
Rationally Designed Novel Phenyloxazoline Synthase Inhibitors: Chemical Synthesis and Biological Evaluation to Accelerate the Discovery of New Antimycobacterial Antibiotics

 , , , ,
, , , , 
Abstract
:
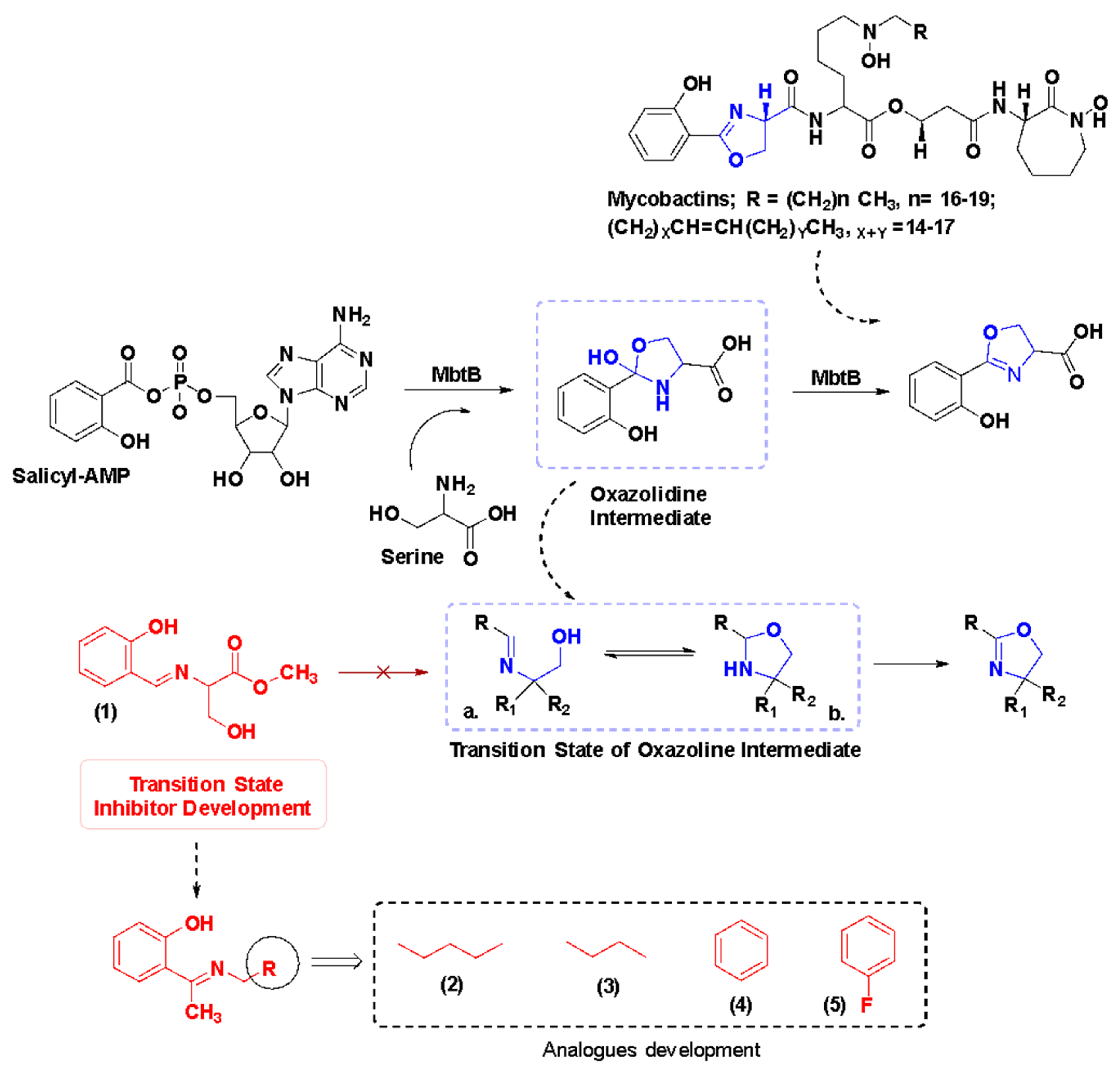
1. Introduction
2. Results and Discussion
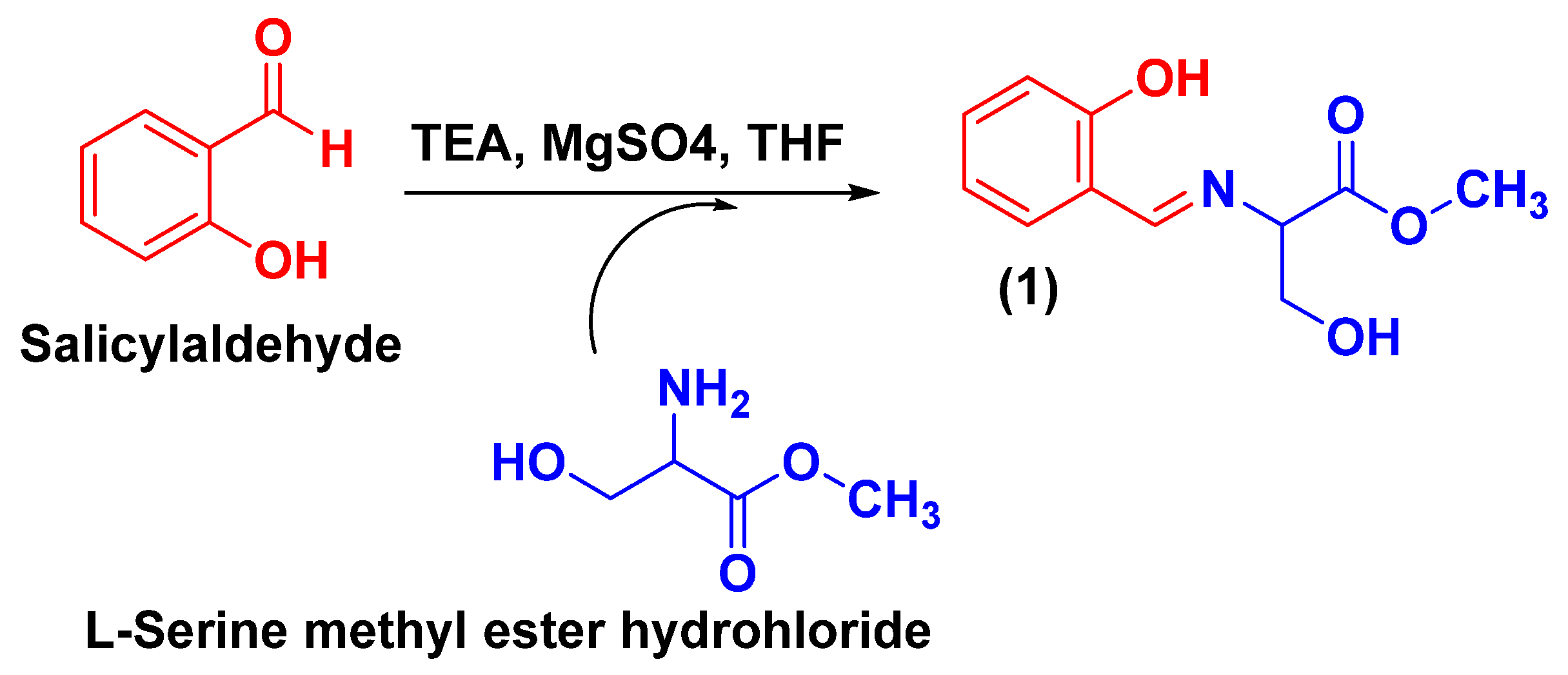
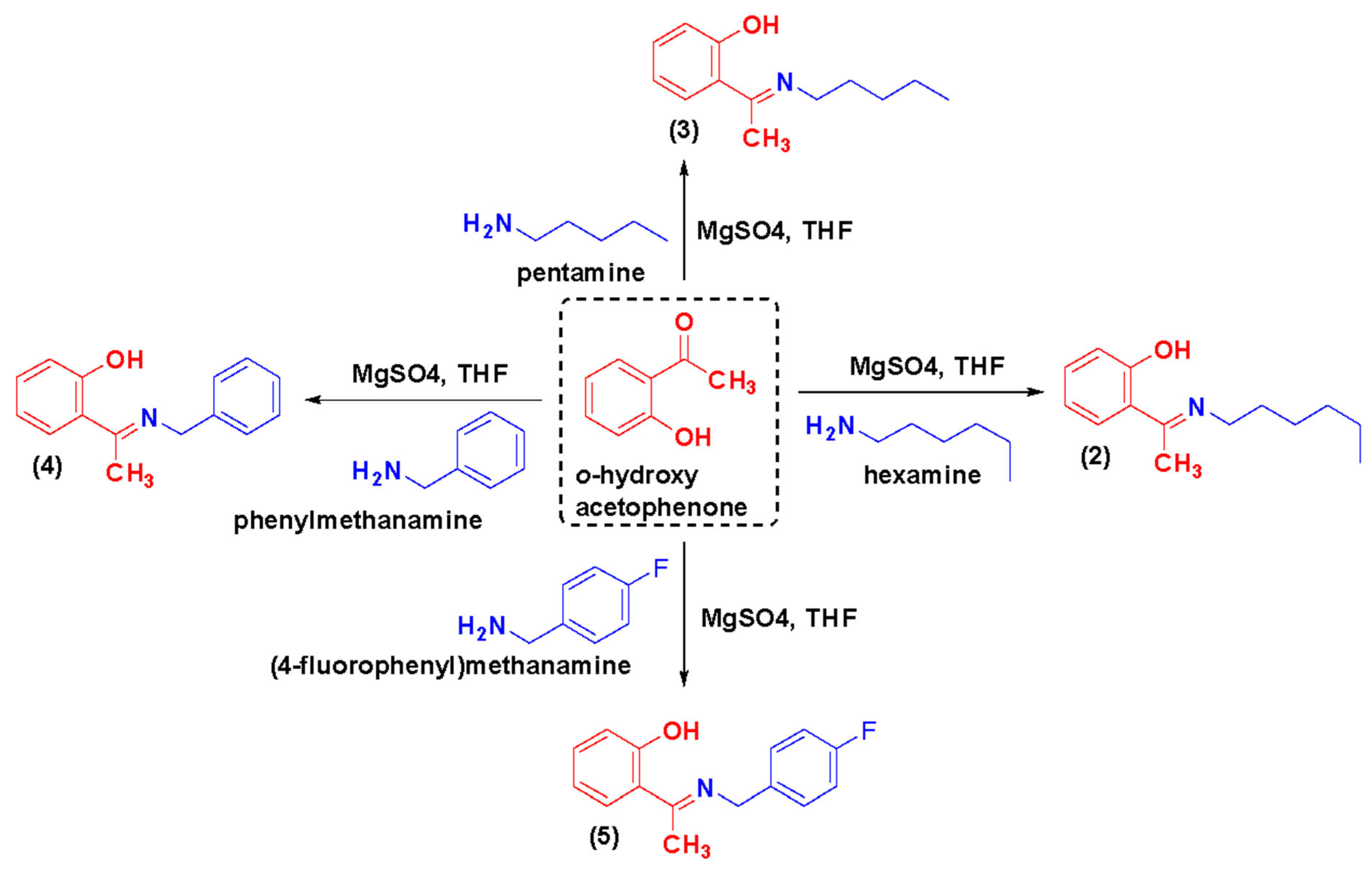
2.1. Chemical Synthesis and Characterization of TSA
2.2. Antimycobacterial Activity Investigations and HIT Identification from the Structure–Activity Relationship (SAR)
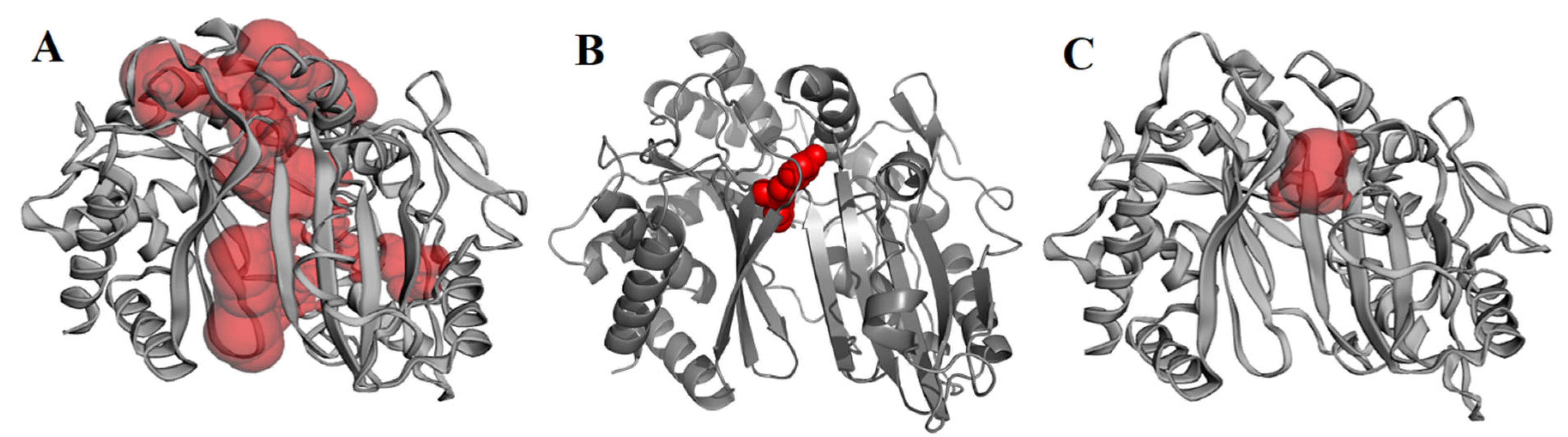
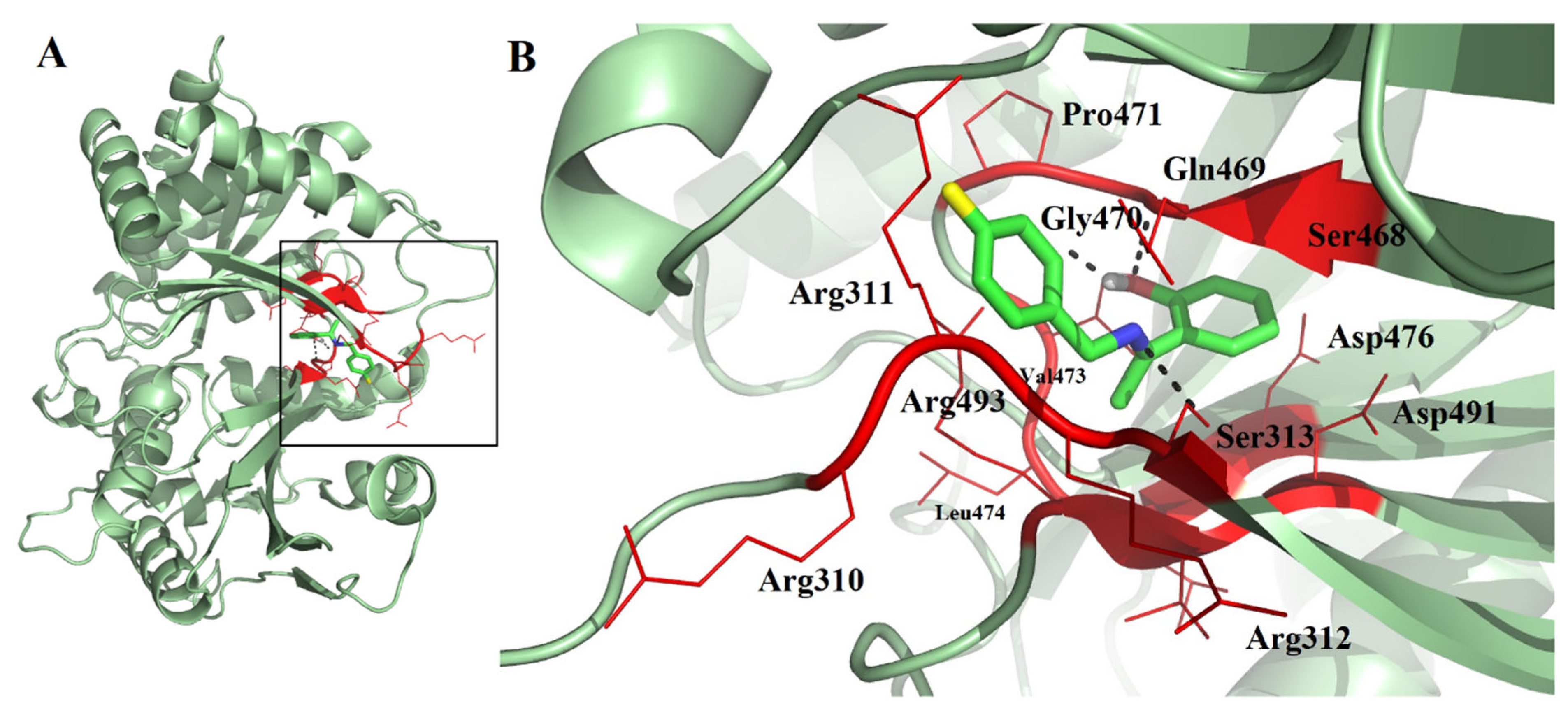
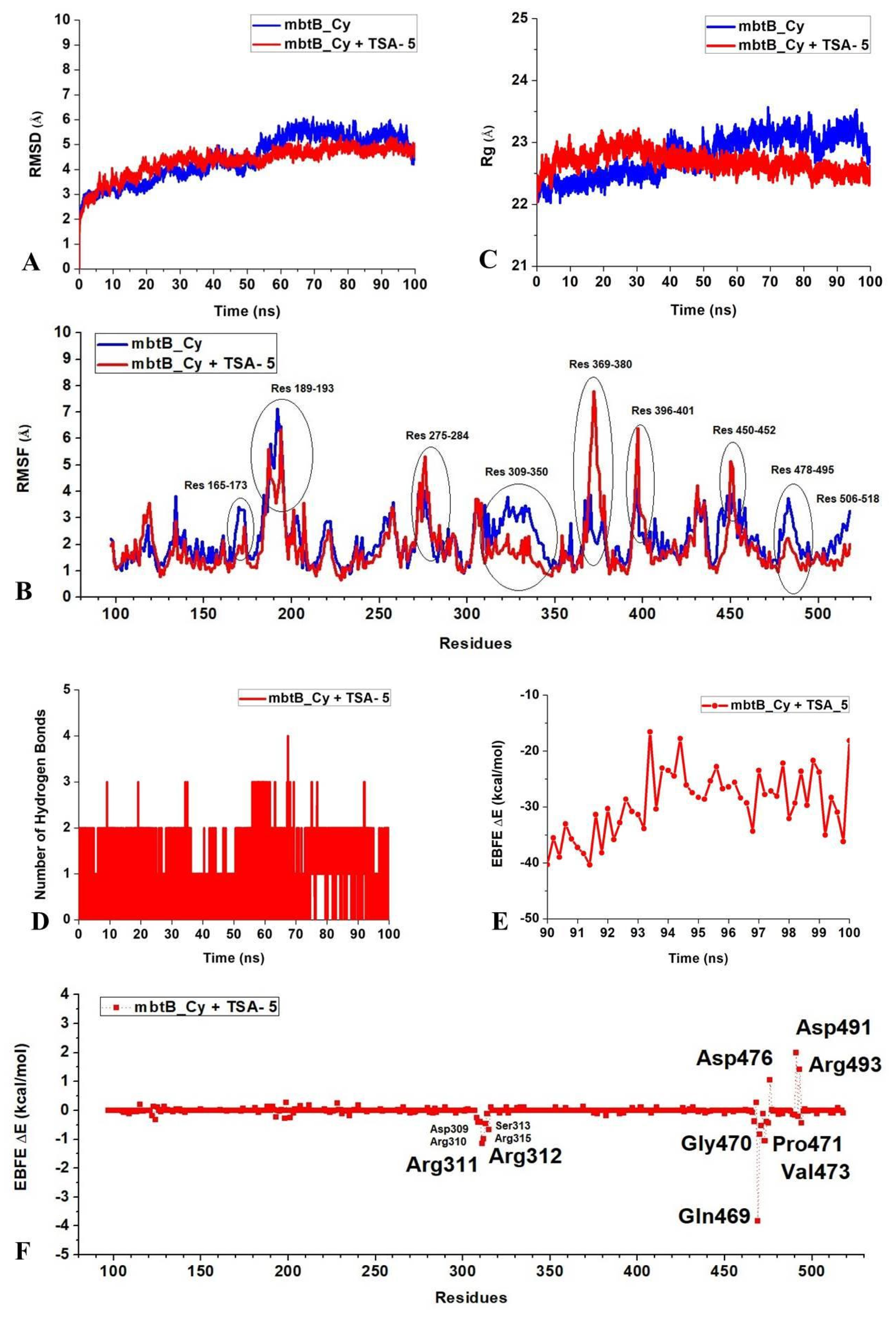
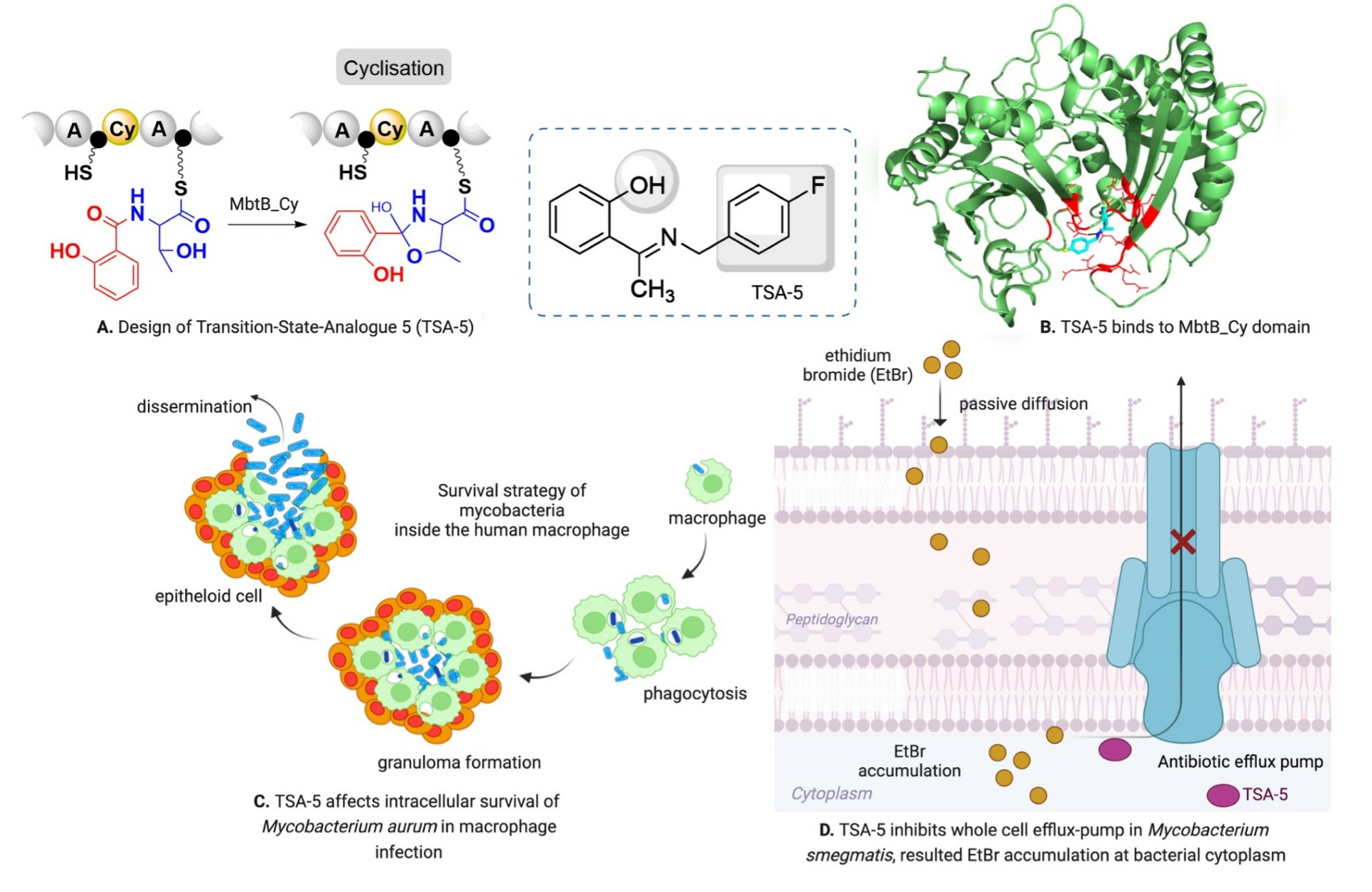
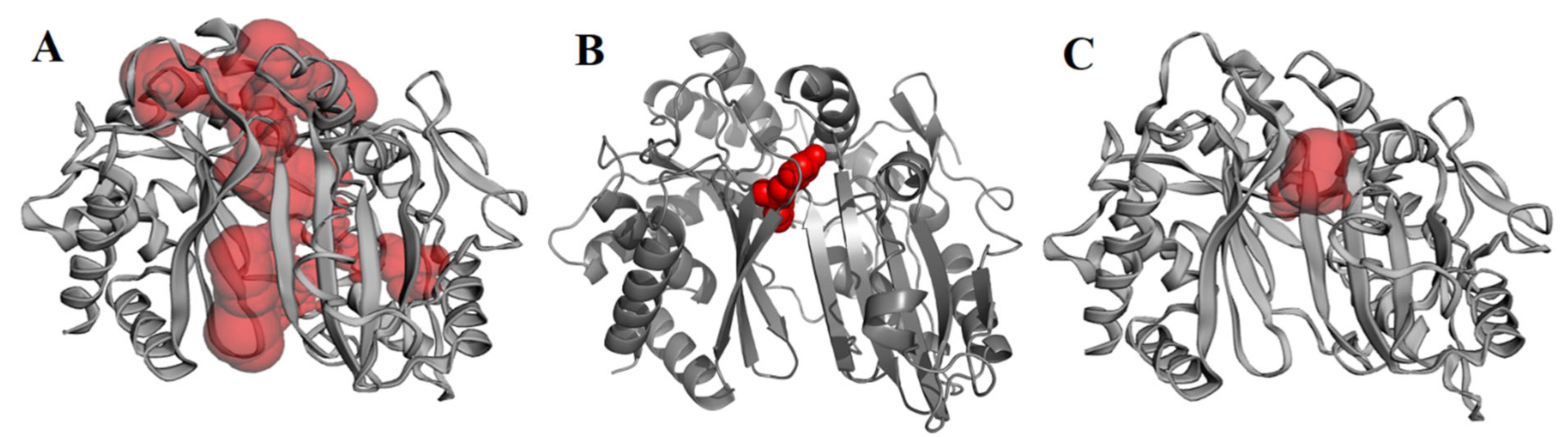
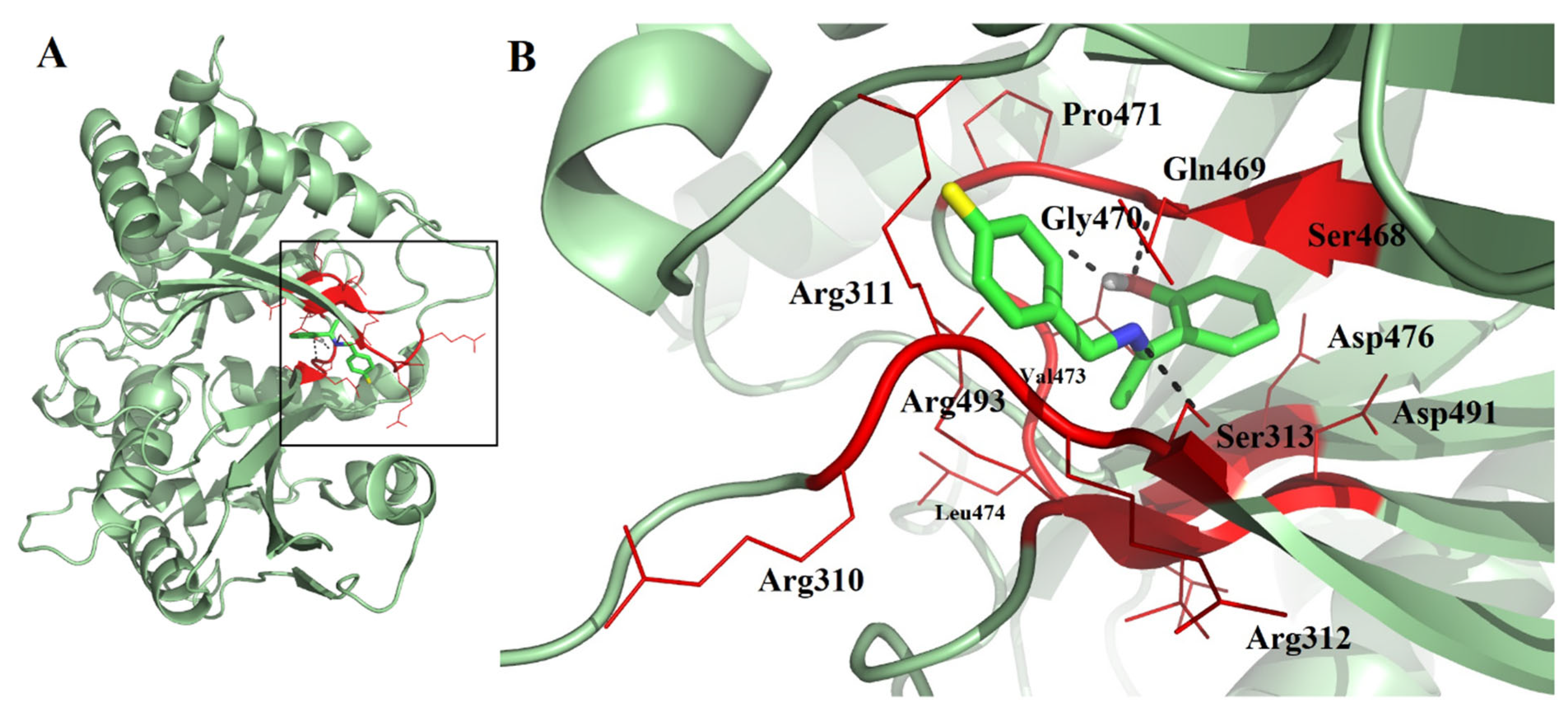
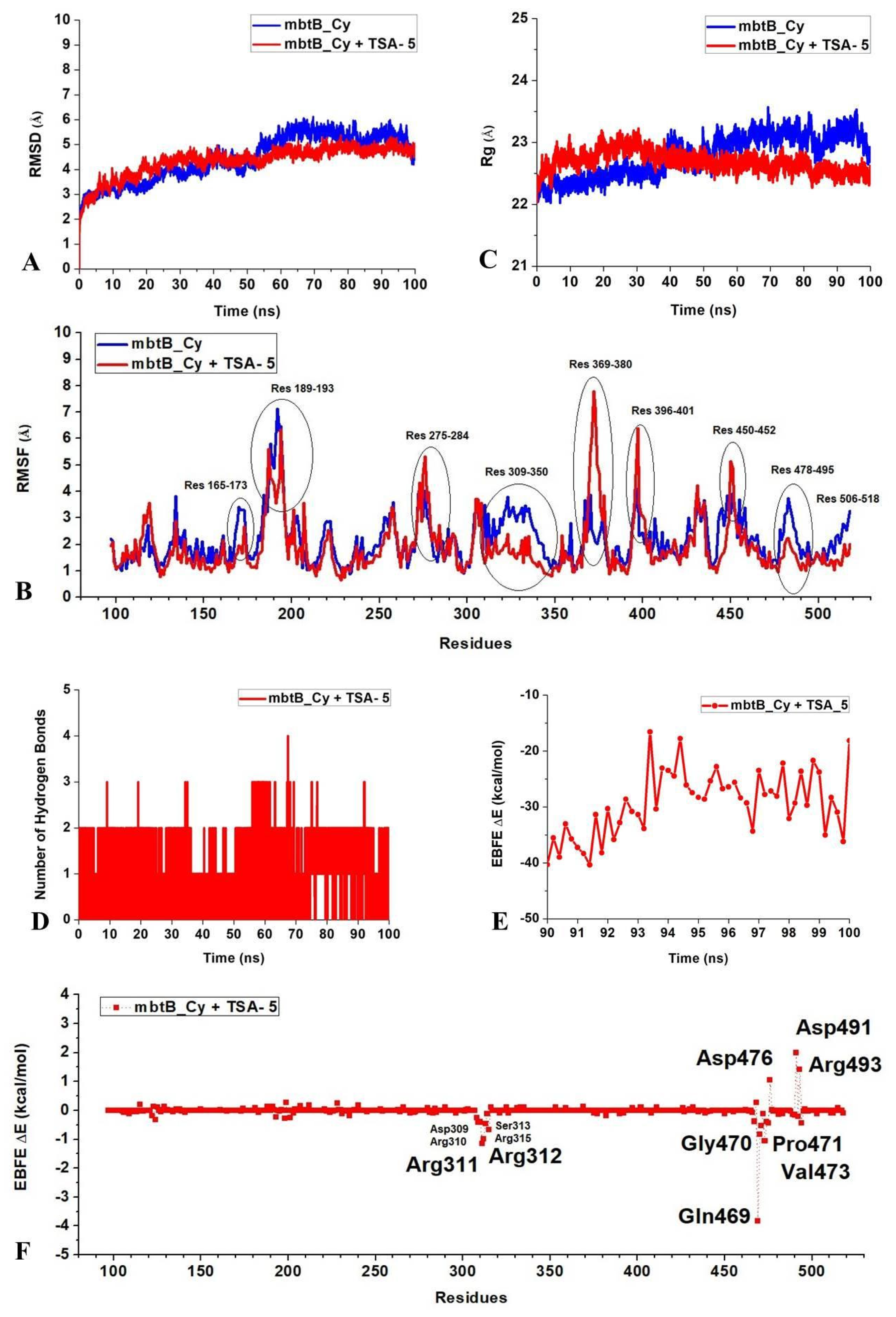
2.3. Molecular Docking, Molecular Dynamics (MD) Simulation, and MMPBSA Analysis to Validate the Putative Drug Target of TSA-5
2.4. Cytotoxicity Profile Evaluation
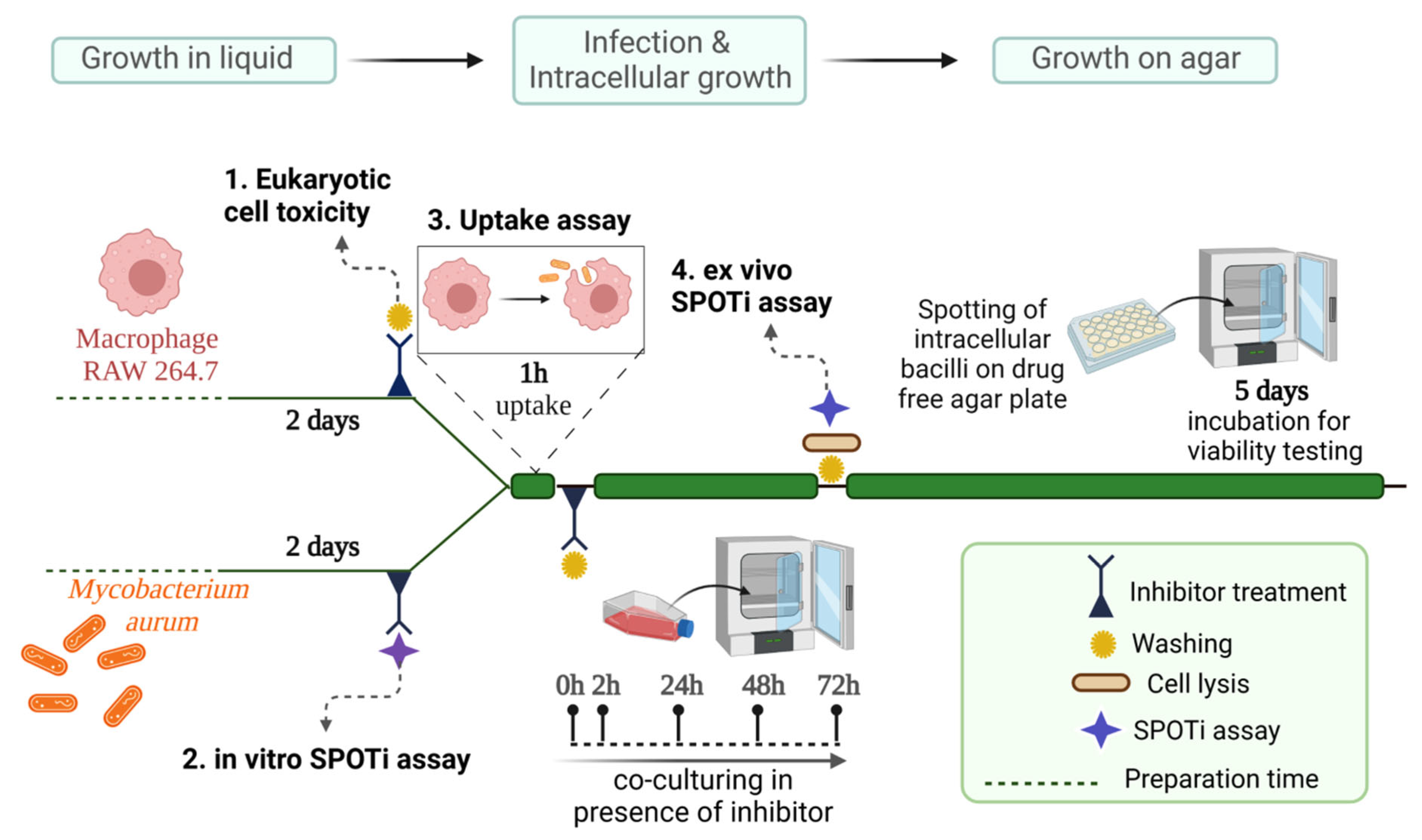
2.5. TSA-5 Affects the Intracellular Survival of Mycobacterium aurum
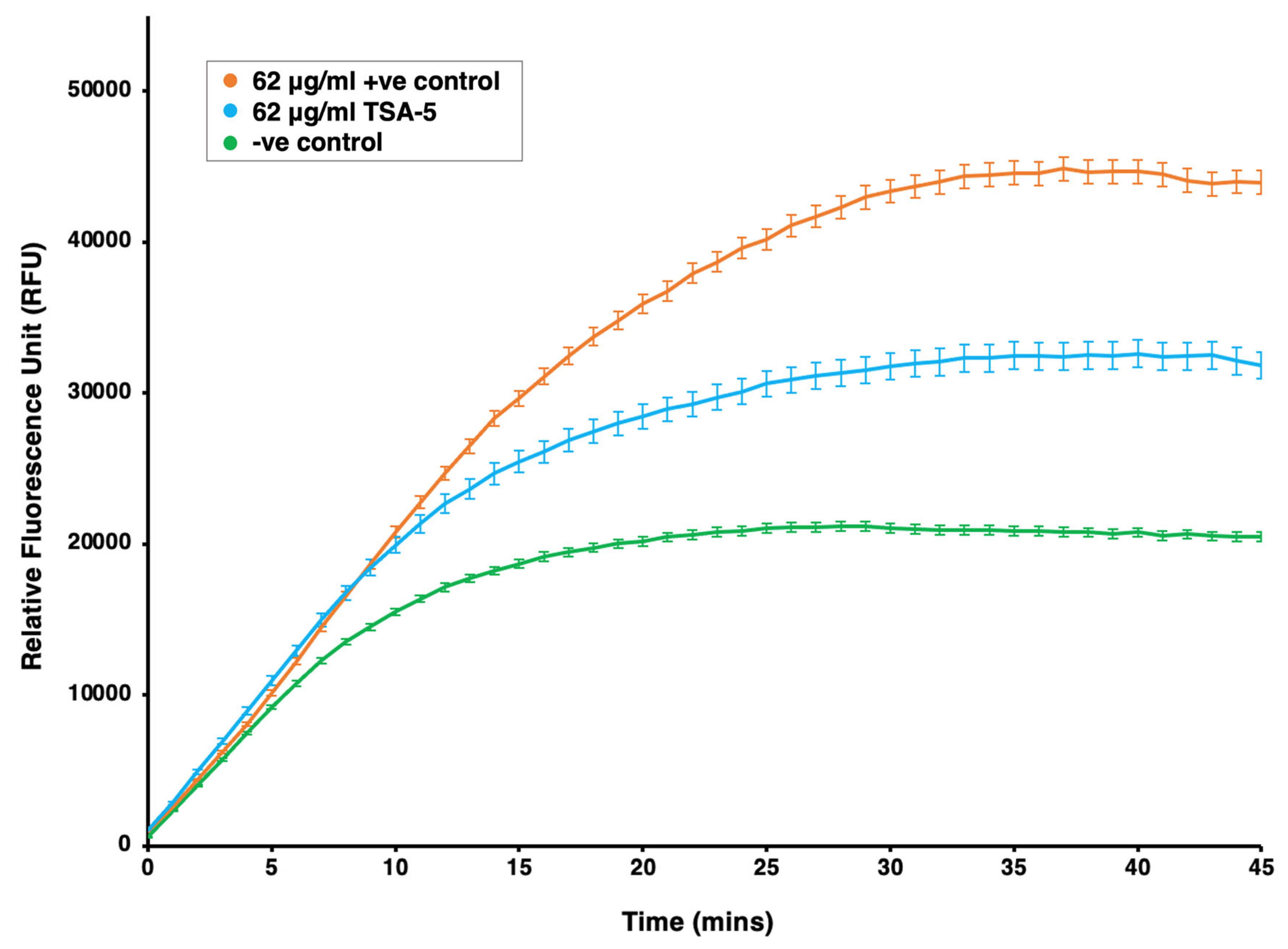
2.6. Investigation of Efflux Pump Inhibition Profile of TSA-5 against Mycobacterium smegmatis
3. Materials and Methods
3.1. Materials and Instruments
3.2. Chemical Synthesis
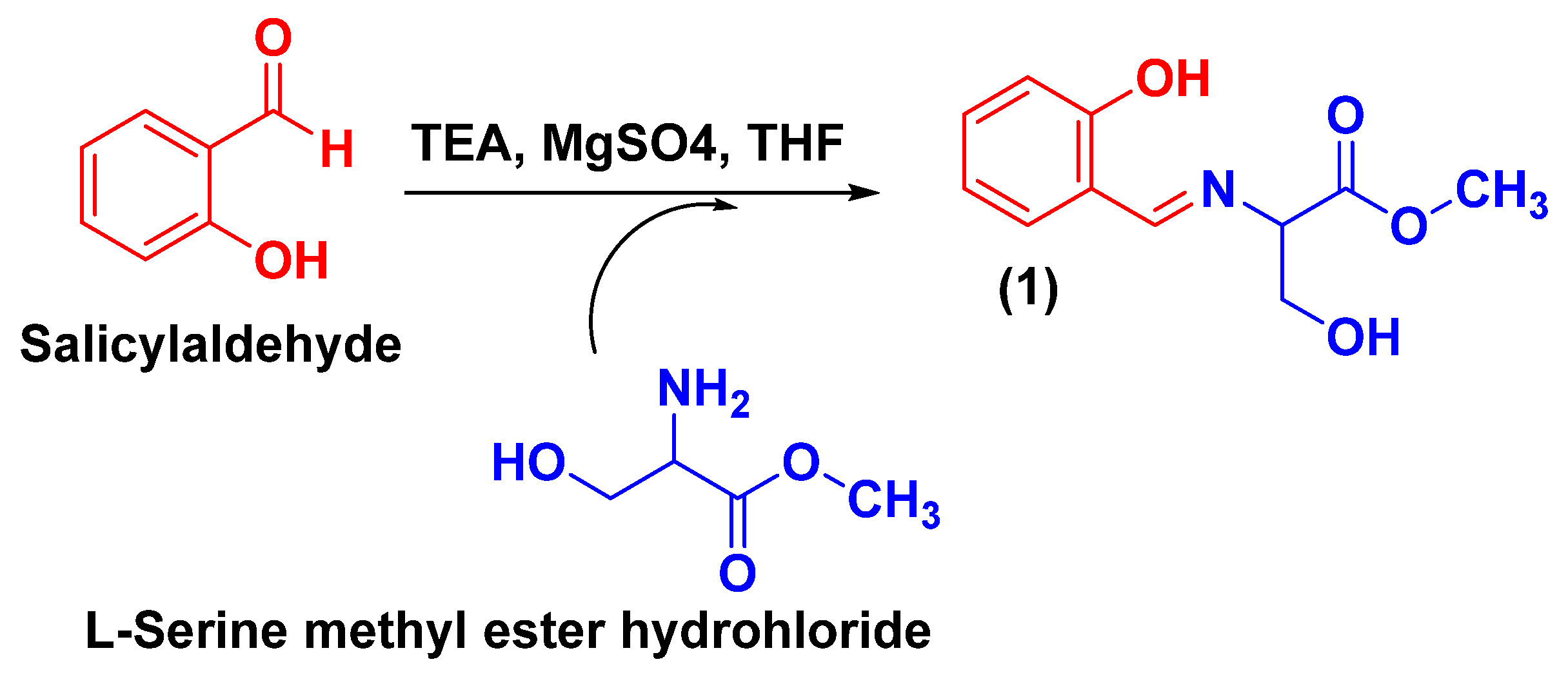
3.2.1. Synthesis of Methyl (E)-3-Hydroxy-2-(((2-hydroxyphenyl)imino)methyl) Propanoate (TSA-1)
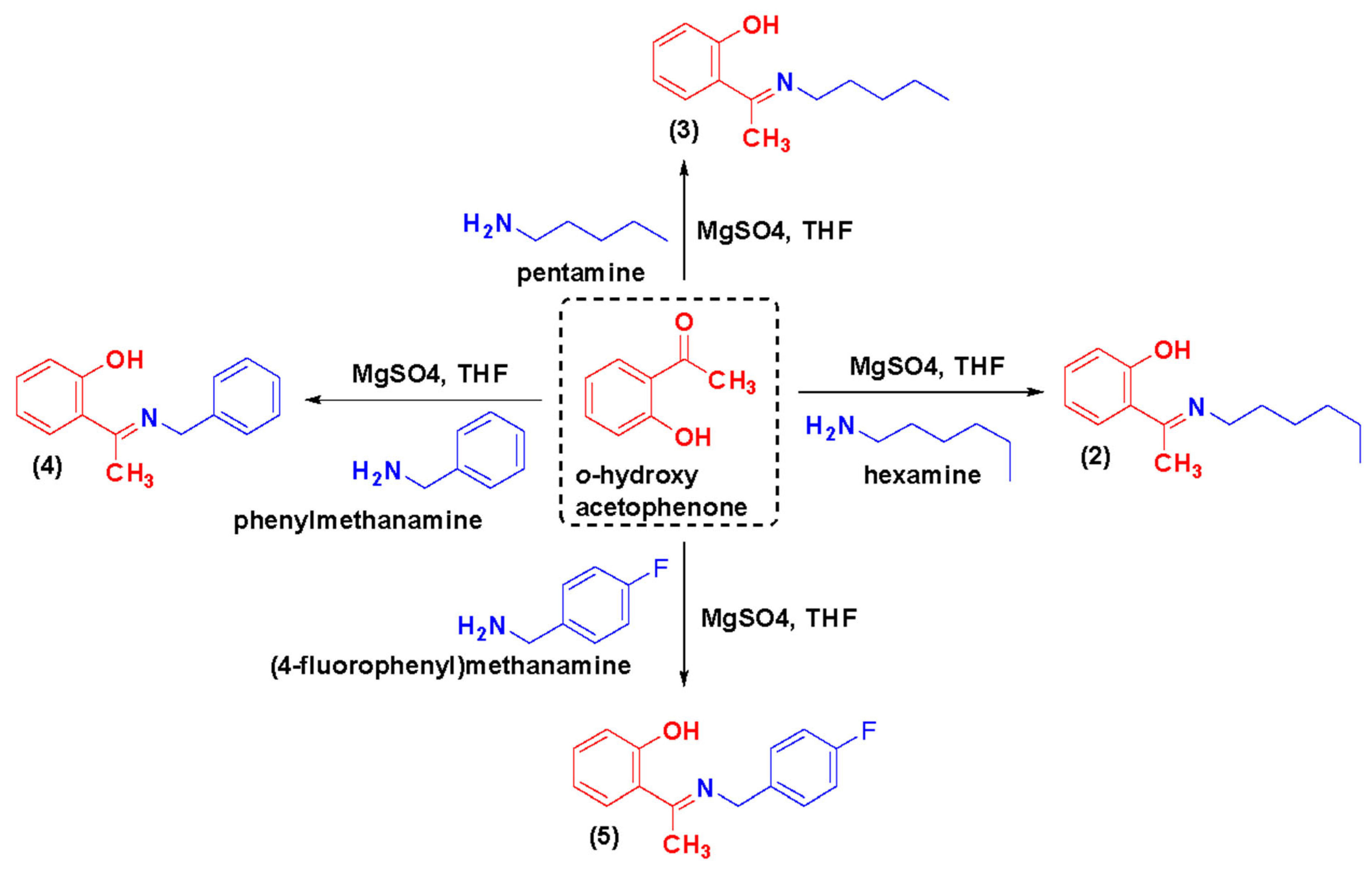
3.2.2. General Procedure for the Synthesis of Azomethines, TSA-(2–5)
3.3. In Vitro Antimycobacterial Activity
3.4. Cytotoxicity Activity
3.5. Antimycobacterial Activity in M. aurum-Infected Murine Macrophage Model
3.6. Whole-Cell Drug Efflux Pump Accumulation Assay against M. smegmatis
3.7. Molecular Docking Studies
3.8. Molecular Dynamics and MMPBSA Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- O’Neill, J. Review on antimicrobial resistance. Antimicrobial resistance: Tackling a crisis for the health and wealth of nations. Rev. Antimicrob. Resist. 2014, 4. Available online: https://cir.nii.ac.jp/crid/1370857593729357568 (accessed on 1 December 2023).
- Katende, B.; Esterhuizen, T.M.; Dippenaar, A.; Warren, R.M. Rifampicin Resistant Tuberculosis in Lesotho: Diagnosis, Treatment Initiation and Outcomes. Sci. Rep. 2020, 10, 1917. [Google Scholar] [CrossRef] [PubMed]
- Quadri, L.E.; Sello, J.; Keating, T.A.; Weinreb, P.H.; Walsh, C.T. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem. Biol. 1998, 5, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Ratledge, C. Iron, mycobacteria and tuberculosis. Tuberculosis 2004, 84, 110–130. [Google Scholar] [CrossRef]
- Shyam, M.; Shilkar, D.; Verma, H.; Dev, A.; Sinha, B.N.; Brucoli, F.; Bhakta, S.; Jayaprakash, V. The Mycobactin Biosynthesis Pathway: A Prospective Therapeutic Target in the Battle against Tuberculosis. J. Med. Chem. 2020, 64, 71–100. [Google Scholar] [CrossRef] [PubMed]
- Shyam, M.; Shilkar, D.; Rakshit, G.; Jayaprakash, V. Approaches for targeting the mycobactin biosynthesis pathway for novel anti-tubercular drug discovery: Where we stand. Expert Opin. Drug Discov. 2022, 17, 699–715. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.D.; Rush, J.S.; Thomas, M.G. Analyses of MbtB, MbtE, and MbtF Suggest Revisions to the Mycobactin Biosynthesis Pathway in Mycobacterium tuberculosis. J. Bacteriol. 2012, 194, 2809–2818. [Google Scholar] [CrossRef] [PubMed]
- Koketsu, K.; Watanabe, K.; Suda, H.; Oguri, H.; Oikawa, H. Reconstruction of the saframycin core scaffold defines dual Pictet-Spengler mechanisms. Nat. Chem. Biol. 2010, 6, 408–410. [Google Scholar] [CrossRef]
- De Voss, J.J.; Rutter, K.; Schroeder, B.G.; Su, H.; Zhu, Y.; Barry, C.E., III. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. USA 2000, 97, 1252–1257. [Google Scholar] [CrossRef]
- Shyam, M.; Verma, H.; Bhattacharje, G.; Mukherjee, P.; Singh, S.; Kamilya, S.; Jalani, P.; Das, S.; Dasgupta, A.; Mondal, A.; et al. Mycobactin analogueues with excellent pharmacokinetic profile demonstrate potent antitubercular specific activity and exceptional efflux pump inhibition. J. Med. Chem. 2022, 65, 234–256. [Google Scholar] [CrossRef]
- Stirrett, K.L.; Ferreras, J.A.; Jayaprakash, V.; Sinha, B.N.; Ren, T.; Quadri, L.E. Small molecules with structural similarities to siderophores as novel antimicrobials against Mycobacterium tuberculosis and Yersinia pestis. Bioorg. Med. Chem. Lett. 2008, 18, 2662–2668. [Google Scholar] [CrossRef] [PubMed]
- Ferreras, J.A.; Gupta, A.; Amin, N.D.; Basu, A.; Sinha, B.N.; Worgall, S.; Jayaprakash, V.; Quadri, L.E. Chemical scaffolds with structural similarities to siderophores of nonribosomal peptide–polyketide origin as novel antimicrobials against Mycobacterium tuberculosis and Yersinia pestis. Bioorg. Med. Chem. Lett. 2011, 21, 6533–6537. [Google Scholar] [CrossRef]
- Craggs, P.D.; de Carvalho, L.P.S. Bottlenecks and opportunities in antibiotic discovery against Mycobacterium tuberculosis. Curr. Opin. Microbiol. 2022, 69, 102191. [Google Scholar] [CrossRef] [PubMed]
- Dawadi, S.; Boshoff, H.I.; Park, S.W.; Schnappinger, D.; Aldrich, C.C. Conformationally constrained cinnolinone nucleoside analogueues as siderophore biosynthesis inhibitors for tuberculosis. ACS Med. Chem. Lett. 2018, 9, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Viswanathan, K.; Dawadi, S.; Duckworth, B.P.; Boshoff, H.I.; Barry, C.E., III; Aldrich, C.C. Synthesis and pharmacokinetic evaluation of siderophore biosynthesis inhibitors for Mycobacterium tuberculosis. J. Med. Chem. 2015, 58, 5459–5475. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Furnham, N.; Thornton, J.M. The Ramachandran plot and protein structure validation. In Biomolecular Forms and Functions: A Celebration of 50 Years of the Ramachandran Map; World Scientific: Singapore, 2013; pp. 62–75. [Google Scholar]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Info. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Gupta, A.; Bhakta, S. An integrated surrogate model for screening of drugs against Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2012, 67, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bhakta, S.; Kundu, S.; Gupta, M.; Srivastava, B.S.; Srivastava, R. Fast-growing, non-infectious and intracellularly surviving drug-resistant Mycobacterium aurum: A model for high-throughput antituberculosis drug screening. J. Antimicrob. Chemother. 2009, 64, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Zhai, W.; Wu, F.; Zhang, Y.; Fu, Y.; Liu, Z. The Immune Escape Mechanisms of Mycobacterium tuberculosis. Int. J. Mol. Sci. 2019, 20, 340. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.O.; Emma Office; Kawate, T.; Orzechowski, M.; Hung, D.T. Large-Scale Chemical-Genetic Strategy Enables the Design of Antimicrobial Combination Chemotherapy in Mycobacteria. ACS Infect. Dis. 2019, 6, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhou, C.-Y.; Wu, Y.-D. Residue-Specific Force Field Based on the Protein Coil Library. RSFF1: Modification of OPLS-AA/L. J. Phys. Chem. B 2014, 118, 6983–6998. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Dodda, L.S.; de Vaca, I.C.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef]
- DeLano, W.L.; Bromberg, S. PyMOL User’s Guide; DeLano Scientific LLC.: San Carlos, CA, USA, 2004; p. 629. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | MIC90 μg/mL | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| M. tuberculosis (Mtb) | M. aurum (Ma) | M. smegmatis (Msmeg) | |||||||
| GAS | GAS-Fe | TSI | GAS | GAS-Fe | TSI | GAS | GAS-Fe | TSI | |
| TSA-1 | 256 | 256 | 1 | 256 | >256 | >1 | >256 | >256 | 1 |
| TSA-2 | >256 | 256 | nd | 256 | 256 | 1 | >256 | >256 | 1 |
| TSA-3 | 256 | >256 | >1 | 256 | 256 | 1 | >256 | >256 | 1 |
| TSA-4 | 256 | >256 | >1 | 128 | 256 | 2 | >256 | >256 | 1 |
| TSA-5 | 32 | >256 | >8 | 64 | 256 | 4 | 256 | >256 | >1 |
| RIF | 0.03 | 0.007 | 0.2 | 0.12 | 0.06 | 0.5 | 8 | 8 | 1 |
| INH | 0.03 | 0.005 | 0.2 | 0.02 | 0.01 | 0.5 | 2 | 2 | 1 |
| Comp. | Cell-Line | GIC90 (μg/mL) | Mtb | Ma | Msmeg | |||
|---|---|---|---|---|---|---|---|---|
| MIC90 | SI | MIC90 | SI | MIC90 | SI | |||
| TSA-5 | RAW 264.7 | 256 | 32 | 8 | 64 | 4 | 256 | 1 |
| THP-1 | 256 | 32 | 8 | 64 | 4 | 256 | 1 | |
| MTX | RAW 264.7 | 2.50 | nd | nd | nd | nd | nd | nd |
| THP-1 | 0.31 | |||||||
| Comp. | Intracellular MIC90 (μg/mL) against M. aurum | GIC90 (μg/mL) RAW 264.7 | Intracellular SI | Extracellular SI |
|---|---|---|---|---|
| TSA-5 | 8 | 512 | 64 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shyam, M.; Bhattacharje, G.; Daniel, C.; Kumar, A.; Yadav, P.; Mukherjee, P.; Singh, S.; Das, A.K.; Narender, T.; Singh, A.; et al. Rationally Designed Novel Phenyloxazoline Synthase Inhibitors: Chemical Synthesis and Biological Evaluation to Accelerate the Discovery of New Antimycobacterial Antibiotics. Molecules 2023, 28, 8115. https://doi.org/10.3390/molecules28248115
Shyam M, Bhattacharje G, Daniel C, Kumar A, Yadav P, Mukherjee P, Singh S, Das AK, Narender T, Singh A, et al. Rationally Designed Novel Phenyloxazoline Synthase Inhibitors: Chemical Synthesis and Biological Evaluation to Accelerate the Discovery of New Antimycobacterial Antibiotics. Molecules. 2023; 28(24):8115. https://doi.org/10.3390/molecules28248115
Chicago/Turabian StyleShyam, Mousumi, Gourab Bhattacharje, Chris Daniel, Amrendra Kumar, Pragya Yadav, Piyali Mukherjee, Samsher Singh, Amit Kumar Das, Tadigoppula Narender, Amit Singh, and et al. 2023. "Rationally Designed Novel Phenyloxazoline Synthase Inhibitors: Chemical Synthesis and Biological Evaluation to Accelerate the Discovery of New Antimycobacterial Antibiotics" Molecules 28, no. 24: 8115. https://doi.org/10.3390/molecules28248115
APA StyleShyam, M., Bhattacharje, G., Daniel, C., Kumar, A., Yadav, P., Mukherjee, P., Singh, S., Das, A. K., Narender, T., Singh, A., Jayaprakash, V., & Bhakta, S. (2023). Rationally Designed Novel Phenyloxazoline Synthase Inhibitors: Chemical Synthesis and Biological Evaluation to Accelerate the Discovery of New Antimycobacterial Antibiotics. Molecules, 28(24), 8115. https://doi.org/10.3390/molecules28248115