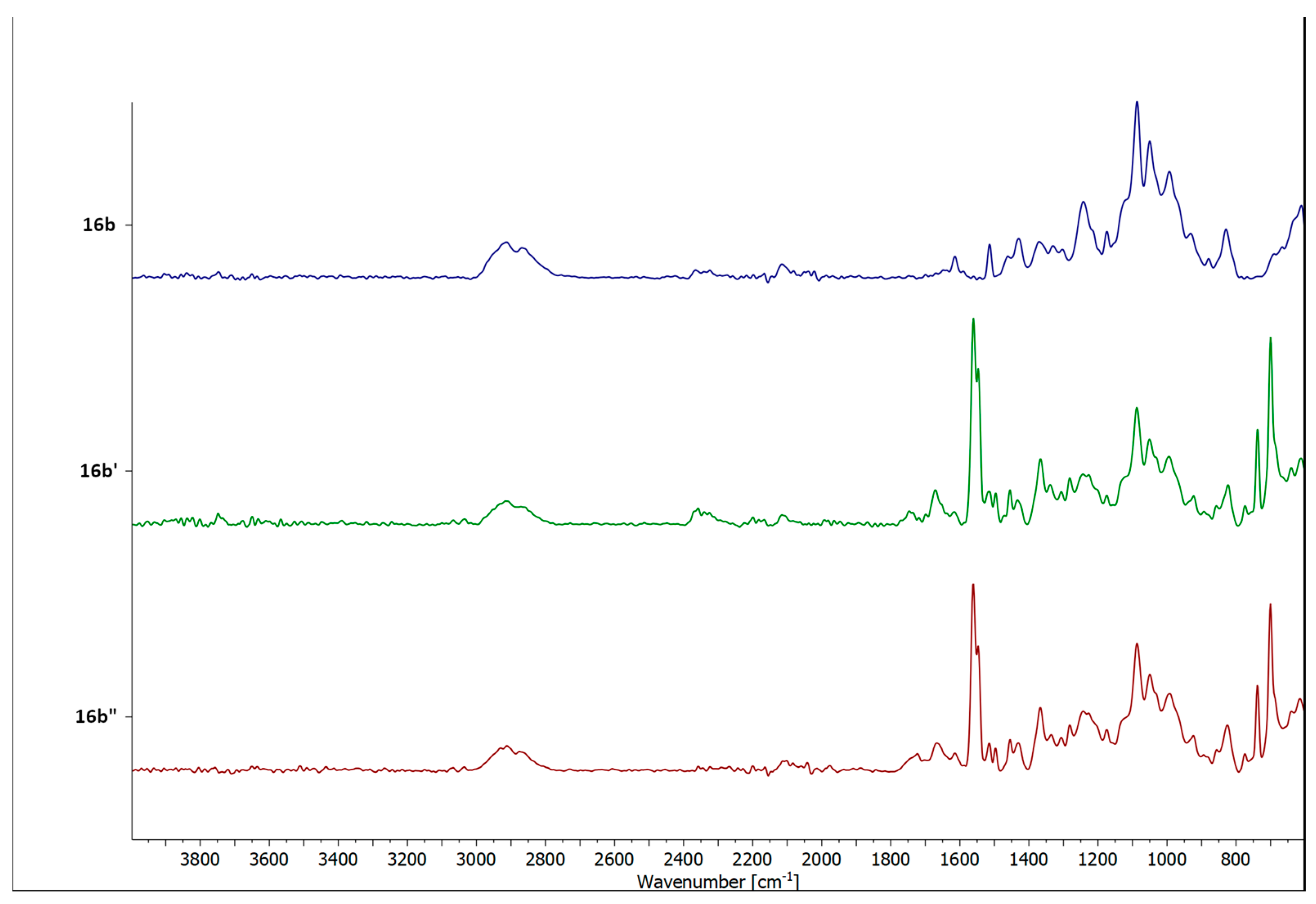
2.5. Catalyst Recovery Experiments
3. Conclusions
4. Materials and Methods
4.1. General
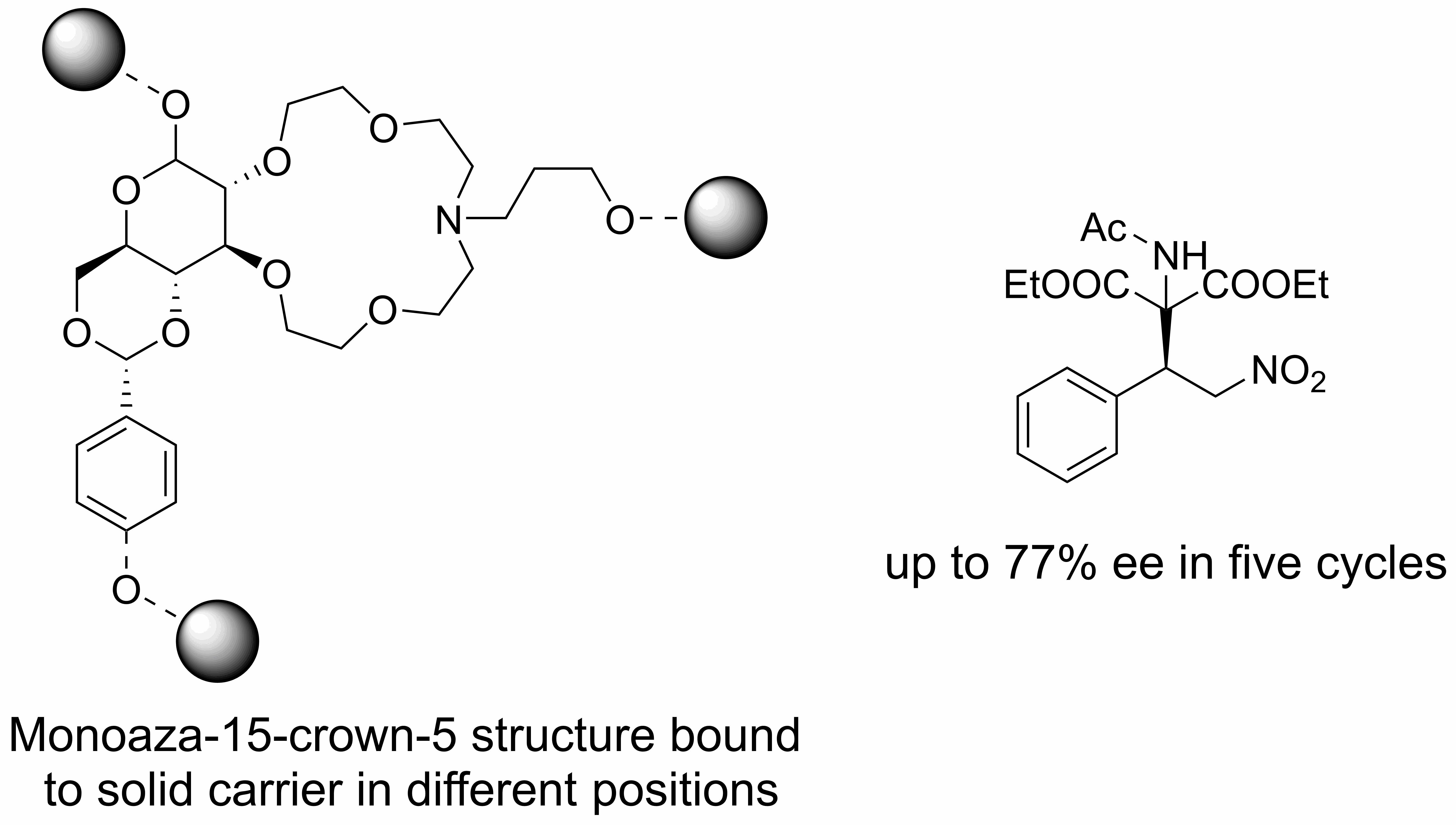
4.2. Methyl-4,6-O-benzylidene-2,3-dideoxy-β-d-glucopyranosido [2,3-h]-N-[3-(propargyloxy)propyl]-1,4,7,10-tetraoxa-13-azacyclopentadecane (1)
- 1H NMR (500 MHz, CDCl3) δ 7.49–7.43 (m, 2H, ArH), 7.40–7.31 (m, 3H, ArH), 5.52 (s, 1H, ArCH), 4.85 (d, J = 3.6 Hz, 1H, H-1), 4.27 (dd, J = 9.9, 4.6 Hz, 1H, H-6a), 4.13 (d, J = 2.3 Hz, 2H, CH2CCH), 3.99–3.51 (m, 18H, 7 × OCH2, H-6b, H-5 H-4, H-3), 3.47 (dd, J = 10.5, 5.2 Hz, 1H, H-2), 3.43 (s, 3H, OCH3), 3.01–2.50 (m, 6H, 3 × NCH2), 2.42 (t, J = 2.3 Hz, 1H, CCH), 1.93–1.68 (m, 2H, CH2CH2CH2).
- 13C NMR (126 MHz, CDCl3) δ 137.56, 129.08, 128.38, 126.18, 101.45, 98.53, 82.39, 80.10, 78.26, 74.37, 72.65, 70.76, 70.63, 70.17, 69.22, 62.38, 58.25, 55.36, 53.39, 29.84.
- FT-IR (ATR, cm−1): 3263.51 (C≡CH), 2929.83, 2900.90, 2860.40, 2794.82, 1450.45, 1371.37, 1282.65, 1253.71, 1139.92, 1120.63, 1085.91, 1056.98, 1028.05, 1010.69, 991.40, 918.10, 871.81, 740.66, 692.44, 677.01, 650.00.
4.3. Propargyl-4,6-O-benzylidene-2,3-bis-O-[(2-chloroethoxy)ethyl]-β-d-glucopyranoside (6)
- 1H NMR (500 MHz, CDCl3) δ 7.51–7.45 (m, 2H, ArH), 7.37 (d, J = 6.7 Hz, 3H, ArH), 5.52 (s, 1H, ArCH), 4.65 (d, J = 7.5 Hz, 1H, H-1), 4.46–4.30 (m, 3H, H-6a, OCH2CCH), 4.06–3.47 (m, 19H, H-3, H-5, H-6b, 6 × OCH2, 2 × CH2Cl), 3.43–3.36 (m, 1H, H-4), 3.31–3.24 (m, 1H, H-2), 2.48 (t, J = 2.5 Hz, 1H, CCH).
- 13C NMR (75 MHz, CDCl3) δ 137.29, 129.08, 128.29, 126.11, 101.62, 101.37, 82.64, 81.64, 80.77, 78.62, 77.25, 75.23, 72.46, 72.40, 71.14, 70.85, 70.81, 68.71, 66.13, 56.36, 42.81, 42.77.
4.4. Propargyl-4,6-O-benzylidene-2,3-bis-O-[(2-idooethoxy)ethyl]-β-d-glucopyranoside (7)
- 1H NMR (500 MHz, CDCl3) δ 7.51–7.45 (m, 2H, ArH), 7.41–7.32 (m, 3H, ArH), 5.53 (s, 1H, ArCH), 4.66 (d, J = 7.7 Hz, 1H, H-1), 4.42 (dd, J = 15.7, 2.4 Hz, 1H, CH2CCH), 4.38 (dd, J = 15.7, 2.4 Hz, 1H, CH2CCH), 4.33 (dd, J = 10.5, 5.0 Hz, 1H, H-6a), 4.04–3.86 (m, 4H, 2 × OCH2), 3.82–3.50 (m, 11H, H-3, H-5, H-6b, 4 × OCH2), 3.39 (br dt, J = 9.6, 4.7 Hz, 1H, H-4), 3.29–3.22 (m, 3H, H-2, CH2I), 3.18 (t, J = 6.9 Hz, 2H, CH2I), 2.49 (t, J = 2.4 Hz, 1H, CCH).
- 13C NMR (126 MHz, CDCl3) δ 137.38, 129.89, 129.14, 126.21, 101.71, 101.45, 82.74, 81.74, 80.86, 75.20, 72.57, 71.91, 71.25, 71.20, 70.89, 70.56, 70.51, 68.80, 66.23, 56.49, 3.16, 3.03.
4.5. Propargyl-4,6-O-benzylidene-2,3-dideoxy-β-d-glucopyranosido [2,3-h]-N-[3-hydroxypropyl]-1,4,7,10-tetraoxa-13-azacyclopentadecane (2)
- 1H NMR (500 MHz, CDCl3) δ 7.50–7.43 (m, 2H, ArH), 7.40–7.31 (m, 3H, ArH), 5.52 (s, 1H, ArCH), 4.94 (br, 1H, OH), 4.66 (d, J = 7.7 Hz, 1H, H-1), 4.40 (dd, J = 15.8, 2.5 Hz, 1H, OCH2CCH), 4.36 (dd, J = 15.8, 2.4 Hz, 1H, OCH2CCH), 4.32 (dd, J = 10.4, 4.9 Hz, 1H, H-6a), 4.02–3.89 (m, 4H, 2 × OCH2), 3.80 (t, J = 5.1 Hz, 2H, OCH2), 3.78–3.49 (m, 11H, H-3, H-5, H-6b, 4 × OCH2), 3.39 (td, J = 9.6, 4.7 Hz, 1H, H-4), 3.29–3.22 (m, 1H, H-2), 2.90–2.80 (m, 2H, NCH2), 2.74–2.66 (m, 4H, 2 × NCH2), 2.46 (t, J = 2.4 Hz, 1H, CCH), 1.75–1.60 (m, 2H, CH2CH2CH2).
- 13C NMR (126 MHz, CDCl3) δ 137.74, 128.96, 128.24, 126.01, 101.96, 101.13, 81.53, 80.92, 78.62, 77.23, 76.96, 75.09, 72.30, 72.17, 70.27, 70.25, 68.69, 68.65, 65.92, 64.18, 56.61, 56.31, 54.22, 54.14, 28.34.
- FT-IR (ATR, cm−1): 3388.88 (OH), 3273.16 (C≡CH), 2933.69, 2868.11, 1450.45, 1371.37, 1352.08, 1244.07, 1176.56, 1076.26, 1043.48, 1041.55, 1008.76, 1006.83, 989.47, 972.11, 883.39, 837.09, 752.23, 692.44, 657.72.
4.6. Methyl-4,6-O-(4-propargyloxybenzylidene)-α-d-glucopyranoside (10)
- 1H NMR (300 MHz, CD3OD) δ 7.43 (d, J = 8.7 Hz, 2H, ArH), 6.96 (d, J = 8.8 Hz, 2H, ArH), 5.53 (s, 1H, ArCH), 4.76–4.68 (m, 3H, ArOCH2, H-1), 4.24–4.13 (m, 1H, H-6a), 3.81 (t, J = 9.3 Hz, 1H, H-3), 3.76–3.66 (m, 2H, H-5, H-6b), 3.51 (dd, J = 9.3, 3.8 Hz, 1H, H-2), 3.46–3.38 (m, 4H, H-4, OCH3), 2.93 (t, J = 2.4 Hz, 1H, CCH).
- 13C NMR (75 MHz, CD3OD) δ 158.79, 131.47, 127.94, 114.58, 102.03, 101.19, 82.00, 75.89, 73.25, 71.16, 69.13, 63.02, 55.77, 54.93, 47.86, 47.58, 47.29. The peak of the propargyl CH was absent.
4.7. Methyl-4,6-O-(4-propargyloxybenzylidene)-2,3-bis-O-[(2-chloroethoxy)ethyl]-α-d-glucopyranoside (11)
- 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 8.4 Hz, 2H, ArH), 6.96 (d, J = 8.5 Hz, 2H, ArH), 5.48 (s, 1H, ArCH), 4.86 (d, J = 3.4 Hz, 1H, H-1), 4.69 (d, J = 1.9 Hz, 2H, ArOCH2), 4.25 (dd, J = 9.9, 4.4 Hz, 1H, H-6a), 3.98–3.47 (m, 21H, H-2, H-3, H-4, H-5, H-6b, 6 × OCH2, 2 × CH2Cl), 3.43 (m, 3H, OCH3), 2.51 (s, 1H, CCH).
- 13C NMR (126 MHz, CDCl3) δ 157.94, 130.87, 127.59, 114.74, 101.41, 99.25, 83.00, 81.80, 80.86, 79.22, 75.74, 72.39, 71.63, 71.47, 71.19, 71.01, 70.86, 69.20, 62.49, 55.96, 55.45, 43.02, 42.87.
4.8. Methyl-4,6-O-(4-propargyloxybenzylidene)-2,3-bis-O-[(2-idooethoxy)ethyl]-α-d-glucopyranoside (12)
- 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 8.7 Hz, 2H, ArH), 6.97 (d, J = 8.7 Hz, 2H, ArH), 5.49 (s, 1H, ArCH), 4.86 (d, J = 3.6 Hz, 1H, H-1), 4.69 (d, J = 2.3 Hz, 2H, ArOCH2), 4.25 (dd, J = 10.0, 4.6 Hz, 1H, H-6a), 3.99–3.47 (m, 17H, H-2, H-3, H-4, H-5, H-6b, 6 × OCH2), 3.43 (s, 3H, OCH3), 3.26 (t, J = 6.6 Hz, 2H, CH2I), 3.15 (t, J = 6.9 Hz, 2H, CH2I), 2.51 (t, J = 2.3 Hz, 1H, CCH).
- 13C NMR (126 MHz, CDCl3) δ 158.28, 130.95, 127.59, 114.76, 101.39, 99.26, 81.79, 80.79, 80.56, 79.26, 75.75, 72.44, 72.04, 71.87, 71.65, 70.79, 70.48, 69.19, 62.50, 55.97, 55.47, 3.19, 3.16.
4.9. Methyl-4,6-O-(4-propargyloxybenzylidene)-2,3-dideoxy-α-d-glucopyranosido [2,3-h]-N-[3-hydroxypropyl]-1,4,7,10-tetraoxa-13-azacyclopentadecane (3)
- 1H NMR (500 MHz, CDCl3) δ 7.43–7.37 (m, 2H, ArH), 6.99–6.93 (m, 2H, ArH), 5.48 (s, 1H, ArCH), 4.83 (d, J = 3.6 Hz, 1H, H-1), 4.69 (d, J = 2.4 Hz, 2H, ArOCH2), 4.28–4.21 (m, 1H, H-6a), 3.96–3.51 (m, 18H, H-3, H-4, H-5, H-6b, 7 × OCH2), 3.47 (dd, J = 9.3, 3.6 Hz, 1H, H-2), 3.42 (s, 3H, OCH3), 2.81 (br, 6H, 3 × NCH2), 2.50 (t, J = 2.4 Hz, 1H, CCH), 1.72 (s, 2H, CH2CH2CH2).
- 13C NMR (126 MHz, CDCl3) δ 158.10, 130.98, 127.53, 114.73, 101.27, 98.55, 82.34, 79.96, 78.55, 78.23, 75.73, 72.65, 70.65, 70.03, 69.16, 62.36, 55.96, 55.34, 54.66, 54.37, 29.84. The peak of the propargyl CH was absent, and some of the OCH2 peaks were not separated.
- FT-IR (ATR, cm−1): 3435.18 (OH), 3238.44 (C≡CH), 2916.33, 2864.25, 1612.47, 1512.17, 1454.31, 1369.44, 1303.86, 1217.07, 1172.70, 1124.48, 1083.98, 1049.26, 1029.97, 989.47, 929.68, 881.46, 823.59, 779.23, 732.94, 665.43.
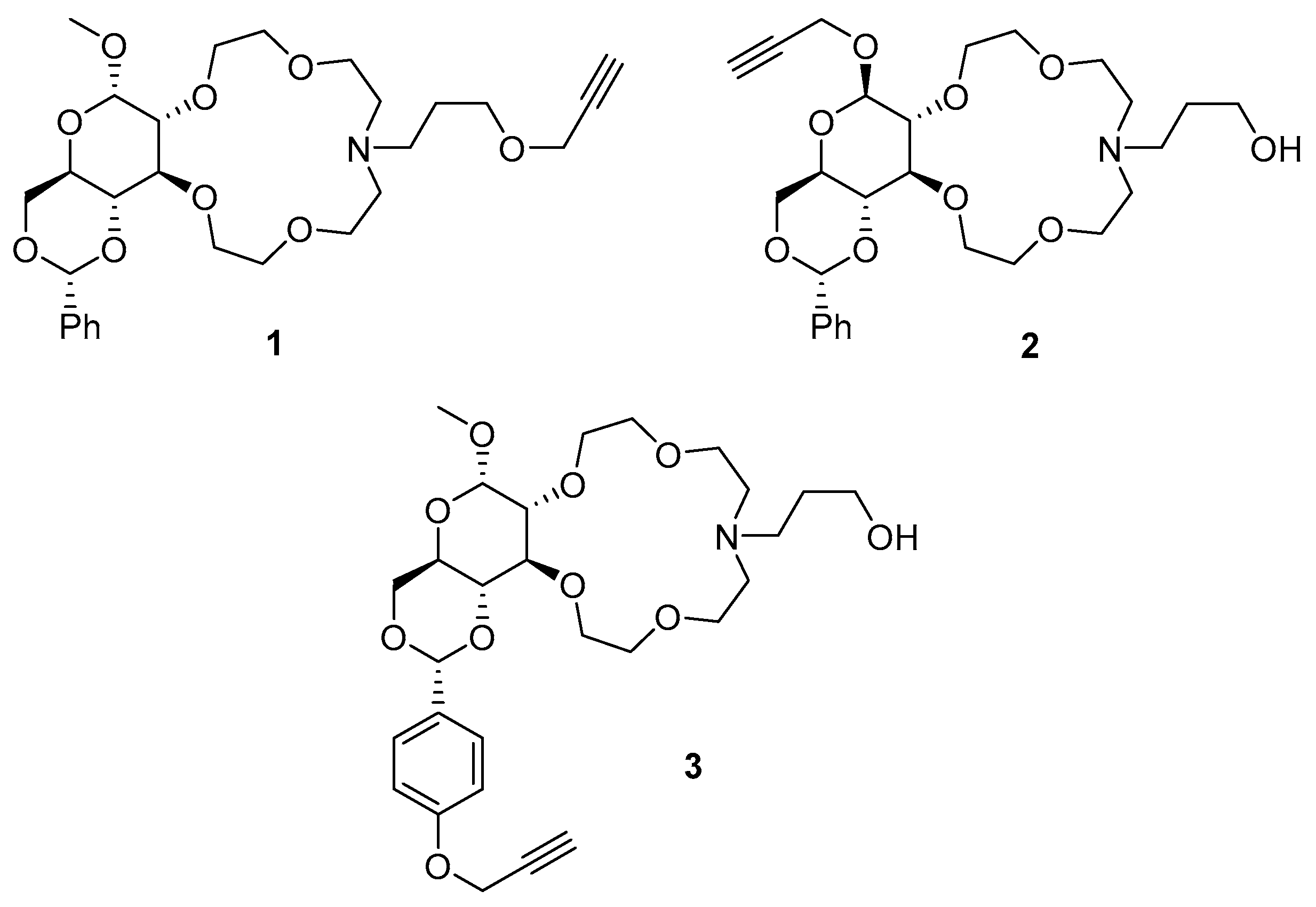
4.10. General Method for Immobilization of Crown Ethers on PVC
4.11. General Method for the Asymmetric Darzens Condensation
4.12. General Method for the Asymetric MIRC Reaction
4.13. General Method for the Asymmetric Michael-Addition
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xiang, S.H.; Tan, B. Advances in asymmetric organocatalysis over the last 10 years. Nat. Commun. 2020, 11, 3786. [Google Scholar] [CrossRef]
- Mancheño, O.G.; Waser, M. Recent Developments and Trends in Asymmetric Organocatalysis. Eur. J. Org. Chem. 2022, 26, e202200950. [Google Scholar] [CrossRef]
- Oliveira, V.d.G.; Cardoso, M.F.d.C.; Forezi, L.d.S.M. Organocatalysis: A Brief Overview on Its Evolution and Applications. Catalysts 2018, 8, 605. [Google Scholar] [CrossRef]
- Albanese, D.C.M.; Penso, M. New Trends in Asymmetric Phase Transfer Catalysis. Eur. J. Org. Chem. 2023, 26, e202300224. [Google Scholar] [CrossRef]
- Liu, S.; Zhu, W. A Minireview of Phase-Transfer Catalysis and Recent Trends. Biomed. J. Sci. Technol. Res. 2022, 45, 36691–36702. [Google Scholar] [CrossRef]
- Ooi, T.; Maruoka, K. Recent Advances in Asymmetric Phase-Transfer Catalysis. Angew. Chem. Int. Ed. 2007, 46, 4222–4266. [Google Scholar] [CrossRef] [PubMed]
- Susam, Z.D.; Tanyeli, C. Recyclable Organocatalysts in Asymmetric Synthesis. Asian J. Org. Chem. 2021, 10, 1251–1266. [Google Scholar] [CrossRef]
- Chinchilla, R.; Mazón, P.; Nájera, C. Asymmetric synthesis of α-amino acids using polymer-supported Cinchona alkaloid-derived ammonium salts as chiral phase-transfer catalysts. Tetrahedron Asymmetry 2000, 11, 3277–3281. [Google Scholar] [CrossRef]
- Chinchilla, R.; Mazón, P.; Nájera, C. Polymer-Supported Cinchona Alkaloid-Derived Ammonium Salts as Recoverable Phase-Transfer Catalysts for the Asymmetric Synthesis of α-Amino Acids. Molecules 2004, 9, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Thierry, B.; Perrard, T.; Audouard, C.; Plaquevent, J.C.; Cahard, D. Solution- and Solid-Phase Approaches in Asymmetric Phase-Transfer Catalysis by Cinchona Alkaloid Derivatives. Synthesis 2001, 2001, 1742–1746. [Google Scholar] [CrossRef]
- Chinchilla, R.; Mazón, P.; Nájera, C. Polystyrene-Anchored Cinchona Ammonium Salts: Easily Recoverable Phase-Transfer Catalysts for the Asymmetric Synthesis of α-Amino Acids. Adv. Synth. Catal. 2004, 346, 1186–1194. [Google Scholar] [CrossRef]
- Shi, Q.; Lee, Y.J.; Kim, M.J.; Park, M.K.; Lee, K.; Song, H.; Cheng, M.; Jeong, B.S.; Park, H.G.; Jew, S.S. Highly efficient polymer supported phase-transfer catalysts containing hydrogen bond inducing functional groups. Tetrahedron Lett. 2008, 49, 1380–1383. [Google Scholar] [CrossRef]
- Itsuno, S.; Paul, D.K.; Ishimoto, M.; Haraguchi, N. Designing Chiral Quaternary Ammonium Polymers: Novel Type of Polymeric Catalyst for Asymmetric Alkylation Reaction. Chem. Lett. 2010, 39, 86–87. [Google Scholar] [CrossRef]
- Parvez, M.M.; Haraguchi, N.; Itsuno, S. Molecular design of chiral quaternary ammonium polymers for asymmetric catalysis applications. Org. Biomol. Chem. 2012, 10, 2870–2877. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.; Ayats, C.; Henseler, A.H.; Pericàs, M.A. A polystyrene-supported 9-amino(9-deoxy)epi quinine derivative for continuous flow asymmetric Michael reactions. Org. Biomol. Chem. 2015, 13, 4204–4209. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.M.; Haraguchi, N.; Itsuno, S. Highly active polymeric organocatalyst: Chiral ionic polymers prepared from 10,11-didehydrogenated cinchonidinium salt. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 621–627. [Google Scholar] [CrossRef]
- Tarí, S.; Avila, A.; Chinchilla, R.; Nájera, C. Enantioselective quaternization of 4-substituted oxazol-5-(4H)-ones using recoverable Cinchona-derived dimeric ammonium salts as phase-transfer organocatalysts. Tetrahedron Asymmetry 2012, 23, 176–180. [Google Scholar] [CrossRef]
- Patil, D.; Chandam, D.; Mulik, A.; Jagdale, S.; Patil, P.; Deshmukh, M. Novel crown ether functionalized imidazolium-based acidic ionic liquid catalyzed synthesis of pyrazole derivatives under solvent-free conditions. Res. Chem. Intermed. 2015, 41, 6843–6858. [Google Scholar] [CrossRef]
- Shirakawa, S.; Tanaka, Y.; Mauroka, K. Development of a Recyclable Fluorous Chiral Phase-Transfer Catalyst: Application to the Catalytic Asymmetric Synthesis of α-Amino Acids. Org. Lett. 2004, 6, 1429–1431. [Google Scholar] [CrossRef]
- Pozzi, G.; Quici, S.; Fish, R. Perfluorocarbon Soluble Crown Ethers as Phase Transfer Catalysts. Adv. Synth. Catal. 2008, 350, 2425–2436. [Google Scholar] [CrossRef]
- Kawamura, M.; Sato, K. Magnetic nanoparticle-supported crown ethers. Chem. Commun. 2007, 3404–3405. [Google Scholar] [CrossRef] [PubMed]
- Sóti, P.L.; Telkes, L.; Rapi, Z.; Tóth, A.; Vigh, T.; Nagy, Z.K.; Bakó, P.; Marosi, G. Synthesis of an Aza Chiral Crown Ether Grafted to Nanofibrous Silica Support and Application in Asymmetric Michael Addition. J. Inorg. Organomet. Polym. Mater. 2014, 24, 713–721. [Google Scholar] [CrossRef]
- Sabah, K.J.; Sead, F.F.; Mohammed, H.J.; Abedul-Hussien, I.H. Novel polymer crafted sugar thiacrown ether and its applications in recovery of metal ions. Carbohydr. Res. 2020, 495, 108057. [Google Scholar] [CrossRef]
- Orbán, I.; Bakó, P.; Rapi, Z. Carbohydrate-Based Azacrown Ethers in Asymmetric Syntheses. Chemistry 2021, 3, 550–577. [Google Scholar] [CrossRef]
- Ouerghui, A.; Elamari, H.; Dardouri, M.; Ncib, S.; Meganem, F.; Girard, C. Chemical modifications of poly(vinyl chloride) to poly(vinyl azide) and “clicked” triazole bearing groups for application in metal cation extraction. React. Funct. Polym. 2016, 100, 191–197. [Google Scholar] [CrossRef]
- Ay, E.; Yenil, N. Click Reactivity of Azide-Modified Polyvinyl Chloride as an Entry to Glycopolymer Scaffolds. Croat. Chem. Acta 2021, 94, 167–176. [Google Scholar] [CrossRef]
- Bakó, P.; Czinege, E.; Bakó, T.; Czugler, M.; Tőke, L. Asymmetric C–C bond forming reactions with chiral crown catalysts derived from d-glucose and d-galactose. Tetrahedron Asymmetry 1999, 10, 4539–4551. [Google Scholar] [CrossRef]
- Mereyala, H.B.; Gurrala, S.R. A highly diastereoselective, practical synthesis of allyl, propargyl 2,3,4,6-tetra-O-acetyl-β-d-gluco, β-d-galactopyranosides and allyl, propargyl heptaacetyl-β-d-lactosides. Carbohydr. Res. 1998, 307, 351–354. [Google Scholar] [CrossRef]
- Giguère, J.B.; Thibeault, D.; Cronier, F.; Marois, J.S.; Auger, M.; Morin, J.F. Synthesis of [2]- and [3]rotaxanes through Sonogashira coupling. Tetrahedron Lett. 2009, 50, 5497–5500. [Google Scholar] [CrossRef]
- Ono, F.; Hirata, O.; Ichimaru, K.; Saruhashi, K.; Watanabe, H.; Shinkai, S. Mild One-Step Synthesis of 4,6-Benzylideneglycopyranosides from Aromatic Aldehydes and Gelation Abilities of the Glucose Derivatives. Eur. J. Org. Chem. 2015, 2015, 6439–6447. [Google Scholar] [CrossRef]
- Takeishi, M.; Okawara, M. Synthesis and reaction of poly(vinyl chloride) containing azide group. J. Polym. Sci. B Polym. Lett. 1969, 7, 201–203. [Google Scholar] [CrossRef]
- Kardos, G.; Soós, T. Tether-Free Immobilized Bifunctional Squaramide Organocatalysts for Batch and Flow Reactions. Eur. J. Org. Chem. 2013, 2013, 4490–4494. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Crown Ether | Concentration, M a | Reaction Time, h | Yield, % b | ee, % c |
| 1 | 1 | 0.33 | 22 | 41 | 61 |
| 2 | 2 | 0.33 | 4 | 37 | 37 |
| 3 | 3 | 0.33 | 7 | 72 | 74 |
| 4 | 1 | 0.10 | 48 | 30 | 18 |
| 5 | 2 | 0.10 | 20 | 25 | 40 |
| 6 | 3 | 0.10 | 20 | 37 | 56 |
 | ||||
|---|---|---|---|---|
| Entry | Immobilized Crown Ether | Reaction Time, d | Yield, % a | ee, % b |
| 1 | 14a (4% substitution) | 13 | 47 | 40 |
| 2 | 15a (4% substitution) | 5 | 66 | 58 |
| 3 | 16a (4% substitution) | 2 | 51 | 77 |
| 4 c | 16a (4% substitution) | 1 | 28 | 70 (54) d |
| 5 | 13 (10% substitution) | 7 | 56 | 46 |
| 6 | 14b (10% substitution) | 2 | 31 | 44 |
| 7 | 15b (10% substitution) | 1 | 22 | 54 |
| 8 e | 15b (10% substitution) | 1 | 41 | 39 |
| 9 | 16b (10% substitution) | 2 | 30 | 48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varga, B.; Ujj, D.; Mátravölgyi, B.; Szolnoki, B.; Koczka, B.; Rapi, Z. Reusable Glucose-Based Crown Ethers Anchored to PVC. Molecules 2023, 28, 7905. https://doi.org/10.3390/molecules28237905
Varga B, Ujj D, Mátravölgyi B, Szolnoki B, Koczka B, Rapi Z. Reusable Glucose-Based Crown Ethers Anchored to PVC. Molecules. 2023; 28(23):7905. https://doi.org/10.3390/molecules28237905
Chicago/Turabian StyleVarga, Bertalan, Dóra Ujj, Béla Mátravölgyi, Beáta Szolnoki, Béla Koczka, and Zsolt Rapi. 2023. "Reusable Glucose-Based Crown Ethers Anchored to PVC" Molecules 28, no. 23: 7905. https://doi.org/10.3390/molecules28237905
APA StyleVarga, B., Ujj, D., Mátravölgyi, B., Szolnoki, B., Koczka, B., & Rapi, Z. (2023). Reusable Glucose-Based Crown Ethers Anchored to PVC. Molecules, 28(23), 7905. https://doi.org/10.3390/molecules28237905