
Asymmetric Mannich/Cyclization Reaction of 2-Benzothiazolimines and 2-Isothiocyano-1-indanones to Construct Chiral Spirocyclic Compounds


Abstract

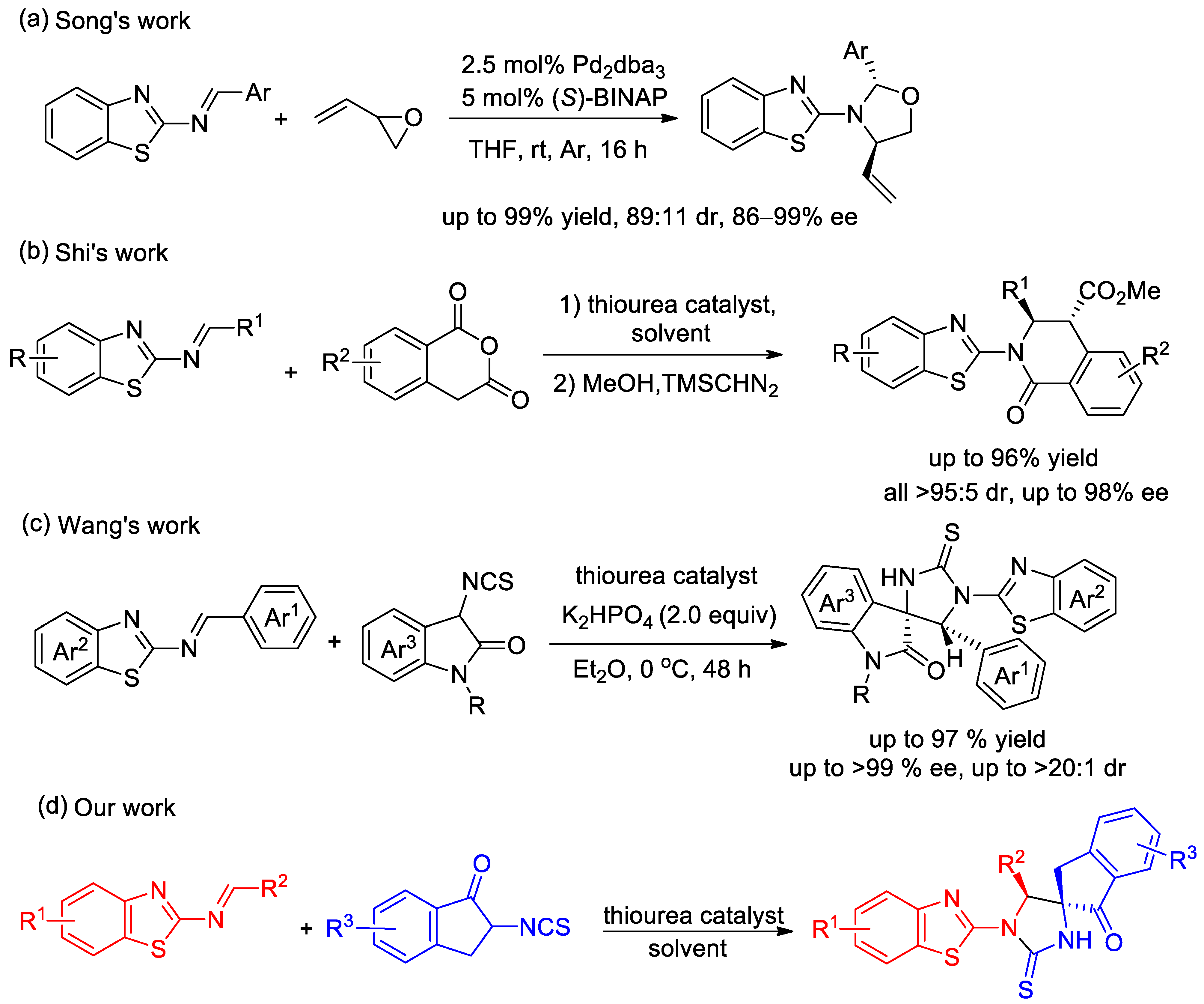
1. Introduction
2. Results and Discussion
2.1. Optimization of Reaction Conditions
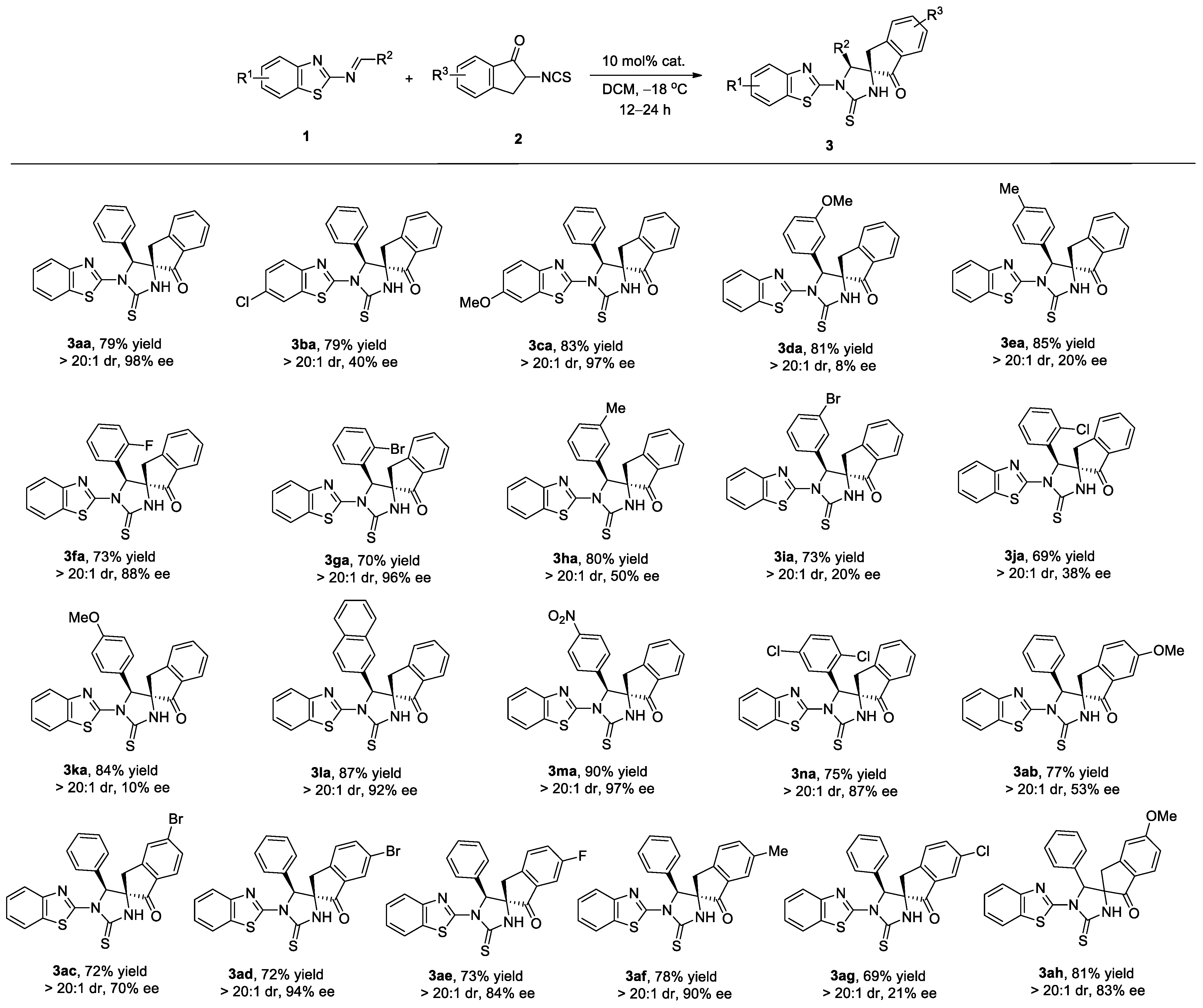
2.2. Substrate Scope
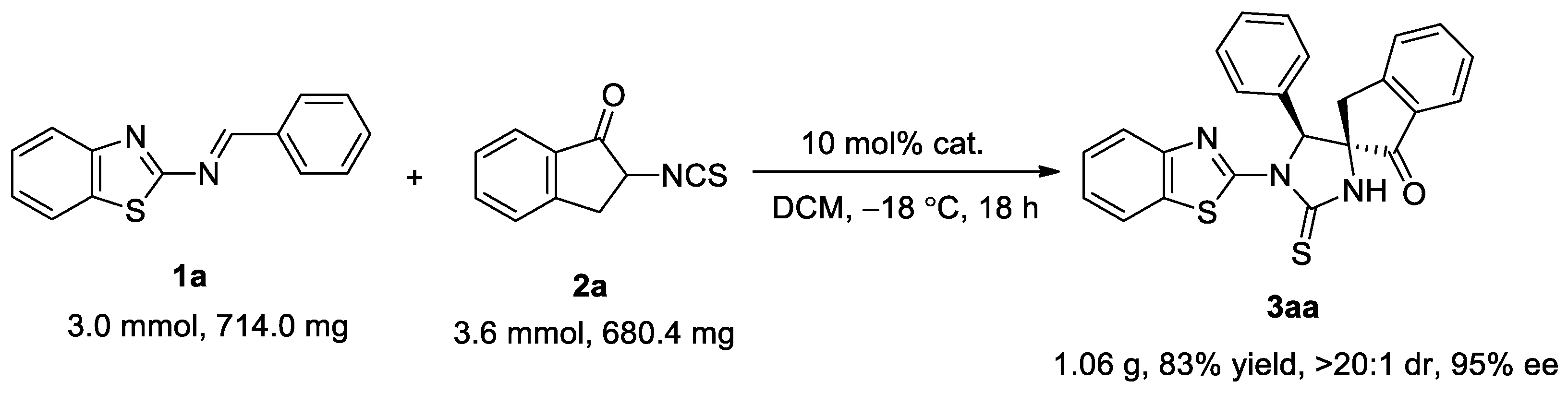
2.3. Scaled-Up Synthesis
2.4. X-ray Diffraction Analysis
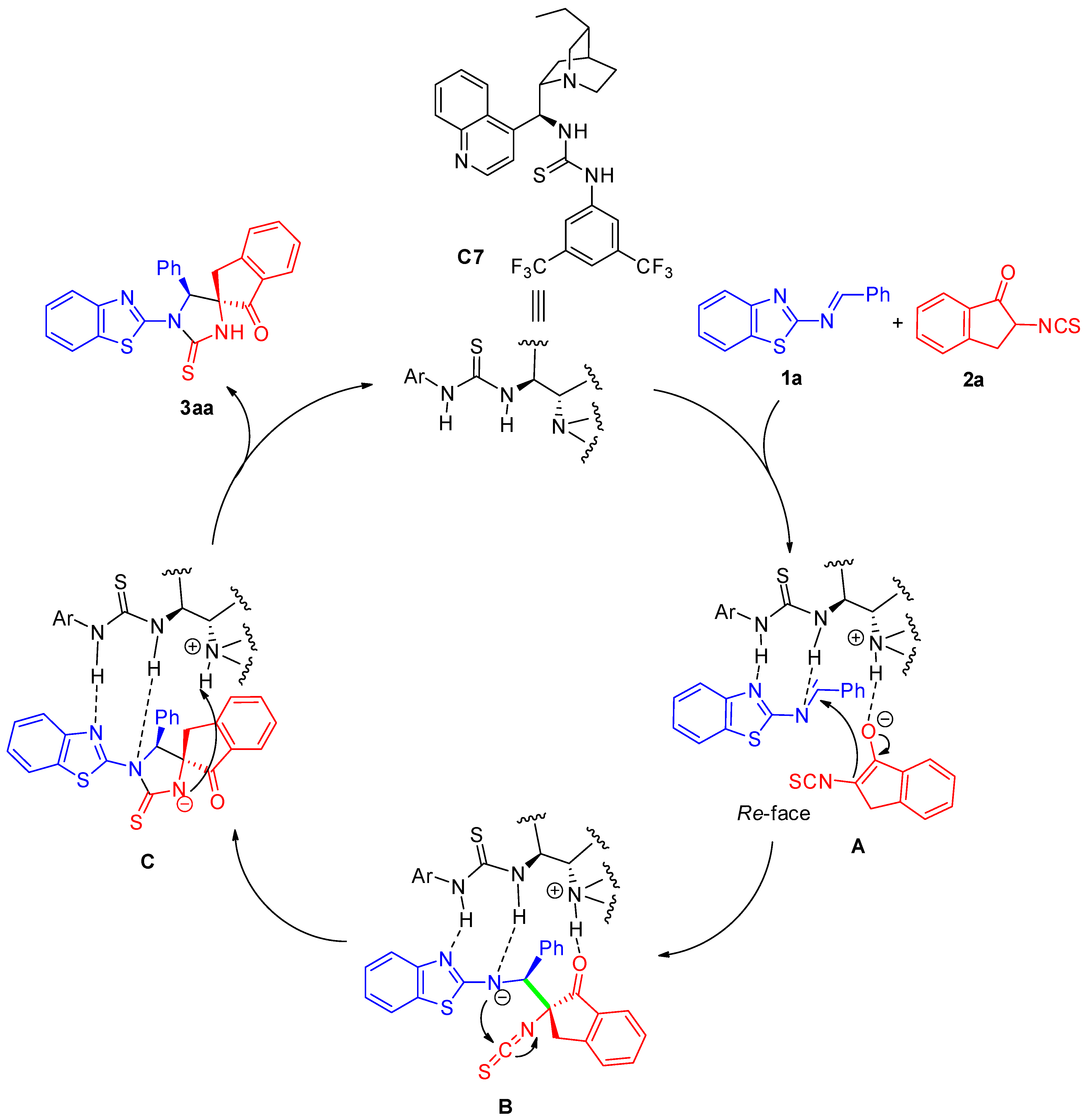
2.5. Plausible Mechanism
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Materials
4.3. Procedure for the Asymmetric Synthesis of Compound 3
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5-phenyl-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3aa). Initially, 1a (23.8 mg, 0.10 mmol) and 2a (22.7 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 31.2 mg (79% yield) of compound 3aa as a white solid, m.p. 239–241 °C. HPLC (Daicel Chiralpak IA, n-hexane/isopropanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 17.1 min (major enantiomer), tR = 8.8 min (minor enantiomer); 98% ee. = −449.0 (c = 0.40, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.82 (d, J = 7.6 Hz, 1H, ArH), 7.70 (d, J = 7.6 Hz, 1H, ArH), 7.62 (t, J = 7.2 Hz, 1H, ArH), 7.54 (d, J = 8.0 Hz, 1H, ArH), 7.44 (t, J = 7.2 Hz, 1H, ArH), 7.38–7.29 (m, 4H, ArH), 7.26 (d, J = 8.0 Hz, 2H, ArH), 7.24–7.18 (m, 4H, ArH + NH), 6.10 (s, 1H, CH), 3.00 (dd, J1 = 17.8 Hz, J2 = 7.6 Hz, 1H, CH2), 2.85 (dd, J = 17.8 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ 201.0, 179.7, 157.5, 149.9, 148.2, 136.4, 136.1, 132.7, 132.5, 129.0, 128.9, 128.7, 127.1, 126.3, 125.7, 125.6, 123.8, 121.2, 120.9, 70.6, 70.3, 34.9 ppm. HRMS (ESI): m/z calcd. for C24H18N3OS2 [M + H]+ 428.0886, found 428.0890.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-6′-methoxy-5-phenyl-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ab). Initially, 1a (23.8 mg, 0.10 mmol) and 2b (26.3mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 30.8 mg (77% yield) of compound 3ab as a white solid, m.p. 246–248 °C. HPLC (Daicel Chiralpak ADH, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 11.6 min (major enantiomer), tR = 10.2 min (minor enantiomer); 53% ee. = −998.4 (c = 0.28, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.73 (d, J = 7.2 Hz, 1H, ArH), 7.56 (d, J = 8.0 Hz, 1H, ArH), 7.36–7.27 (m, 4H, ArH), 7.25–7.19 (m, 5H, ArH), 7.17 (d, J = 8.4 Hz, 1H, ArH), 6.84 (s, 1H, NH), 6.11 (s, 1H, CH), 3.84 (s, 3H, CH3), 2.87 (d, J = 17.6 Hz, 1H, CH2), 2.78 (d, J = 17.6 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ 200.7, 169.3, 166.2, 160.1, 151.5, 143.6, 134.7, 133.2, 131.2, 130.2, 129.3, 129.0, 128.8, 127.1, 126.4, 125.9, 125.7, 122.2, 120.9, 118.7, 105.8, 89.4, 55.7, 35.3 ppm. HRMS (ESI): m/z calcd. for C25H20N3O2S2 [M + H]+ 458.0991, found 458.0953.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5′-bromo-5-phenyl-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ac). Initially, 1a (23.8 mg, 0.10 mmol) and 2c (31.9 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 27.9 mg (72% yield) of compound 3ac as a white solid, m.p. 243–245 °C. HPLC (Daicel Chiralpak ADH, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 9.8 min (major enantiomer), tR = 12.5 min (minor enantiomer); 70% ee. = −238.7 (c = 0.15, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.86 (d, J = 7.6 Hz, 1H, ArH), 7.73 (d, = 7.6 Hz, 1H, ArH), 7.65 (td, J = 7.6, 1.2 Hz, 1H, ArH), 7.56 (d, J = 8.0 Hz, 1H, ArH), 7.47 (t, J = 7.6 Hz, 1H, ArH), 7.38–7.29 (m, 4H, ArH), 7.25–7.19 (m, 3H, ArH), 6.83 (s, 1H, NH), 6.12 (s, 1H, CH), 2.95 (d, J = 17.8 Hz, 1H, CH2), 2.87 (d, J = 17.8 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ 201.0, 179.8, 157.5, 149.9, 148.3, 136.4, 136.1, 132.8, 132.6, 130.9, 129.0, 128.9, 128.7, 126.3, 125.7, 125.6, 123.8, 122.3, 121.2, 120.9, 70.6, 70.3, 34.9 ppm. HRMS (ESI): m/z calcd. for C24H17N3O3S2 [M-Br]+ 428.0886, found 428.0853.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-6′-bromo-5-phenyl-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ad). Initially, 1a (23.8 mg, 0.10 mmol) and 2d (31.9mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 28.0mg (72% yield) of compound 3ad as a white solid, m.p. 257–259 °C. HPLC (Daicel Chiralpak IA, n-hexane/isopropanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 15.6 min (major enantiomer), tR = 8.3 min (minor enantiomer); 94% ee. = −776.2 (c = 0.50, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.81 (d, J = 7.6 Hz, 1H, ArH), 7.70 (d, J = 8.0 Hz, 1H, ArH), 7.61 (t, J = 7.6 Hz, 1H, ArH), 7.54 (d, J = 8.0 Hz, 1H, ArH), 7.42 (t, J = 7.4 Hz, 1H, ArH), 7.35–7.29 (m, 4H, ArH), 7.27–7.18 (m, 4H, ArH + NH), 6.10 (s, 1H, CH), 2.98 (d, J = 17.8 Hz, 1H, CH2), 2.84 (d, J = 17.8 Hz, 1H, CH2) ppm. 13C NMR (176 MHz, CDCl3) δ 201.1, 179.6, 157.5, 149.9, 148.2, 136.4, 136.0, 132.7, 132.5, 129.0, 128.9, 128.6, 126.3, 125.64, 125.57, 123.7, 121.1, 120.8, 70.7, 70.2, 34.9 ppm. HRMS (ESI): m/z calcd. for C24H17BrN3O3S2 [M + H]+ 505.9991, found 505.9969.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-6′-fluoro-5-phenyl-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ae). Initially, 1a (23.8 mg, 0.10 mmol) and 2e (24.8 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 29.5 mg (73% yield) of compound 3ae as a white solid, m.p. 277–279 °C. HPLC (Daicel Chiralpak ADH, n-hexane/isopropanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 24.0 min (major enantiomer), tR = 14.7 min (minor enantiomer); 84% ee. = −418.6 (c = 0.55, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.73 (d, J = 8.0 Hz, 1H, ArH), 7.56 (d, J = 8.0 Hz, 1H, ArH), 7.47 (dd, J = 7.0, 2.6 Hz, 1H, ArH), 7.39–7.31 (m, 4H, ArH), 7.30–7.26 (m, 2H, ArH), 7.24–7.20 (m, 3H, ArH), 6.99 (s, 1H, NH), 6.11 (s, 1H, CH), 2.92 (d, J = 17.6 Hz, 1H, CH2), 2.83 (d, J = 18.0 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ 200.4, 179.6, 162.7 (1JC–F = 249.0 Hz), 157.4, 148.1, 145.4, 135.9, 134.2 (3JC–F = 7.4 Hz), 132.6, 129.1 (4JC–F = 5.5 Hz), 127.8 (3JC–F = 7.9 Hz), 125.7, 124.2 (2JC–F = 23.5 Hz), 123.8, 121.2, 120.9, 111.3 (2JC–F = 22.0 Hz), 77.2, 71.3, 70.2, 34.4 ppm. HRMS (ESI): m/z calcd. for C24H17FN3OS2 [M + H]+ 446.0792, found 446.0763.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-6′-methyl-5-phenyl-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3af). Initially, 1a (23.8 mg, 0.10 mmol) and 2f (24.4 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 31.5 mg (78% yield) of compound 3af as a white solid, m.p. 249–251 °C. HPLC (Daicel Chiralpak ADH, n-hexane/isopropanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 18.6 min (major enantiomer), tR = 12.8 min (minor enantiomer); 90% ee. = −330.0 (c = 0.33, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.73 (d, J = 8.0 Hz, 1H, ArH), 7.64 (s, 1H, ArH), 7.55 (d, J = 8.0 Hz, 1H, ArH), 7.46 (d, J = 7.6 Hz, 1H, ArH), 7.36–7.27 (m, 4H, ArH), 7.23–7.15 (m, 4H, ArH), 6.69 (s, 1H, NH), 6.11 (s, 1H, CH), 2.88 (d, J = 17.6 Hz, 1H, CH2), 2.80 (d, J = 17.6 Hz, 1H, CH2), 2.43 (s, 3H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 201.0, 179.7, 157.5, 148.3, 147.2, 138.9, 137.7, 136.2, 132.7, 132.6, 129.0, 128.9, 126.0, 125.6, 125.4, 123.7, 121.2, 120.9, 71.0, 70.3, 34.6, 21.1 ppm. HRMS (ESI): m/z calcd. for C25H20N3OS2 [M + H]+ 442.1042, found 442.1008.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-6′-chloro-5-phenyl-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ag). Initially, 1a (23.8 mg, 0.10 mmol) and 2g (26.8 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 26.9 mg (69% yield) of compound 3ag as a white solid, m.p. 239–241 °C. HPLC (Daicel Chiralpak IA, n-hexane/isopropanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 15.7 min (major enantiomer), tR = 8.3 min (minor enantiomer); 21% ee. = −576.9 (c = 0.35, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 7.6 Hz, 1H, ArH), 7.72 (d, J = 7.6 Hz, 1H, ArH), 7.65 (t, J = 7.6 Hz, 1H, ArH), 7.55 (d, J = 8.0 Hz, 1H, ArH), 7.46 (t, J = 7.6 Hz, 1H, ArH), 7.37–7.29 (m, 4H, ArH), 7.23–7.19 (m, 3H, ArH), 7.07 (s, 1H, NH), 6.11 (s, 1H, CH), 2.97 (d, J = 17.6 Hz, 1H, CH2), 2.86 (d, J = 17.6 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ 200.9, 179.7, 157.4, 149.8, 148.2, 136.4, 136.1, 132.7, 132.6, 129.0, 128.9, 128.8, 126.4, 125.7, 125.6, 123.8, 121.2, 120.9, 70.6, 70.3, 34.9 ppm. HRMS (ESI): m/z calcd. for C24H17ClN3OS2 [M + H]+ 462.0497, found 462.0494.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5′-methoxy-5-phenyl-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ah). Initially, 1a (23.8 mg, 0.10 mmol) and 2h (26.3mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 30.8 mg (77% yield) of compound 3ah as a white solid, m.p. 236–238 °C. HPLC (Daicel Chiralpak IC, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 9.04 min (major enantiomer), tR = 12.26 min (minor enantiomer); 83% ee. = −94.2 (c = 0.55, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.72 (d, J = 8.0 Hz, 1H, ArH), 7.55 (d, J = 8.0 Hz, 1H, ArH), 7.36–7.27 (m, 4H, ArH), 7.22 (d, J = 7.8 Hz, 5H, ArH), 7.17 (d, J = 7.8 Hz, 1H, ArH), 7.06 (s, 1H, NH), 6.10 (s, 1H, CH), 3.83 (s, 3H, CH3), 2.88 (d, J = 17.4 Hz, 1H, CH2), 2.77 (d, J = 17.4 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ 200.9, 179.8, 160.3, 157.5, 148.3, 142.7, 136.2, 133.7, 132.8, 129.0, 128.4, 127.1, 125.9, 125.7, 123.8, 121.2, 120.9, 106.5, 71.3, 70.4, 55.8, 34.4 ppm. HRMS (ESI): m/z calcd. for C25H20N3O2S2 [M + H]+ 458.0992, found 458.0959.
- (2′R,5S)-1-(6-Chlorobenzo[d]thiazol-2-yl)-5-phenyl-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ba). Initially, 1b (27.2 mg, 0.10 mmol) and 2a (22.7mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 30.2 mg (79% yield) of compound 3ba as a white solid, m.p. 243–245 °C. HPLC (Daicel Chiralpak IC, n-hexane/isopropanol = 90:10, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 25.3 min (major enantiomer), tR = 48.9 min (minor enantiomer); 40% ee. = −94.2 (c = 0.55, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.85 (d, J = 8.0 Hz, 1H, ArH), 7.68–7.63 (m, 2H, ArH), 7.49–7.44 m, 2H, ArH), 7.38–7.32 (m, 3H, ArH), 7.29 (d, J = 7.6 Hz, 1H, ArH), 7.25–7.20 (m, 3H, ArH), 6.90 (s, 1H, NH), 6.07 (s, 1H, CH), 2.96 (d, J = 18.0 Hz, 1H, CH2), 2.87 (d, J = 18.0 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ 200.9, 179.7, 157.8, 149.9, 146.9, 136.6, 135.9, 133.9, 132.5, 129.14, 129.11, 129.0, 128.8, 126.4, 126.3, 125.7, 122.0, 120.4, 70.6, 70.3, 34.9 ppm. HRMS (ESI): m/z calcd. for C24H17ClN3OS2 [M + H]+ 462.0497, found 462.0459.
- (2′R,5S)-1-(6-Methoxybenzo[d]thiazol-2-yl)-5-phenyl-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ca). Initially, 1c (26.8 mg, 0.10 mmol) and 2a (22.7 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 37.9 mg (83% yield) of compound 3ca as a white solid, m.p. 257–259 °C. HPLC (Daicel Chiralpak IC, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 12.2 min (major enantiomer), tR = 20.1 min (minor enantiomer); 97% ee. = −94.7 (c = 0.48, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.82 (d, J = 7.6 Hz, 1H, ArH), 7.74–7.45 (m, 2H, ArH), 7.46–7.41 (m, 2H, ArH), 7.33–7.27 (m, 3H, ArH + NH), 7.22–7.13 (m, 4H, ArH), 6.87 (dd, J = 8.8, 2.8 Hz, 1H, ArH), 6.06 (s, 1H, CH), 3.81 (s, 3H, CH3), 2.96 (d, J = 18.0 Hz, 1H, CH2), 2.83 (d, J = 18.0 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ 200.9, 179.6, 156.7, 155.5, 149.8, 142.6, 136.4, 136.1, 134.0, 132.6, 129.0, 128.9, 128.7, 126.3, 125.6, 121.8, 114.6, 103.8, 70.6, 70.3, 55.8, 35.0 ppm. HRMS (ESI): m/z calcd. for C25H20N3O2S2 [M + H]+ 458.0991, found 458.0961.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5-(3-methoxyphenyl)-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3da). Initially, 1d (26.8 mg, 0.10 mmol) and 2a (22.7 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 30.5 mg (81% yield) of compound 3da as a white solid, m.p. 197–199 °C. HPLC (Daicel Chiralpak IIC, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 7.5 min (major enantiomer), tR = 9.4 min (minor enantiomer); 8% ee. = −27.1 (c = 0.70, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.86 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 7.6 Hz, 1H, ArH), 7.68–7.63 (m, 1H, ArH), 7.58 (d, J = 8.0 Hz, 1H, ArH), 7.47 (t, J = 7.6 Hz, 1H, ArH), 7.32–7.27 (m, 2H, ArH), 7.25–7.20 (m, 2H, ArH), 6.85–6.80 (m, 2H, ArH), 6.74 (t, J = 2.0 Hz, 1H, ArH), 6.66 (s, 1H, NH), 6.08 (s, 1H, CH), 3.68 (s, 3H, CH3), 2.96 (d, J = 18.0 Hz, 1H, CH2), 2.96 (d, J = 18.0 Hz, 1H, CH2), 2.91 (d, J = 18.0 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ 201.0, 179.6, 160.0, 157.5, 150.0, 148.3, 137.6, 136.4, 132.8, 132.5, 130.2, 128.7, 126.4, 125.64, 125.60, 123.8, 121.2, 120.9, 114.2, 70.6, 70.2, 55.2, 34.9 ppm. HRMS (ESI): m/z calcd. for C25H20N3O2S2 [M + H]+ 458.0991, found 458.0956.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5-(4-methylphenyl)-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ea). Initially, 1e (25.2 mg, 0.10 mmol) and 2a (22.7 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 32.0 mg (85% yield) of compound 3ea as a white solid, m.p. 234–236 °C. HPLC (Daicel Chiralpak IC, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 7.0 min (major enantiomer), tR = 9.3 min (minor enantiomer); 20% ee. = −52.8 (c = 0.30, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.85 (d, J = 7.6 Hz, 1H, ArH), 7.73 (d, J = 8.0 Hz, 1H, ArH), 7.65 (t, J = 7.6 Hz, 1H, ArH), 7.57 (d, J = 8.0 Hz, 1H, ArH), 7.47 (t, J = 7.6 Hz, 1H, ArH), 7.31–7.26 (m, 3H, ArH), 7.23–7.19 (m, 1H, ArH), 7.14–7.10 (m, 3H, ArH), 6.67 (s, 1H, NH), 6.09 (s, 1H, CH), 2.95 (d, J = 18.0 Hz, 1H, CH2), 2.90 (d, J = 17.6 Hz, 1H, CH2), 2.31 (s, 3H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 201.0, 179.8, 157.5, 149.9, 148.3, 138.8, 136.4, 133.1, 132.8, 132.6, 129.7, 128.7, 126.4, 125.6, 123.7, 121.2, 120.9, 70.7, 70.2, 34.9, 21.2 ppm. HRMS (ESI): m/z calcd. for C25H20N3OS2 [M + H]+ 442.1042, found 442.1007.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5-(2-fluorophenyl)-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3fa). Initially, 1f (25.6 mg, 0.10 mmol) and 2a (22.7 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 27.9mg (73% yield) of compound 3fa as a white solid, m.p. 228–220 °C. HPLC (Daicel Chiralpak IC, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 6.9 min (major enantiomer), tR = 7.8 min (minor enantiomer); 88% ee. = −321.5 (c = 0.80, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.82 (d, J = 7.6 Hz, 1H, ArH), 7.73 (d, J = 8.0 Hz, 1H, ArH), 7.63 (t, J = 7.6 Hz, 1H, ArH), 7.57 (d, J = 8.0 Hz, 1H, ArH), 7.43 (d, J = 7.6 Hz, 1H, ArH), 7.32–7.27 (m, 3H, ArH), 7.24–7.15 (m, 3H, ArH + NH), 7.11 (d, J = 7.2 Hz, 1H, ArH), 7.07 (d, J = 8.8 Hz, 1H, ArH), 6.53 (s, 1H, CH), 3.03 (d, J = 18.0 Hz, 1H, CH2), 2.96 (d, J = 18.0 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ 200.7, 179.5, 160.4 (1JC–F = 246.3 Hz), 157.0, 150.0, 148.2, 136.5, 132.7, 132.3, 130.4 (3JC–F = 8.0 Hz), 128.6, 126.8, 126.2, 125.7, 125.6, 125.1 (4JC–F = 3.2 Hz), 123.8, 123.4 (2JC–F = 13.4 Hz), 121.3, 120.8, 115.5 (2JC–F = 21.1 Hz), 70.2, 63.3, 34.8 ppm. HRMS (ESI): m/z calcd. for C24H17FN3OS2 [M + H]+ 446.0792, found 446.0759.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5-(2-bromophenyl)-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ga). Initially, 1g (31.6 mg, 0.10 mmol) and 2a (22.7 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 25.8 mg (70% yield) of compound 3ga as a white solid, m.p. 210–212 °C. HPLC (Daicel Chiralpak IC, n-hexane/isopropanol = 95:5, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 24.6 min (major enantiomer), tR = 27.2 min (minor enantiomer); 96% ee. = −371.4 (c = 0.48, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.84 (d, J = 7.6 Hz, 1H, ArH), 7.73 (d, J = 7.6 Hz, 1H, ArH), 7.67–7.62 (m, 1H, ArH), 7.59–7.56 (m, 2H, ArH), 7.45 (t, J = 7.6 Hz, 1H, ArH), 7.34–7.27 (m, 3H, ArH), 7.25–7.15 (m, 3H, ArH), 6.91 (s, 1H, NH), 6.66 (s, 1H, CH), 2.96 (ABq, J = 18.4 Hz, 2H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ 200.7, 179.3, 156.9, 150.6, 148.3, 136.5, 135.6, 133.0, 132.7, 132.4, 130.1, 128.7, 128.4, 127.0, 126.3, 125.7, 125.5, 124.0, 123.8, 121.5, 120.8, 70.1, 69.0, 35.2 ppm. HRMS (ESI): m/z calcd. for C24H1779BrN3OS2 [M + H]+ 505.9991, found 505.9948; calcd. for C24H1781BrN3OS2 [M + H]+ 507.9971, found 507.9930.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5-(3-methylphenyl)-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ha). Initially, 1h (25.2 mg, 0.10 mmol) and 2a (22.7 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 29.8 mg (80% yield) of compound 3ha as a white solid, m.p. 167–169 °C. HPLC (Daicel Chiralpak IC, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 7.6 min (major enantiomer), tR = 10.3 min (minor enantiomer); 50% ee. = −128.5 (c = 1.25, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.81 (t, J = 8.2 Hz, 1H, ArH), 7.70 (d, J = 8.0 Hz, 1H, ArH), 7.63–7.54 (m, 2H, ArH), 7.45–7.39 (m, 2H, ArH), 7.29–7.26 (m, 2H, ArH), 7.22–7.17 (m, 2H, ArH), 7.10 (d, J = 7.6 Hz, 1H, ArH), 7.04 (d, J = 6.8 Hz, 2H, ArH + NH), 6.09 (s, 1H, CH), 2.98 (dd, J = 17.8, 7.2 Hz, 1H, CH2), 2.84 (d, J = 17.6 Hz, 1H, CH2), 2.30 (s, 3H, CH3) ppm. 13C NMR (100 MHz, CDCl3) δ 201.3, 179.5, 157.5, 150.1, 148.2, 138.8, 136.3, 135.9, 132.7, 132.4, 129.7, 128.8, 128.6, 128.5, 126.3, 126.2, 125.6, 125.5, 123.6, 121.14, 121.12, 120.8, 70.7, 70.3, 34.8, 21.4 ppm. HRMS (ESI): m/z calcd. for C25H20N3OS2 [M + H]+ 442.1043, found 442.1010.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5-(3-bromophenyl)-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ia). Initially, 1i (31.6 mg, 0.10 mmol) and 2a (22.7 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 29.6 mg (73% yield) of compound 3ia as a white solid, m.p. 134–136 °C. HPLC (Daicel Chiralpak IC, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 6.9 min (major enantiomer), tR = 8.8 min (minor enantiomer); 20% ee. = −8.1 (c = 0.15, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.86–7.83 (m, 1H, ArH), 7.73 (d, J = 8.0 Hz, 1H, ArH), 7.86–7.64 (m, 1H, ArH), 7.58 (d, J = 8.0 Hz, 1H, ArH), 7.47 (t, J = 7.8 Hz, 1H, ArH), 7.39 (s, 1H, ArH), 7.31 (t, J = 7.4 Hz, 2H, ArH), 7.24–7.18 (m, 2H, ArH), 7.00 (s, 1H, NH), 6.06 (s, 1H, CH), 3.01 (d, J = 17.6 Hz, 1H, CH2) 2.88 (d, J = 18.0 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ 200.7, 179.4, 157.2, 149.8, 148.1, 138.4, 136.6, 136.3, 132.7, 132.3, 132.1, 131.8, 130.6, 128.8, 127.0, 126.4, 125.7, 125.6, 123.9, 121.2, 120.9, 70.5, 69.5, 34.9 ppm. HRMS (ESI): m/z calcd. for C24H1779BrN3OS2 [M + H]+ 505.9991, found 505.9943; calcd. for C24H1781BrN3OS2 [M + H]+ 507.9971, found 507.9927.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5-(2-chlorophenyl)-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ja). Initially, 1j (27.2 mg, 0.10 mmol) and 2a (22.7 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 26.9 mg (69% yield) of compound 3ja as a white solid, m.p. 227–229 °C. HPLC (Daicel Chiralpak ADH, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 20.1 min (major enantiomer), tR = 17.6 min (minor enantiomer); 38% ee. = −39.4 (c = 1.60, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.85 (d, J = 7.6 Hz, 1H, ArH), 7.75–7.64 (m, 4H, ArH), 7.55–7.46 (m, 2H, ArH), 7.38 (d, J = 8.0 Hz, 2H, ArH), 7.32–7.28 (m, 2H, ArH), 7.24 (t, J = 7.6 Hz, 1H, ArH), 6.93 (s, 1H, NH), 6.15 (s, 1H, CH), 3.05–2.73 (m, 2H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ 200.6, 179.5, 157.0, 150.5, 148.3, 136.5, 134.0, 133.8, 132.8, 132.4, 129.8, 129.7, 128.7, 127.9, 126.8, 126.3, 125.7, 125.5, 123.8, 121.5, 120.9, 70.1, 66.7, 35.1 ppm. HRMS (ESI): m/z calcd. for C24H17ClN3OS2 [M + H]+ 462.0497, found 462.0461.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5-(4-methoxyphenyl)-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ka). Initially, 1k (26.8 mg, 0.10 mmol) and 2a (22.7 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 32.4 mg (84% yield) of compound 3ka as a white solid, m.p. 242–245 °C. HPLC (Daicel Chiralpak ADH, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 54.9 min (major enantiomer), tR = 18.1 min (minor enantiomer); 10% ee. = −50.4 (c = 1.25, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.78 (d, J = 7.6 Hz, 1H, ArH), 7.69 (d, J = 7.6 Hz, 1H, ArH), 7.63–7.54 (m, 3H, ArH), 7.40 (t, J = 7.2 Hz, 1H, ArH), 7.30–7.25 (m, 2H, ArH), 7.21–7.14 (m, 3H, ArH), 6.84 (s, 1H, ArH), 6.82 (s, 1H, NH), 6.04 (s, 1H, CH), 3.74 (s, 3H, CH3), 3.02 (d, J = 17.6 Hz, 1H, CH2), 2.89 (d, J = 17.6 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ 201.3, 179.5, 159.8, 157.5, 150.0, 148.2, 136.3, 132.7, 132.5, 131.1, 128.5, 128.0, 126.3, 125.6, 125.5, 123.7, 121.1, 120.8, 114.3, 70.8, 70.0, 55.2, 34.7 ppm. HRMS (ESI): m/z calcd. for C25H20N3O2S2 [M + H]+ 458.0991, found 458.0957.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5-(naphthalen-2-yl)-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3la). Initially, 1l (28.8 mg, 0.10 mmol) and 2a (22.7 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 33.8 mg (87% yield) of compound 3la as a white solid, m.p. 162–164 °C. HPLC (Daicel Chiralpak IC, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 8.1 min (major enantiomer), tR = 11.3 min (minor enantiomer); 92% ee. = −371.0 (c = 0.40, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.85–7.78 (m, 4H, ArH), 7.74 (s, 1H, ArH), 7.69 (d, J = 7.6 Hz 1H, ArH), 7.62–7.58 (m, 1H, ArH), 7.52–7.42 (m, 4H), 7.35–7.27 (m, 2H, ArH), 7.24–7.14 (m, 3H, ArH), 7.10 (s, 1H, NH), 6.29 (s, 1H, CH), 2.98 (d, J = 17.8 Hz, 1H, CH2), 2.87 (d, J = 17.8 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, CDCl3): δ 201.1, 179.7, 157.5, 149.9, 148.2, 136.4, 133.5, 133.4, 133.1, 132.7, 132.5, 129.2, 128.7, 128.2, 127.7, 126.62, 126.59, 126.3, 125.6, 123.8, 121.1, 120.8, 70.7, 70.5, 35.1 ppm. HRMS (ESI): m/z calcd. for C28H20N3OS2 [M + H]+ 478.1043, found 478.1006.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5-(4-nitrophenyl)-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3ma). Initially, 1m (28.3 mg, 0.10 mmol) and 2a (22.7 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 34.5 mg (90% yield) of compound 3ma as a white solid, m.p. 182–184 °C. HPLC (Daicel Chiralpak IC, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 12.0 min (major enantiomer), tR = 16.9 min (minor enantiomer); 97% ee. = −633.4 (c = 0.43, CH2Cl2). 1H NMR (400 MHz, DMSO-d6): δ 10.45 (s, 1H, NH), 8.29 (d, J = 9.2 Hz, 2H, ArH), 7.95 (d, J = 7.2 Hz, 1H, ArH), 7.80 (d, J = 7.6 Hz, 1H, ArH), 7.76–7.72 (m, 1H, ArH), 7.68–7.61 (m, 2H, ArH), 7.54–7.48 (m, 3H, ArH), 7.36–7.32 (m, 1H, ArH), 7.29–7.25 (m, 1H, ArH), 6.42 (s, 1H, CH), 3.01 (d, J = 17.8 Hz, 1H, CH2), 2.81 (d, J = 17.8 Hz, 1H, CH2) ppm. 13C NMR (100 MHz, DMSO-d6 + CD3OD): δ 201.5, 178.3, 157.5, 151.2, 147.8, 147.7, 144.5, 136.4, 132.9, 132.0, 128.5, 126.8, 126.4, 124.8, 124.2, 124.0, 121.6, 120.6, 69.5, 67.9, 34.5 ppm. HRMS (ESI): m/z calcd. for C24H17N4O3S2 [M + H]+ 473.0737, found 473.0707.
- (2′R,5S)-1-(Benzo[d]thiazol-2-yl)-5-(2,5-dichlorophenyl)-2-thioxospiro[imidazolidine-4,2′-inden]-1′(3′H)-one (3na). Initially, 1n (30.6 mg, 0.10 mmol) and 2a (22.7 mg, 0.12 mmol) were purified by silica gel (200–300 mesh) column chromatography using petroleum ether/ethyl acetate (2/1) as eluent to obtain 30.9 mg (75% yield) of compound 3na as a white solid, m.p. 268–270 °C. HPLC (Daicel Chiralpak ADH, n-hexane/isopropanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): major diastereomer: tR = 23.99min (major enantiomer), tR = 38.01 min (minor enantiomer); 87% ee. = −275.1 (c = 0.64, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 7.6 Hz, 1H, ArH), 7.73 (d, J = 8.0 Hz, 1H, ArH), 7.63 (t, J = 7.2 Hz, 1H, ArH), 7.57 (d, J = 8.0 Hz, 1H, ArH), 7.44–7.41 (m, 2H, ArH), 7.35–7.28 (m, 3H, ArH + NH), 7.25–7.18 (m, 2H, ArH), 7.11 (d, J = 8.4 Hz, 1H, ArH), 6.58 (s, 1H, CH), 3.02 (d, J = 18.0 Hz, 1H, CH2), 2.91 (d, J = 18.0 Hz, 1H, CH2) ppm. 13C NMR (176 MHz, CDCl3): δ 200.4, 179.2, 156.8, 150.3, 148.2, 136.6, 135.1, 134.5, 132.72, 132.68, 132.2, 129.6, 128.8, 128.3, 127.8, 126.3, 125.8, 125.6, 124.0, 121.5, 120.9, 70.0, 66.2, 35.0 ppm. HRMS (ESI): m/z calcd. for C24H16Cl2N3OS2 [M + H]+ 496.0107, found 496.0091.
4.4. Procedure for the Scaled-Up Synthesis of Compound 3aa
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vicini, P.; Geronikaki, A.; Incerti, M.; Busonera, B.; Poni, C.; Cabras, A.; Colla, P.L. Synthesis and biological evaluation of benzo[d]isothiazole, benzothiazole and thiazole schiff bases. Bioorgan. Med. Chem. 2003, 11, 4785–4789. [Google Scholar] [CrossRef] [PubMed]
- Malamas, M.S.; Manas, E.S.; McDevitt, R.E.; Gunawan, I.; Xu, Z.B.; Collini, M.D.; Miller, C.P.; Dinh, T.; Henderson, R.A.; Keith, J.C.; et al. Design and synthesis of aryl diphenolic azoles as potent andselective estrogen receptor-beta ligands. J. Med. Chem. 2004, 47, 5021–5040. [Google Scholar] [CrossRef]
- Damalanka, V.C.; Wildman, S.A.; Janetka, J.W. Piperidine carbamate peptidomimetic inhibitors of the serine proteases HGFA, matriptase and hepsin. Med. Chem. Commun. 2019, 10, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Patil, M.R.; Patil, S.A.; Budagumpi, A. A comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. Eur. J. Med. Chem. 2015, 89, 207–251. [Google Scholar] [CrossRef]
- Rouf, A.; Tanyeli, C. Bioactive thiazole and benzothiazole derivatives. Eur. J. Med. Chem. 2014, 97, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Sulthana, S.; Pandian, P. A review on indole and benzothiazole derivatives its importance. J. Drug. Deliv. Therap. 2019, 9, 505–509. [Google Scholar] [CrossRef]
- Noël, S.; Cadet, S.; Gras, E.; Hureau, C. The benzazole scaffold: A SWAT to combat Alzheimer’s disease. Chem. Soc. Rev. 2013, 42, 7747–7762. [Google Scholar] [CrossRef]
- Massari, S.; Daelemans, D.; Barreca, M.L.; Knezevich, A.; Sabatini, S.; Cecchetti, V.; Marcello, A.; Pannecouque, C.; Tabarrini, O. A 1,8-naphthyridone derivative targets the HIV-1 Tat-mediated transcription and potently inhibits the HIV-1 replication. J. Med. Chem. 2010, 53, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.K.; Colvin, C.J.; Needle, D.B.; Medie, F.M.; Champion, P.A.D.; Abramovitch, R.B. The carbonic anhydrase inhibitor Ethoxzolamide inhibits the Mycobacterium tuberculosis PhoPR regulon and Esx-1 secretion and attenuates virulence. Anti. Agents Chem. 2015, 59, 4436–4445. [Google Scholar] [CrossRef]
- Doble, A. The pharmacology and mechanism of action of riluzole. Neurology 1996, 47, 233S–241S. [Google Scholar] [CrossRef]
- Bryson, H.M.; Fulton, B.; Benfield, P. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in amyotrophic lateral sclerosis. Drugs 1996, 52, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergström, M.; Savitcheva, I.; Huang, G.F.; Estrada, S.; et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 2004, 55, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Zhang, H.; Zhang, L.P.; Zhao, Y.N.; Chen, S.K.; Peng, X.Y.; Li, Q.; Yuan, M.G.; Hu, X.P. Zopolrestat as a human glyoxalase I inhibitor and its structural basis. Chem. Med. Chem. 2013, 8, 1462–1464. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, N.P.; Vekariya, R.H.; Borad, M.A.; Patel, H.D. Recent advances in the synthesis of 2-substituted benzothiazoles: A review. RSC Adv. 2014, 4, 60176–60208. [Google Scholar] [CrossRef]
- He, H.X.; Yang, W.; Du, D.M. Enantioselective aza-Henry reaction of imines bearing a benzothiazole moiety catalyzed by a cinchona-based squaramide. Adv. Synth. Catal. 2013, 355, 1137–1148. [Google Scholar] [CrossRef]
- Li, W.H.; Wang, Y.F.; Xu, D.Q. Asymmetric synthesis of β-amino ketones by using cinchona alkaloid-based chiral phase transfer catalysts. Org. Bio. Chem. 2018, 16, 8704–8709. [Google Scholar] [CrossRef]
- He, H.X.; Yang, W.; Du, D.M. Organocatalytic enantioselective Strecker reaction of imines containing a thiazole moiety by using a cinchona-based squaramide catalyst. Eur. J. Org. Chem. 2014, 2014, 6190–6199. [Google Scholar] [CrossRef]
- Ni, Q.J.; Song, X.X.; Xiong, J.W.; Ender, D.; Raabe, G. Regio- and stereoselective synthesis of benzothiazolo- pyrimidinones via an NHC-catalyzed Mannich/lactamization domino reaction. Chem. Commun. 2015, 51, 1263–1266. [Google Scholar] [CrossRef] [PubMed]
- Jarrige, L.; Glavac, D.; Levitre, G.; Retailleau, P.; Bernadat, G.; Neuville, L.; Masson, G. Chiral phosphoric acid-catalyzed enantioselective construction of structurally diverse benzothiazolopyrimidines. Chem. Sci. 2019, 10, 3765–3769. [Google Scholar] [CrossRef]
- Ni, Q.J.; Wang, X.Y.; Xu, F.F.; Chen, X.Y.; Song, X.X. Organocatalytic asymmetric [4+2] cyclization of 2-benzothiazolimines with azlactones: Access to chiral benzothiazolopyrimidine derivatives. Chem. Commun. 2020, 56, 3155–3158. [Google Scholar] [CrossRef]
- Ke, C.Q.; Liu, Z.Z.; Ruan, S.; Feng, X.M.; Liu, X.H. Organocatalytic asymmetric synthesis of benzothiazolopyrimidines via a [4+2] cycloaddition of azlactones with 2-benzothiazolimines. Org. Chem. Front. 2021, 8, 5705–5709. [Google Scholar] [CrossRef]
- Chen, X.P.; Hou, K.Q.; Zhou, F.; Chan, A.S.C.; Xiong, X.F. Organocatalytic asymmetric synthesis of benzothiazolopyrimidines via [4+2] cyclization of 2-benzothiazolimines and aldehydes. J. Org. Chem. 2021, 86, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Song, X.X.; Gu, M.J.; Chen, X.Y.; Xu, L. Highly Stereoselective palladium-catalyzed [3+2] cycloaddition of vinyl epoxides and N-benzothiazolimines. Asian J. Org. Chem. 2019, 8, 2180–2183. [Google Scholar] [CrossRef]
- Liu, S.J.; Chen, Z.H.; Chen, J.Y.; Ni, S.F.; Zhang, Y.C.; Shi, F. Rational design of axially chiral styrene-based organocatalysts and their application in catalytic asymmetric (2+4) cyclizations. Angew. Chem. Int. Ed. 2022, 61, e202112226. [Google Scholar] [CrossRef] [PubMed]
- Li, K.H.; Lu, D.M.; Tan, J.P.; Ren, X.; Wang, T. Chemoselective annulation of 2-benzothiazolimines enabled by bifunctional phosphonium salt: Highly enantioselective synthesis of indole-fused cyclic thiourea scaffolds. Asian J. Org. Chem. 2022, 11, e202200368. [Google Scholar] [CrossRef]
- CCDC 2346984 (for 3ab) Contains the Supplementary Crystallographic Data for this Paper. These Data Can Be Obtained Free of Charge via. Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 9 April 2024).
- Zhao, B.L.; Du, D.M. Catalytic Asymmetric Mannich/cyclization of 2-isothiocyanato-1-indanones: An approach to the synthesis of bispirocyclic indanone−thioimidazolidine−oxindoles. Org. Lett. 2018, 20, 3797–3800. [Google Scholar] [CrossRef]
- Yoneda, N.; Fukata, Y.; Asano, K.; Matsubara, S. Asymmetric synthesis of spiroketals with aminothiourea catalysts. Angew. Chem. Int. Ed. 2015, 54, 15497–15500. [Google Scholar] [CrossRef]
- Vakulya, B.; Varga, S.; Csámpai, A.; Soos, T. Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional cinchona organocatalysts. Org. Lett. 2005, 7, 1967–1969. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst | Yield b (%) | dr c | ee d (%) |
| 1 | CH2Cl2 | C1 | 75 | >20:1 | 83 |
| 2 | CH2Cl2 | C2 | 78 | >20:1 | 92 |
| 3 | CH2Cl2 | C3 | 84 | >20:1 | 82 |
| 4 | CH2Cl2 | C4 | 80 | >20:1 | 79 |
| 5 | CH2Cl2 | C5 | 76 | >20:1 | 52 |
| 6 | CH2Cl2 | C6 | 71 | >20:1 | 7 |
| 7 | CH2Cl2 | C7 | 83 | >20:1 | 95 |
| 8 | CH2Cl2 | C8 | 80 | >20:1 | 89 |
| 9 | CH2Cl2 | C9 | 81 | >20:1 | 52 |
| 10 | CH2Cl2 | C10 | 86 | >20:1 | 87 |
| 11 | CH2Cl2 | C11 | 78 | >20:1 | 73 |
| 12 | CH2Cl2 | C12 | 77 | >20:1 | 95 |
| 13 | CH2Cl2 | C13 | 73 | >20:1 | 75 |
| 14 | CH2Cl2 | C14 | 71 | >20:1 | 4 |
| 15 | dioxane | C7 | 61 | >20:1 | 74 |
| 16 | EtOAc | C7 | 76 | >20:1 | 65 |
| 17 | DCE | C7 | 80 | >20:1 | 71 |
| 18 | MeCN | C7 | 81 | >20:1 | 43 |
| 19 | THF | C7 | 83 | >20:1 | 54 |
| 20 | MTBE | C7 | 77 | >20:1 | 84 |
| 21 e | CH2Cl2 | C7 | 79 | >20:1 | 67 |
| 22 f | CH2Cl2 | C7 | 83 | >20:1 | 79 |
| 23 g | CH2Cl2 | C7 | 85 | >20:1 | 98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.; Du, D.-M. Asymmetric Mannich/Cyclization Reaction of 2-Benzothiazolimines and 2-Isothiocyano-1-indanones to Construct Chiral Spirocyclic Compounds. Molecules 2024, 29, 2958. https://doi.org/10.3390/molecules29132958
Zheng Y, Du D-M. Asymmetric Mannich/Cyclization Reaction of 2-Benzothiazolimines and 2-Isothiocyano-1-indanones to Construct Chiral Spirocyclic Compounds. Molecules. 2024; 29(13):2958. https://doi.org/10.3390/molecules29132958
Chicago/Turabian StyleZheng, Yao, and Da-Ming Du. 2024. "Asymmetric Mannich/Cyclization Reaction of 2-Benzothiazolimines and 2-Isothiocyano-1-indanones to Construct Chiral Spirocyclic Compounds" Molecules 29, no. 13: 2958. https://doi.org/10.3390/molecules29132958
APA StyleZheng, Y., & Du, D.-M. (2024). Asymmetric Mannich/Cyclization Reaction of 2-Benzothiazolimines and 2-Isothiocyano-1-indanones to Construct Chiral Spirocyclic Compounds. Molecules, 29(13), 2958. https://doi.org/10.3390/molecules29132958