
Unveiling the Potential of B3O3 Nanoflake as Effective Transporter for the Antiviral Drug Favipiravir: Density Functional Theory Analysis

 , , and
, , and
Abstract
:1. Introduction
2. Results
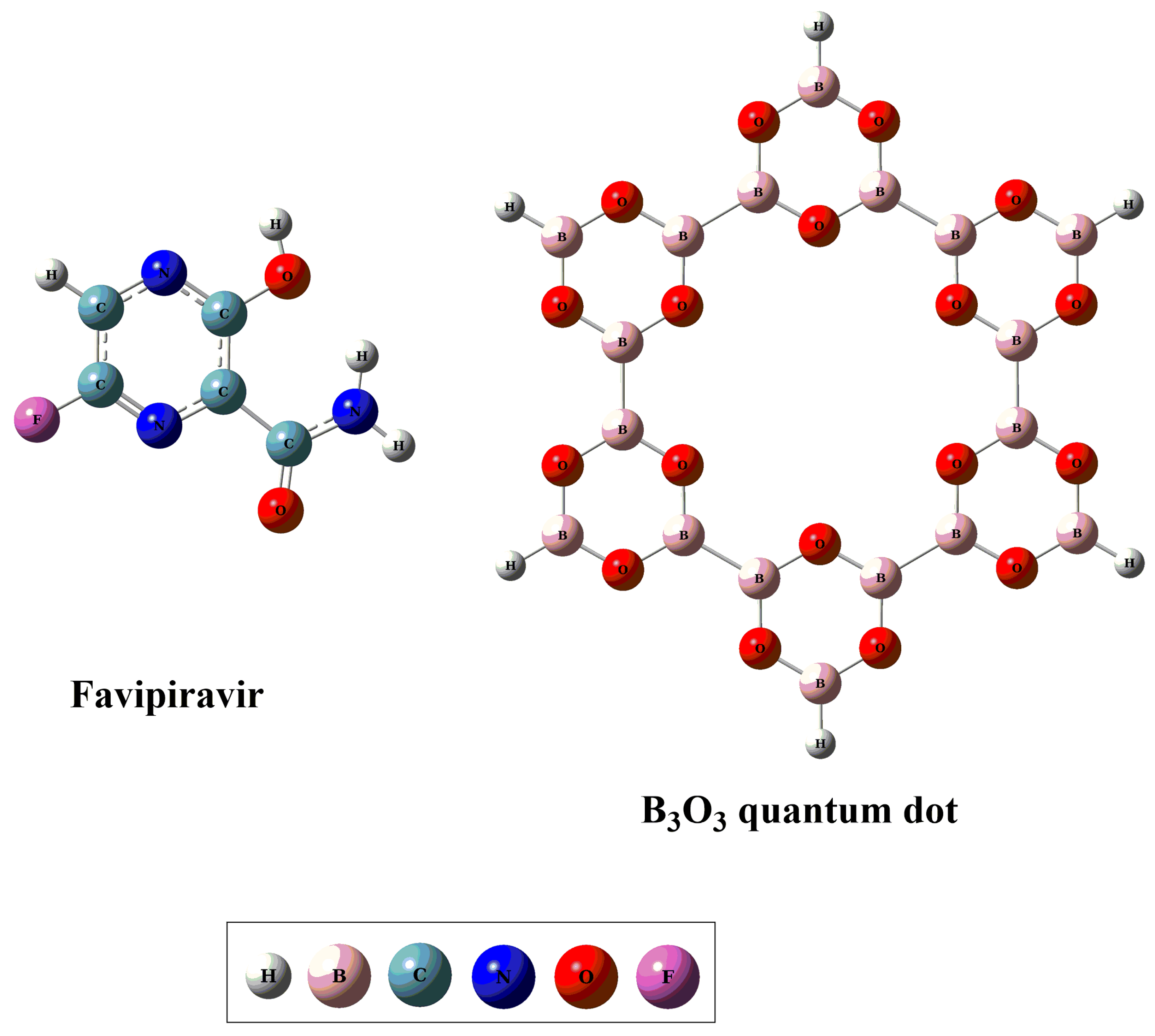
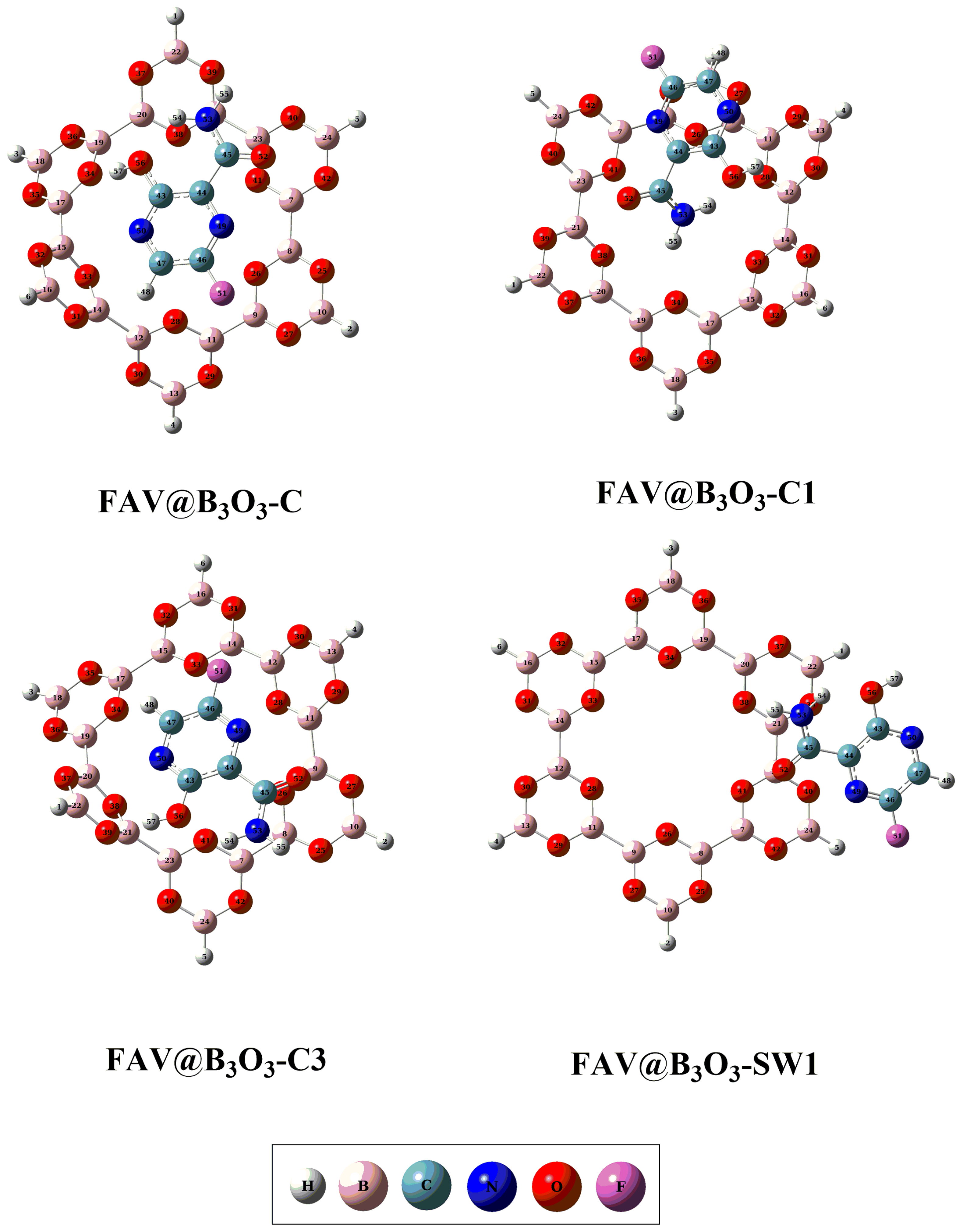
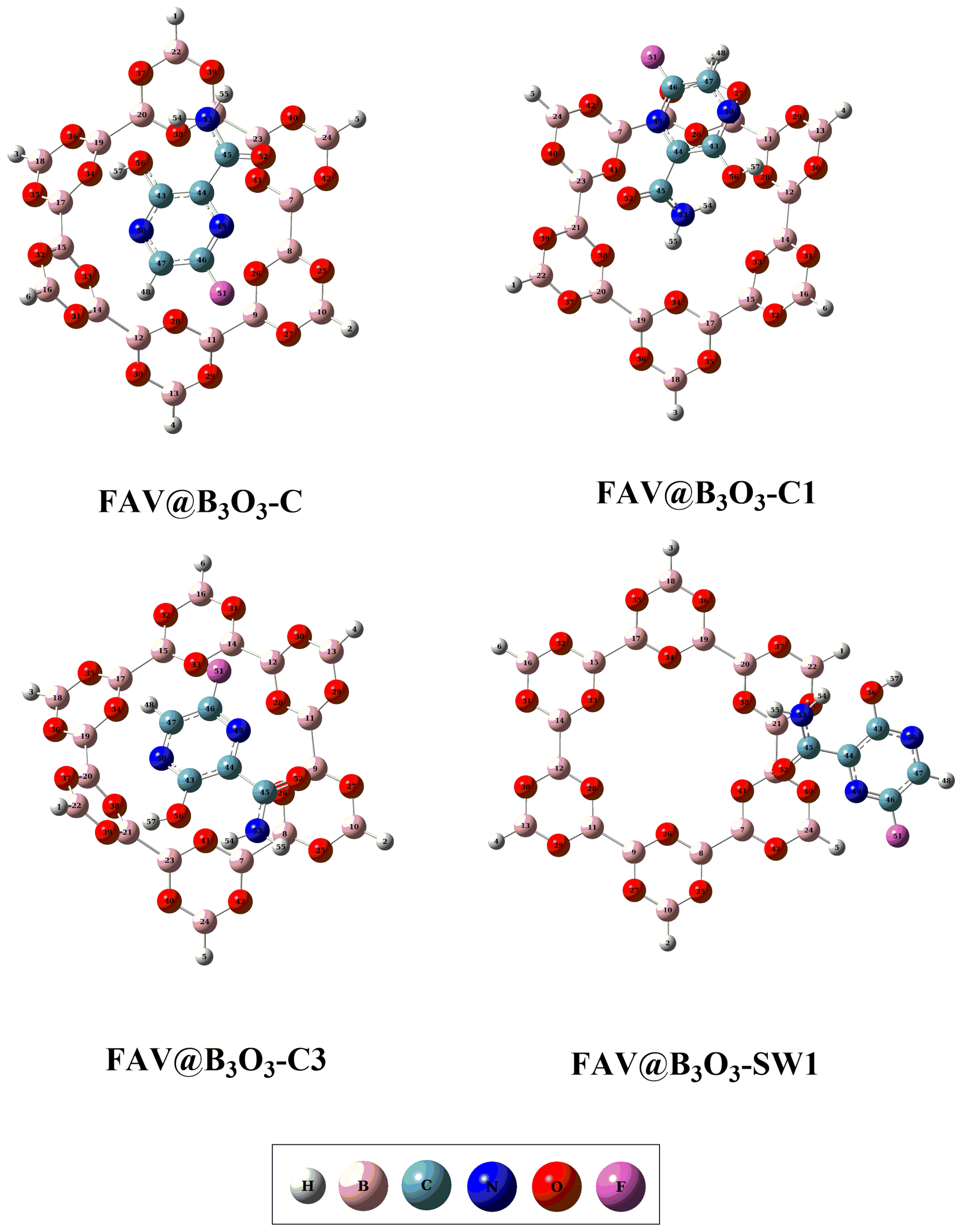
2.1. Geometric and Energetic Analysis
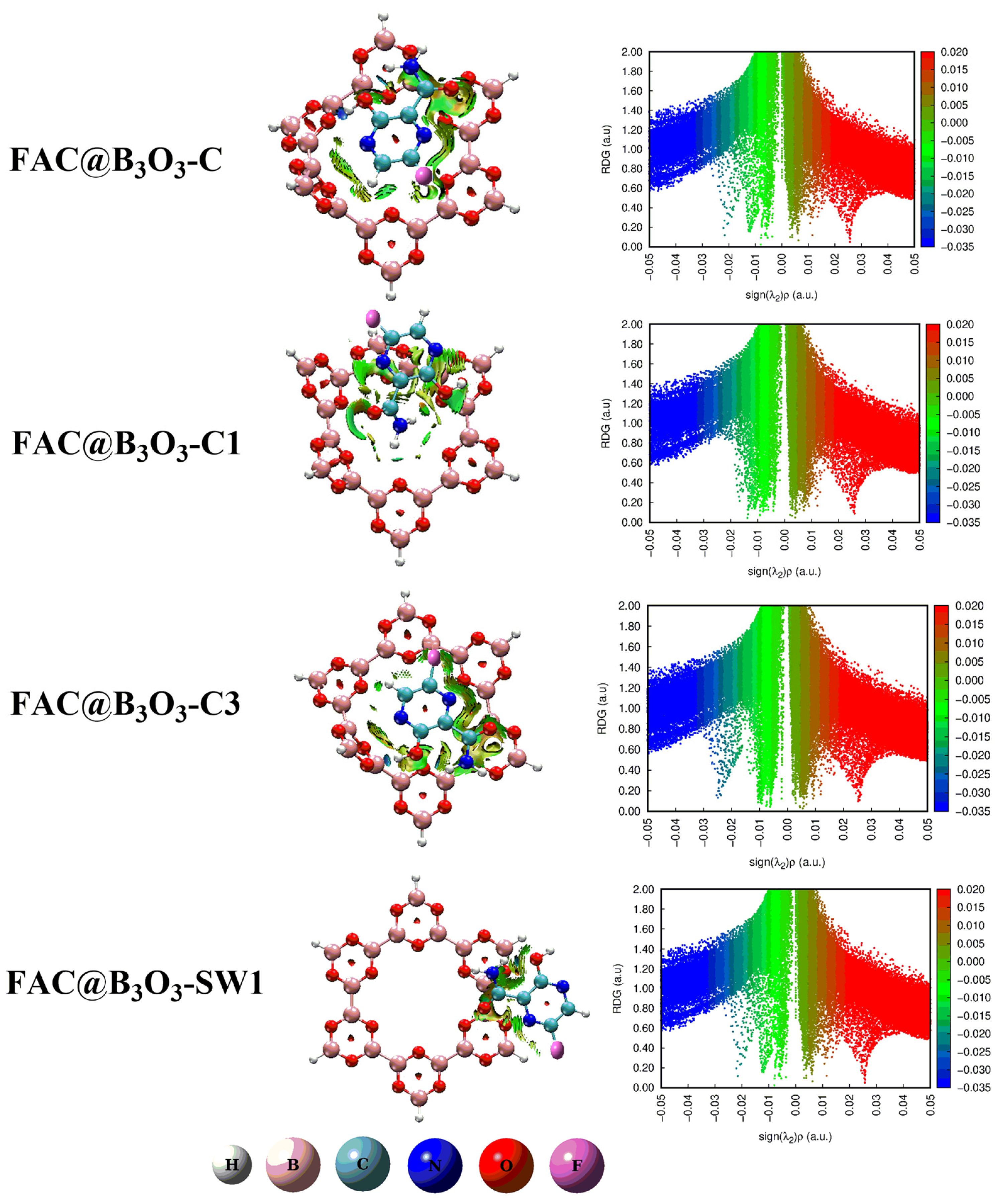
2.2. Noncovalent Interactions (NCI) Analysis
2.3. Quantum Theory of Atoms in Molecules (QTAIM) Analysis
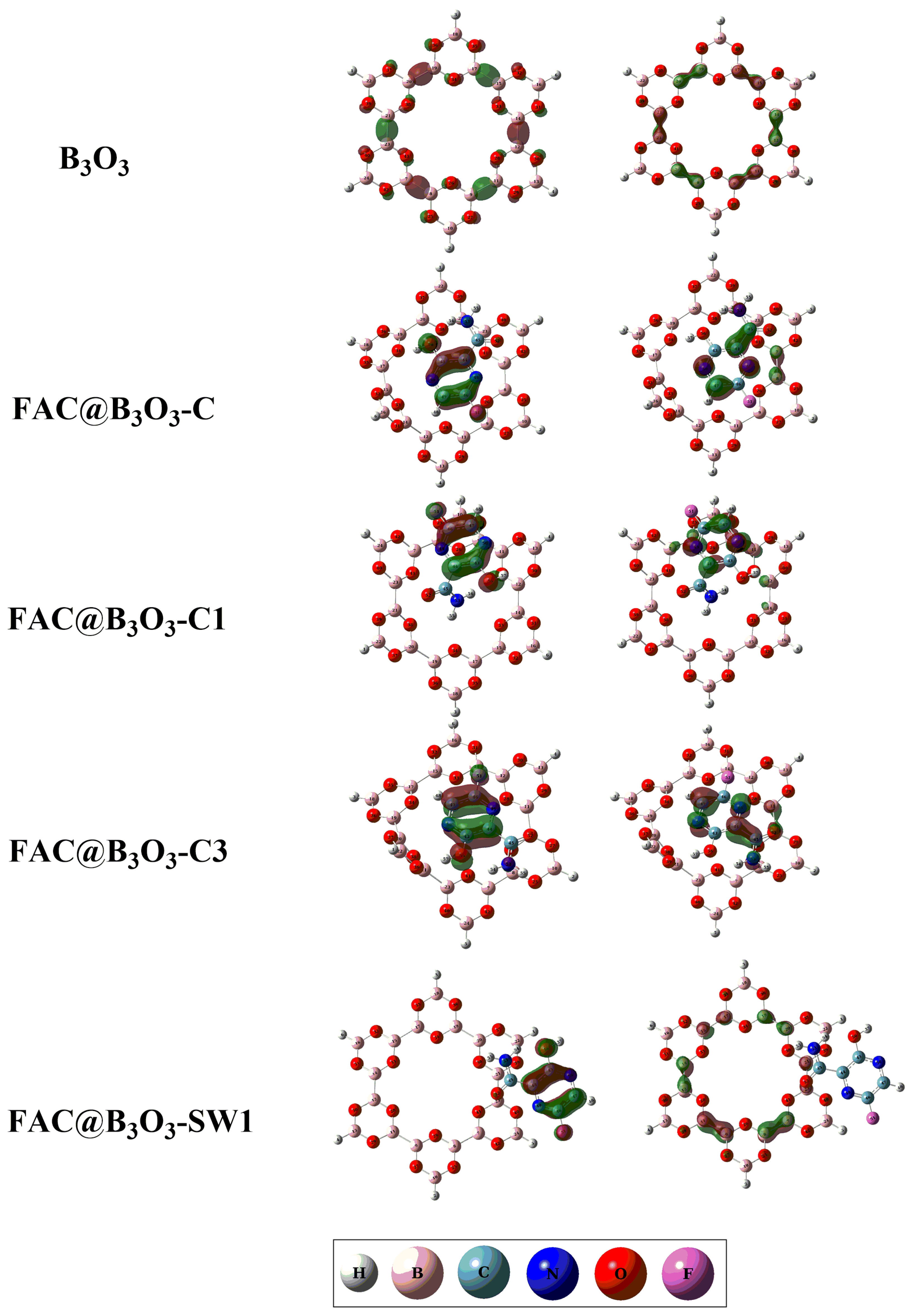
2.4. Electronic Properties
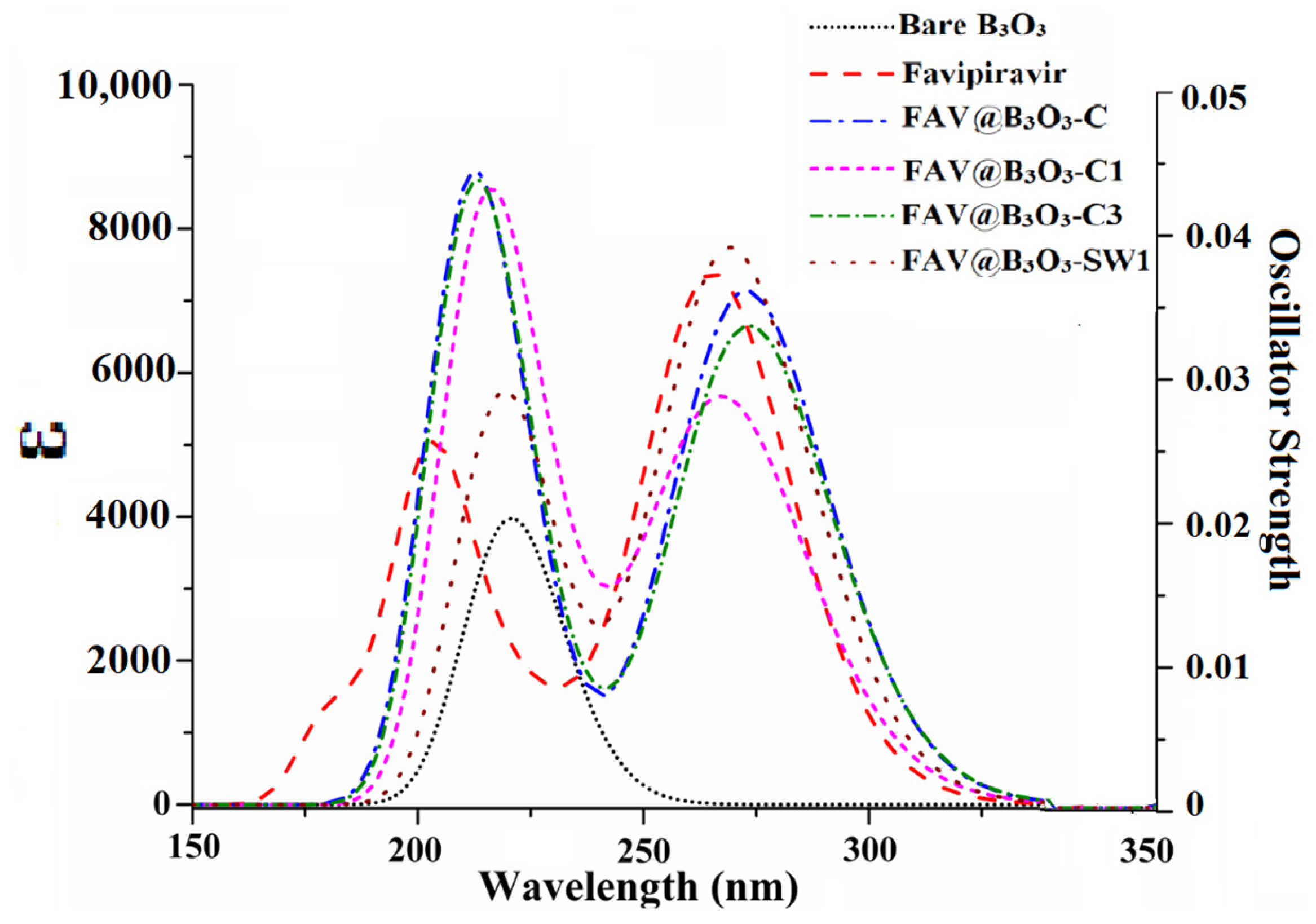
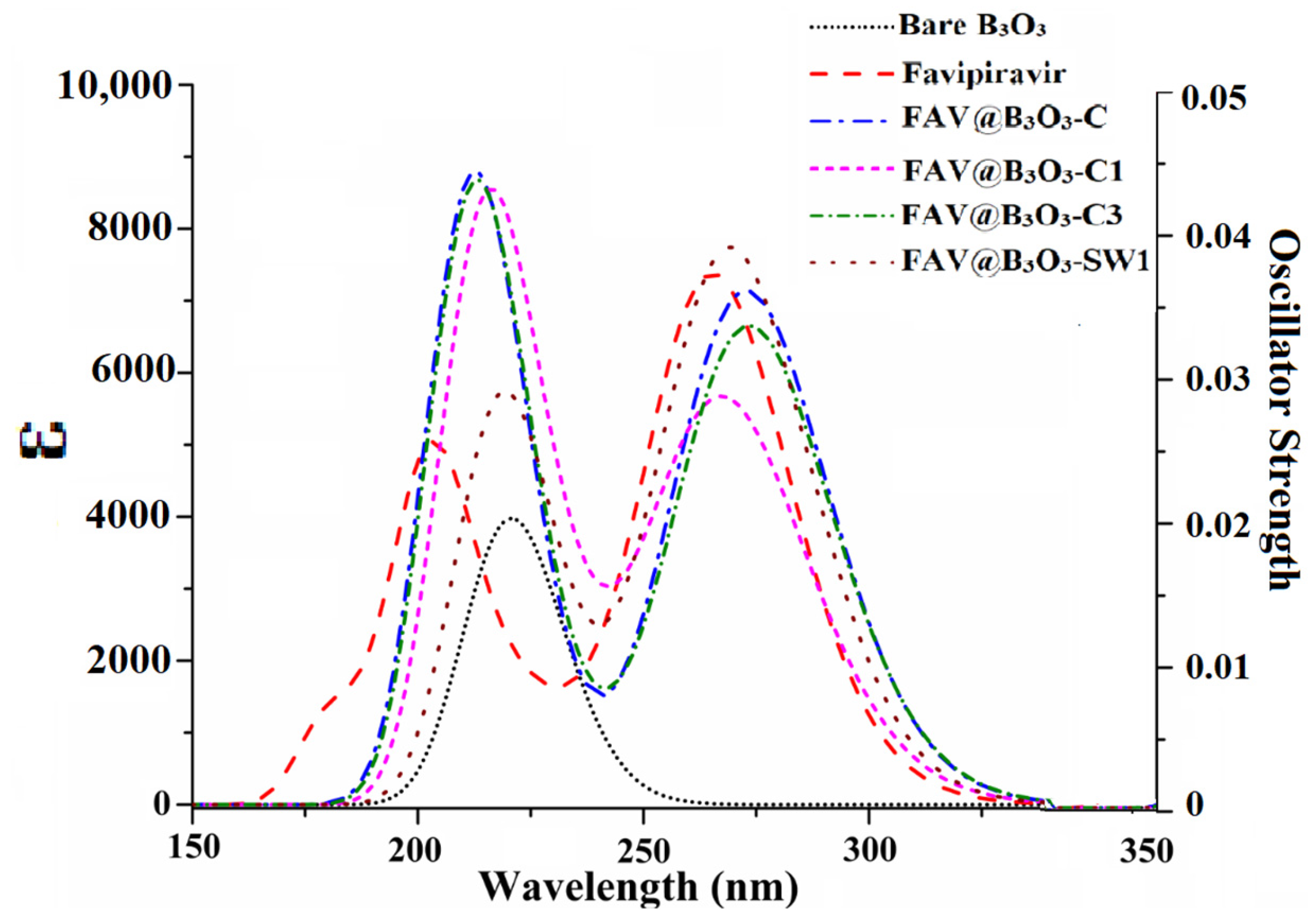
2.5. UV-VIS Analysis
3. Materials and Methods
Computational Methodology
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agrawal, U.; Raju, R.; Udwadia, Z.F. Favipiravir: A New and Emerging Antiviral Option in COVID-19. Med. J. Armed Forces India 2020, 76, 370–376. [Google Scholar] [CrossRef]
- Furuta, Y.; Gowen, B.B.; Takahashi, K.; Shiraki, K.; Smee, D.F.; Barnard, D.L. Favipiravir (T-705), a Novel Viral RNA Polymerase Inhibitor. Antivir. Res. 2013, 100, 446–454. [Google Scholar] [CrossRef]
- Pandey, A.; Yadav, S. Essentials of COVID-19 and Treatment Approaches. In Data Science for COVID-19; Elsevier: Amsterdam, The Netherlands, 2022; pp. 397–422. [Google Scholar]
- Sissoko, D.; Laouenan, C.; Folkesson, E.; M’Lebing, A.-B.; Beavogui, A.-H.; Baize, S.; Camara, A.-M.; Maes, P.; Shepherd, S.; Danel, C.; et al. Experimental Treatment with Favipiravir for Ebola Virus Disease (the JIKI Trial): A Historically Controlled, Single-Arm Proof-of-Concept Trial in Guinea. PLoS Med. 2016, 13, e1001967. [Google Scholar] [CrossRef]
- Chen, P.; Nirula, A.; Heller, B.; Gottlieb, R.L.; Boscia, J.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with COVID-19. N. Engl. J. Med. 2021, 384, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, K.; Agarwal, A.; Jaiswal, N.; Dahiya, N.; Ahuja, A.; Mahajan, S.; Tong, L.; Duggal, M.; Singh, M.; Agrawal, R.; et al. Ocular Surface Manifestations of Coronavirus Disease 2019 (COVID-19): A Systematic Review and Meta-Analysis. PLoS ONE 2020, 15, e0241661. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Multifunctional, Stimuli-Sensitive Nanoparticulate Systems for Drug Delivery. Nat. Rev. Drug Discov. 2014, 13, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Bryan, C.J.; Mintz, J.; Clemans, T.A.; Leeson, B.; Burch, T.S.; Williams, S.R.; Maney, E.; Rudd, M.D. Effect of Crisis Response Planning vs. Contracts for Safety on Suicide Risk in U.S. Army Soldiers: A Randomized Clinical Trial. J. Affect. Disord. 2017, 212, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Wang, L.; Zeng, S. Inter-Organizational Knowledge Acquisition and Firms’ Radical Innovation: A Moderated Mediation Analysis. J. Bus. Res. 2018, 90, 295–306. [Google Scholar] [CrossRef]
- Mariadoss, A.V.A.; Saravanakumar, K.; Sathiyaseelan, A.; Venkatachalam, K.; Wang, M.-H. Folic Acid Functionalized Starch Encapsulated Green Synthesized Copper Oxide Nanoparticles for Targeted Drug Delivery in Breast Cancer Therapy. Int. J. Biol. Macromol. 2020, 164, 2073–2084. [Google Scholar] [CrossRef]
- Nagarajan, V.; Chandiramouli, R. NiO Nanocone as a CO Sensor: DFT Investigation. Struct. Chem. 2014, 25, 1765–1771. [Google Scholar] [CrossRef]
- Chandiramouli, R.; Nagarajan, V. Borospherene Nanostructure as CO and NO Sensor—A First-Principles Study. Vacuum 2017, 142, 13–20. [Google Scholar] [CrossRef]
- Chandiramouli, R.; Jeyaprakash, B.G. Review of CdO Thin Films. Solid State Sci. 2013, 16, 102–110. [Google Scholar] [CrossRef]
- Bhuvaneswari, R.; Nagarajan, V.; Chandiramouli, R. Arsenene Nanoribbons for Sensing NH3 and PH3 Gas Molecules—A First-Principles Perspective. Appl. Surf. Sci. 2019, 469, 173–180. [Google Scholar] [CrossRef]
- Padash, R.; Sobhani-Nasab, A.; Rahimi-Nasrabadi, M.; Mirmotahari, M.; Ehrlich, H.; Rad, A.S.; Peyravi, M. Is It Possible to Use X12Y12 (X = Al, B, and Y = N, P) Nanocages for Drug-Delivery Systems? A DFT Study on the Adsorption Property of 4-Aminopyridine Drug. Appl. Phys. A 2018, 124, 582. [Google Scholar] [CrossRef]
- Rahimi, R.; Solimannejad, M. B3O3 Monolayer with Dual Application in Sensing of COVID-19 Biomarkers and Drug Delivery for Treatment Purposes: A Periodic DFT Study. J. Mol. Liq. 2022, 354, 118855. [Google Scholar] [CrossRef]
- Vatanparast, M.; Shariatinia, Z. Hexagonal Boron Nitride Nanosheet as Novel Drug Delivery System for Anticancer Drugs: Insights from DFT Calculations and Molecular Dynamics Simulations. J. Mol. Graph. Model. 2019, 89, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Sakr, A.K.; Al-Hamarneh, I.F.; Gomaa, H.; Abdel Aal, M.M.; Hanfi, M.Y.; Sayyed, M.I.; Khandaler, M.U.; Cheira, M.F. Removal of Uranium from Nuclear Effluent Using Regenerated Bleaching Earth Steeped in Β-naphthol. Radiat. Phys. Chem. 2022, 200, 110204. [Google Scholar] [CrossRef]
- Bhuvaneswari, R.; Princy Maria, J.; Nagarajan, V.; Chandiramouli, R. Graphdiyne Nanosheets as a Sensing Medium for Formaldehyde and Formic Acid—A First-Principles Outlook. Comput. Theor. Chem. 2020, 1176, 112751. [Google Scholar] [CrossRef]
- Mujtaba Munir, M.A.; Yousaf, B.; Ali, M.U.; Dan, C.; Abbas, Q.; Arif, M.; Yang, X. In Situ Synthesis of Micro-Plastics Embedded Sewage-Sludge Co-Pyrolyzed Biochar: Implications for the Remediation of Cr and Pb Availability and Enzymatic Activities from the Contaminated Soil. J. Clean. Prod. 2021, 302, 127005. [Google Scholar] [CrossRef]
- Perveen, S.; Akram, M.; Nasar, A.; Arshad-Ayaz, A.; Naseem, A. Vaccination-hesitancy and Vaccination-inequality as Challenges in Pakistan’s COVID-19 Response. J. Community Psychol. 2022, 50, 666–683. [Google Scholar] [CrossRef]
- Zeenat; Elahi, A.; Bukhari, D.A.; Shamim, S.; Rehman, A. Plastics Degradation by Microbes: A Sustainable Approach. J. King Saud Univ.—Sci. 2021, 33, 101538. [Google Scholar] [CrossRef]
- Srimathi, U.; Nagarajan, V.; Chandiramouli, R. Investigation on Graphdiyne Nanosheet in Adsorption of Sorafenib and Regorafenib Drugs: A DFT Approach. J. Mol. Liq. 2019, 277, 776–785. [Google Scholar] [CrossRef]
- Yang, F.; Wang, X.; Li, M.; Liu, X.; Zhao, X.; Zhang, D.; Zhang, Y.; Yang, J.; Li, Y. Templated Synthesis of Single-Walled Carbon Nanotubes with Specific Structure. Acc. Chem. Res. 2016, 49, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Shen, B.; Cui, Y.; Chen, Y.; Wang, J.; Wang, L.; Kang, Y.; Zhao, X.; Si, W.; Li, W.; et al. Generation of Gene-Modified Cynomolgus Monkey via Cas9/RNA-Mediated Gene Targeting in One-Cell Embryos. Cell 2014, 156, 836–843. [Google Scholar] [CrossRef]
- Schwierz, F. Graphene Transistors. Nat. Nanotechnol. 2010, 5, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Singh Raman, R.K.; Tiwari, A. Graphene: The Thinnest Known Coating for Corrosion Protection. JOM 2014, 66, 637–642. [Google Scholar] [CrossRef]
- Barzan Talab, M.; Hasan Muttashar, H.; Faraj, J.; Abdullaha, S.A.H.; Hachim, S.K.; Adel, M.; Kadhim, M.M.; Mahdi Rheima, A. Inspection the Potential of B3O3 Monolayer as a Carrier for Flutamide Anticancer Delivery System. Comput. Theor. Chem. 2022, 1217, 113886. [Google Scholar] [CrossRef]
- Li, H.-Y.; Zhao, S.-N.; Zang, S.-Q.; Li, J. Functional Metal–Organic Frameworks as Effective Sensors of Gases and Volatile Compounds. Chem. Soc. Rev. 2020, 49, 6364–6401. [Google Scholar] [CrossRef]
- Li, M.; Cushing, S.K.; Wu, N. Plasmon-Enhanced Optical Sensors: A Review. Analyst 2015, 140, 386–406. [Google Scholar] [CrossRef]
- Florensa, M.; Llenas, M.; Medina-Gutiérrez, E.; Sandoval, S.; Tobías-Rossell, G. Key Parameters for the Rational Design, Synthesis, and Functionalization of Biocompatible Mesoporous Silica Nanoparticles. Pharmaceutics 2022, 14, 2703. [Google Scholar] [CrossRef]
- Vatanparast, M.; Shariatinia, Z. Revealing the Role of Different Nitrogen Functionalities in the Drug Delivery Performance of Graphene Quantum Dots: A Combined Density Functional Theory and Molecular Dynamics Approach. J. Mater. Chem. B 2019, 7, 6156–6171. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Jin, C.; Jin, W.; Hou, X. Target-Oriented Synthesis of Borate Derivatives Featuring Isolated [B3O3] Six-Membered Rings as Structural Features. Inorg. Chem. 2023, 62, 9209–9216. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Li, A.; Liu, J.-C.; Li, Z.; Chen, W.; Gong, Y.; Zhang, Q.; Cheong, W.-C.; Wang, Y.; Zheng, L.; et al. Direct Observation of Noble Metal Nanoparticles Transforming to Thermally Stable Single Atoms. Nat. Nanotechnol. 2018, 13, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Stredansky, M.; Sala, A.; Fontanot, T.; Costantini, R.; Africh, C.; Comelli, G.; Floreano, L.; Morgante, A.; Cossaro, A. On-Surface Synthesis of a 2D Boroxine Framework: A Route to a Novel 2D Material? Chem. Commun. 2018, 54, 3971–3973. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Xing, J.; Quan, L.N.; de Arquer, F.P.G.; Gong, X.; Lu, J.; Xie, L.; Zhao, W.; Zhang, D.; Yan, C.; et al. Perovskite Light-Emitting Diodes with External Quantum Efficiency Exceeding 20 per Cent. Nature 2018, 562, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gu, J.; Zhang, H.; Wang, Y.; Chen, Z. Porous Hexagonal Boron Oxide Monolayer with Robust Wide Band Gap: A Computational Study. FlatChem 2018, 9, 27–32. [Google Scholar] [CrossRef]
- Nauman Zahid, M.; Asif, M.; Sajid, H.; Kosar, N.; Akbar Shahid, M.; Allangawi, A.; Ayub, K.; Azeem, M.; Mahmood, T. Therapeutic Efficiency of B3O3 Quantum Dot as a Targeted Drug Delivery System toward Foscarnet Anti-HIV Drug. Comput. Theor. Chem. 2023, 1224, 114107. [Google Scholar] [CrossRef]
- Rahimi, R.; Solimannejad, M.; Ehsanfar, Z. First-Principles Studies on Two-Dimensional B3O3 Adsorbent as a Potential Drug Delivery Platform for TEPA Anticancer Drug. J. Mol. Model. 2021, 27, 347. [Google Scholar] [CrossRef]
- Carrera Espinoza, M.J.; Lin, K.-S.; Weng, M.-T.; Kunene, S.C.; Lin, Y.-S.; Liu, S.-Y. Magnetic Boron Nitride Nanosheets-Based on PH-Responsive Smart Nanocarriers for the Delivery of Doxorubicin for Liver Cancer Treatment. Colloids Surf. B Biointerfaces 2023, 222, 113129. [Google Scholar] [CrossRef]
- Saha, S.K.; Murmu, M.; Murmu, N.C.; Obot, I.B.; Banerjee, P. Molecular Level Insights for the Corrosion Inhibition Effectiveness of Three Amine Derivatives on the Carbon Steel Surface in the Adverse Medium: A Combined Density Functional Theory and Molecular Dynamics Simulation Study. Surf. Interfaces 2018, 10, 65–73. [Google Scholar] [CrossRef]
- Trabelsi, S.; Tlili, M.; Abdelmoulahi, H.; Bouazizi, S.; Nasr, S.; González, M.A.; Bellissent-Funel, M.-C.; Darpentigny, J. Intermolecular Interactions in an Equimolar Methanol-Water Mixture: Neutron Scattering, DFT, NBO, AIM, and MD Investigations. J. Mol. Liq. 2022, 349, 118131. [Google Scholar] [CrossRef]
- Farías-Rico, J.A.; Ruud Selin, F.; Myronidi, I.; Frühauf, M.; von Heijne, G. Effects of Protein Size, Thermodynamic Stability, and Net Charge on Cotranslational Folding on the Ribosome. Proc. Natl. Acad. Sci. USA 2018, 115, E9280–E9287. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Wang, T.; An, Y.; Dai, X.; Xia, C. VS2 Nanosheet as a Promising Candidate of Recycle and Reuse NO2 Gas Sensor and Capturer: A DFT Study. J. Phys. Condens. Matter 2021, 33, 165501. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Ghalla, H.; Issaoui, N.; Bardak, F.; Atac, A. Intermolecular Interactions and Molecular Docking Investigations on 4-Methoxybenzaldehyde. Comput. Mater. Sci. 2018, 149, 291–300. [Google Scholar] [CrossRef]
- Issaoui, N.; Abdessalem, K.; Ghalla, H.; Yaghmour, S.J.; Calvo, F.; Oujia, B. Theoretical Investigation of the Relative Stability of Na + He n (n = 2–24) Clusters: Many-Body versus Delocalization Effects. J. Chem. Phys. 2014, 141, 174316. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Chemical Bonds without Bonding Electron Density? Does the Difference Electron-Density Analysis Suffice for a Description of the Chemical Bond? Angew. Chem. Int. Ed. Engl. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Jenkins, S.; Morrison, I. The Chemical Character of the Intermolecular Bonds of Seven Phases of Ice as Revealed by Ab Initio Calculation of Electron Densities. Chem. Phys. Lett. 2000, 317, 97–102. [Google Scholar] [CrossRef]
- Khan, S.; Yar, M.; Kosar, N.; Ayub, K.; Arshad, M.; Zahid, M.N.; Mahmood, T. First-Principles Study for Exploring the Adsorption Behavior of G-Series Nerve Agents on Graphdyine Surface. Comput. Theor. Chem. 2020, 1191, 113043. [Google Scholar] [CrossRef]
- Asif, M.; Sajid, H.; Kosar, N.; Mahmood, T. Effect of Fluorination on the Adsorption Properties of Aromatic Heterocycles toward Methyl Halides: A Quantum Chemical Study. Comput. Theor. Chem. 2021, 1204, 113394. [Google Scholar] [CrossRef]
- Perveen, M.; Nazir, S.; Arshad, A.W.; Khan, M.I.; Shamim, M.; Ayub, K.; Khan, M.A.; Iqbal, J. Therapeutic Potential of Graphitic Carbon Nitride as a Drug Delivery System for Cisplatin (Anticancer Drug): A DFT Approach. Biophys. Chem. 2020, 267, 106461. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, J.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Schlegel, H.B.; Scalmani, G.; Barone, V.; Mennucci, B. Gaussian 09, Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. GaussView 5.0; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Su, H.; Wang, H.; Wang, H.; Lu, Y.; Zhu, Z. Description of Noncovalent Interactions Involving Π-system with High Precision: An Assessment of RPA, MP2, and DFT-D Methods. J. Comput. Chem. 2019, 40, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, P.; Karmakar, A.; Deb, J.; Sarkar, U.; Seikh, M.M. Non-Covalent Interactions between Epinephrine and Nitroaromatic Compounds: A DFT Study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 228, 117827. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Pang, H.; Li, H.; Wang, X. A Density Functional Theory Study on Complexation Processes and Intermolecular Interactions of Triptycene-Derived Oxacalixarenes. Theor. Chem. Acc. 2019, 138, 113. [Google Scholar] [CrossRef]
- Turi, L.; Dannenberg, J.J. Correcting for Basis Set Superposition Error in Aggregates Containing More than Two Molecules: Ambiguities in the Calculation of the Counterpoise Correction. J. Phys. Chem. 1993, 97, 2488–2490. [Google Scholar] [CrossRef]
- Hasan, P. Antimonachismus v České Osvícenské Společnosti. Cornova 2013, 3, 83–100. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- De Castro, E.A.S.; de Oliveira, D.A.B.; Farias, S.A.S.; Gargano, R.; Martins, J.B.L. Structure and Electronic Properties of Azadirachtin. J. Mol. Model. 2014, 20, 2084. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Zhang, J.; Grant, C. Ab Initio Calculation of Ionization Potential and Electron Affinity of Six Common Explosive Compounds. Rep. Theor. Chem. 2012, 1, 11–19. [Google Scholar] [CrossRef]
- Alp, M.; Yurdakul, S. Experimental and Theoretical Vibrational Spectroscopic, Quantum Chemical Analysis, and Electronic Properties Investigations of Novel Ruthenium Complexes (RuLCl2·2H2O; L: 4,4′-Dimethoxy-2,2′-Bipyridine, 4,4′-Dimethyl-2,2′-Bipyridine). Polyhedron 2023, 234, 116322. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complexes | Eint (kcal/mol) | Aint | dint (Å) | Ecp (kcal/mol) |
|---|---|---|---|---|
| FAV@B3O3-C | −23.84 | H57—O34 | 1.84 | −20.08 |
| H48—O28 | 2.70 | |||
| FAV@B3O3-C1 | −26.98 | H55—O34 | 2.13 | −22.59 |
| H54—O28 | 2.27 | |||
| FAV@B3O3-C3 | −24.47 | H57—O39 | 1.94 | −20.70 |
| H48—O34 | 2.57 | |||
| FAV@B3O3-SW1 | −12.55 | O52—B23 | 2.71 | −10.66 |
| N49—O40 | 3.01 |
| BCPs | Ana-Surface | ρ | ∇2ρ | G | V | H |
|---|---|---|---|---|---|---|
| FAV@B3O3-C | ||||||
| 78 | N49-O26 | 0.60 × 10−2 | 0.21 × 10−1 | 0.44 × 10−2 | −0.36 × 10−2 | 0.79 × 10−3 |
| 130 | H57-O34 | 0.31 × 10−1 | 0.94 × 10−1 | 0.23 × 10−1 | −0.23 × 10−1 | 0.92 × 10−4 |
| FAV@B3O3-C1 | ||||||
| 81 | N49-B7 | 0.80 × 10−2 | 0.24 × 10−1 | 0.52 × 10−2 | −0.44 × 10−2 | 0.79 × 10−3 |
| 95 | H55-O34 | 0.17 × 10−1 | 0.52 × 10−1 | 0.13 × 10−1 | −0.13 × 10−1 | −0.26 × 10−3 |
| 132 | O56-B9 | 0.12 × 10−1 | 0.33 × 10−1 | 0.80 × 10−2 | −0.77 × 10−2 | 0.22 × 10−3 |
| FAV@B3O3-C3 | ||||||
| 24 | H48-O34 | 0.42 × 100 | −0.18 × 102 | 0.68 × 10−2 | −0.46 × 101 | −0.46 × 101 |
| 65 | H57-O39 | 0.25 × 10−1 | 0.71 × 10−1 | 0.18 × 10−1 | −0.19 × 10−1 | 0.49 × 103 |
| 126 | O56-O41 | 0.11 × 10−1 | 0.35 × 10−1 | 0.82 × 10−2 | −0.77 × 10−2 | 0.52 × 103 |
| FAV@B3O3-SW1 | ||||||
| 66 | O56-H1 | 0.54 × 10−2 | 0.19 × 10−1 | 0.38 × 10−2 | −0.29 × 10−2 | 0.93 × 10−3 |
| 109 | N49-O40 | 0.79 × 10−2 | 0.26 × 10−1 | 0.59 × 10−2 | −0.53 × 10−2 | 0.60 × 10−3 |
| 119 | F51-H5 | 0.36 × 10−2 | 0.16 × 10−1 | 0.31 × 10−2 | −0.20 × 10−2 | 0.11 × 10−2 |
| Complexes | EHOMO | ELUMO | EL-H | s | η | μ | ω |
|---|---|---|---|---|---|---|---|
| B3O3 | −10.42 | −0.46 | 9.96 | 0.10 | 4.98 | −5.44 | 2.96 |
| Favipiravir | −9.51 | −0.72 | 8.79 | 0.11 | 4.40 | −5.11 | 2.97 |
| FAV@B3O3-C | −9.53 | −0.89 | 8.64 | 0.12 | 4.32 | −5.21 | 3.14 |
| FAV@B3O3-C1 | −9.54 | −0.85 | 8.69 | 0.11 | 4.34 | −5.20 | 3.11 |
| FAV@B3O3-C3 | −9.59 | −0.97 | 8.62 | 0.12 | 4.31 | −5.28 | 3.23 |
| FAV@B3O3-SW1 | −9.59 | −0.88 | 8.71 | 0.11 | 4.36 | −5.23 | 3.13 |
| Complex | Wavelength (nm) | Oscillating Strength (fo) | Excitation energy (eV) |
|---|---|---|---|
| B3O3 | 221 | 0.098 | 5.62 |
| Favipiravir | 265 | 0.179 | 4.67 |
| FAV@B3O3-C | 273 | 0.170 | 4.54 |
| FAV@B3O3-C1 | 266 | 0.119 | 4.66 |
| FAV@B3O3-C3 | 274 | 0.154 | 4.53 |
| FAV@B3O3-SW1 | 269 | 0.182 | 4.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahid, M.N.; Kosar, N.; Sajid, H.; Ibrahim, K.E.; Gatasheh, M.K.; Mahmood, T. Unveiling the Potential of B3O3 Nanoflake as Effective Transporter for the Antiviral Drug Favipiravir: Density Functional Theory Analysis. Molecules 2023, 28, 8092. https://doi.org/10.3390/molecules28248092
Zahid MN, Kosar N, Sajid H, Ibrahim KE, Gatasheh MK, Mahmood T. Unveiling the Potential of B3O3 Nanoflake as Effective Transporter for the Antiviral Drug Favipiravir: Density Functional Theory Analysis. Molecules. 2023; 28(24):8092. https://doi.org/10.3390/molecules28248092
Chicago/Turabian StyleZahid, Muhammad Nauman, Naveen Kosar, Hasnain Sajid, Khalid Elfaki Ibrahim, Mansour K. Gatasheh, and Tariq Mahmood. 2023. "Unveiling the Potential of B3O3 Nanoflake as Effective Transporter for the Antiviral Drug Favipiravir: Density Functional Theory Analysis" Molecules 28, no. 24: 8092. https://doi.org/10.3390/molecules28248092
APA StyleZahid, M. N., Kosar, N., Sajid, H., Ibrahim, K. E., Gatasheh, M. K., & Mahmood, T. (2023). Unveiling the Potential of B3O3 Nanoflake as Effective Transporter for the Antiviral Drug Favipiravir: Density Functional Theory Analysis. Molecules, 28(24), 8092. https://doi.org/10.3390/molecules28248092