
Step-by-Step Replacement of Cyano Groups by Tricyanovinyls—The Influence on the Acidity

Abstract
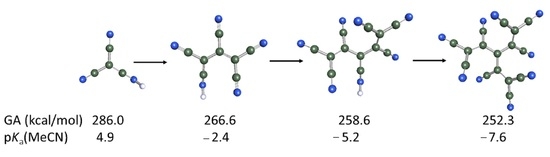
:
1. Introduction
Compounds under Study
2. Results and Discussion
2.1. Gas-Phase Acidities

{kind=link}
{kind=link}
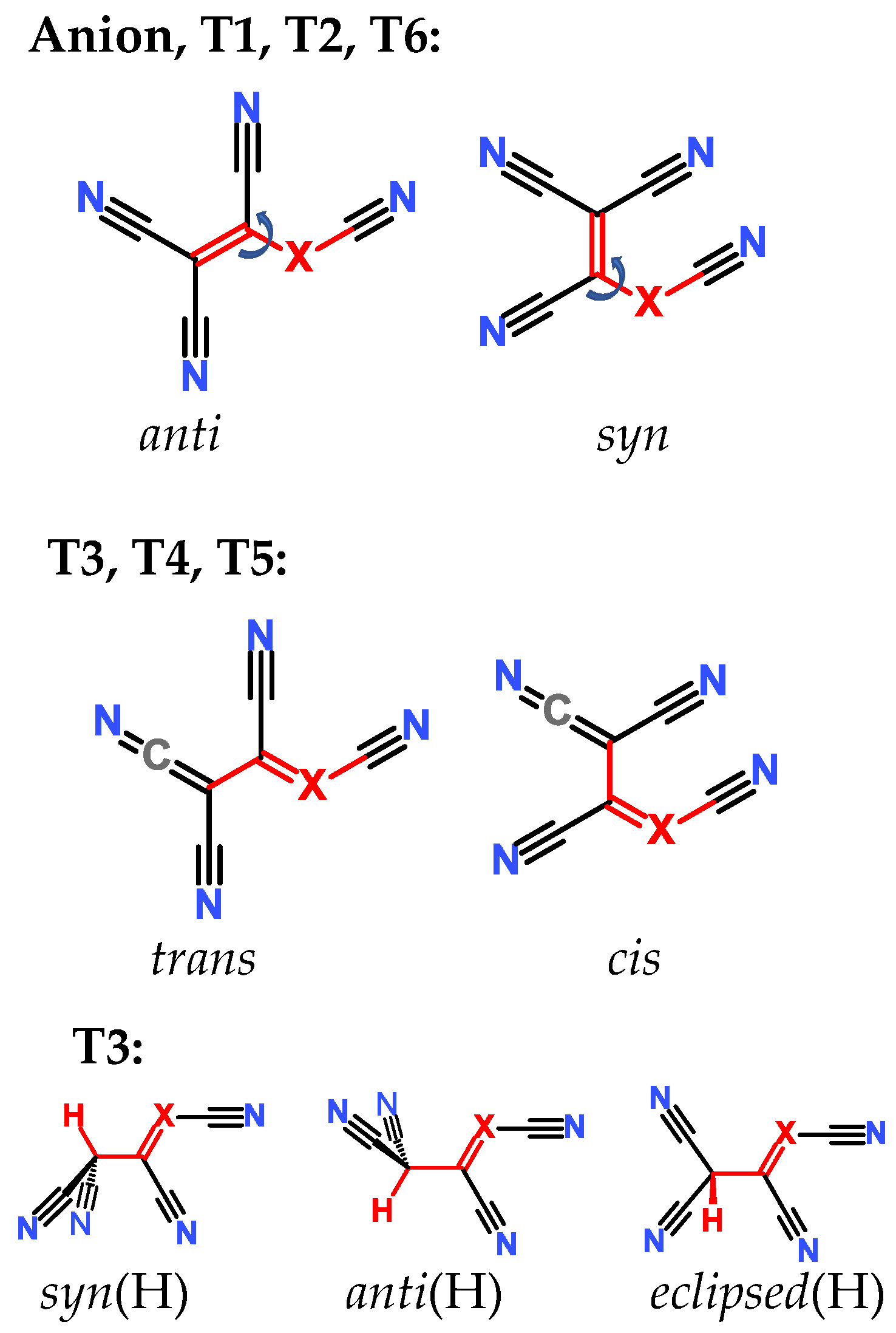{kind=link}
{kind=link}
{kind=link}
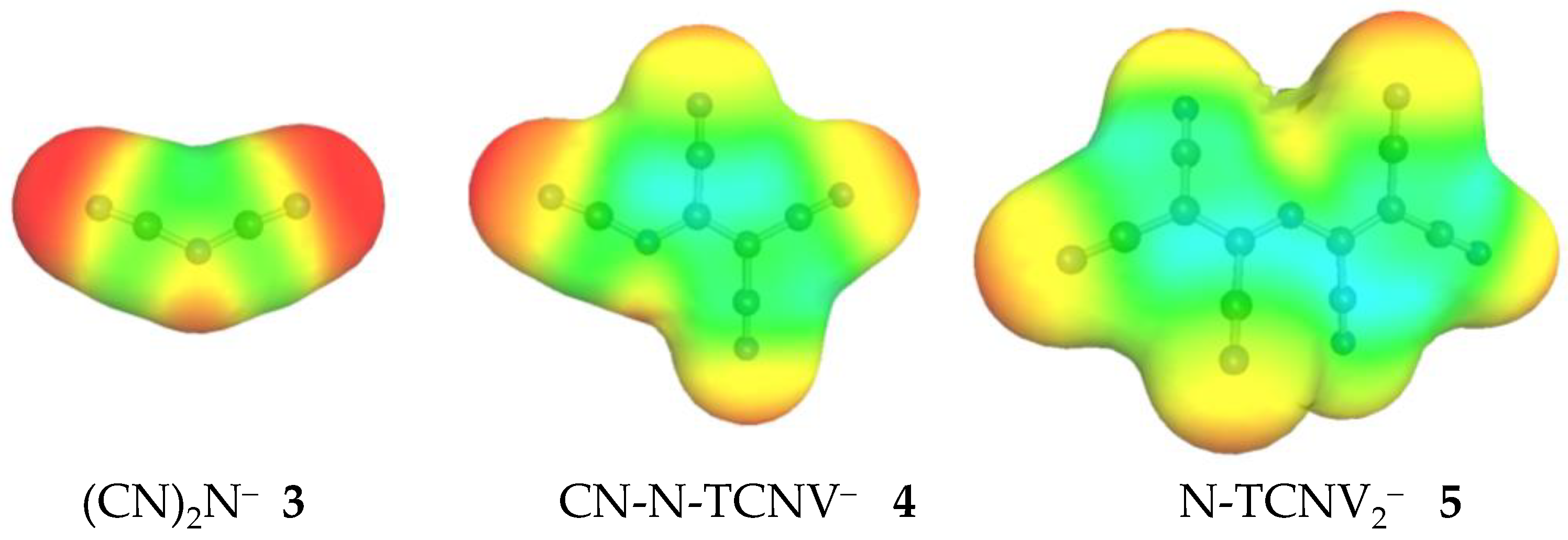{kind=link}
{kind=link}
{kind=link}
{kind=link}
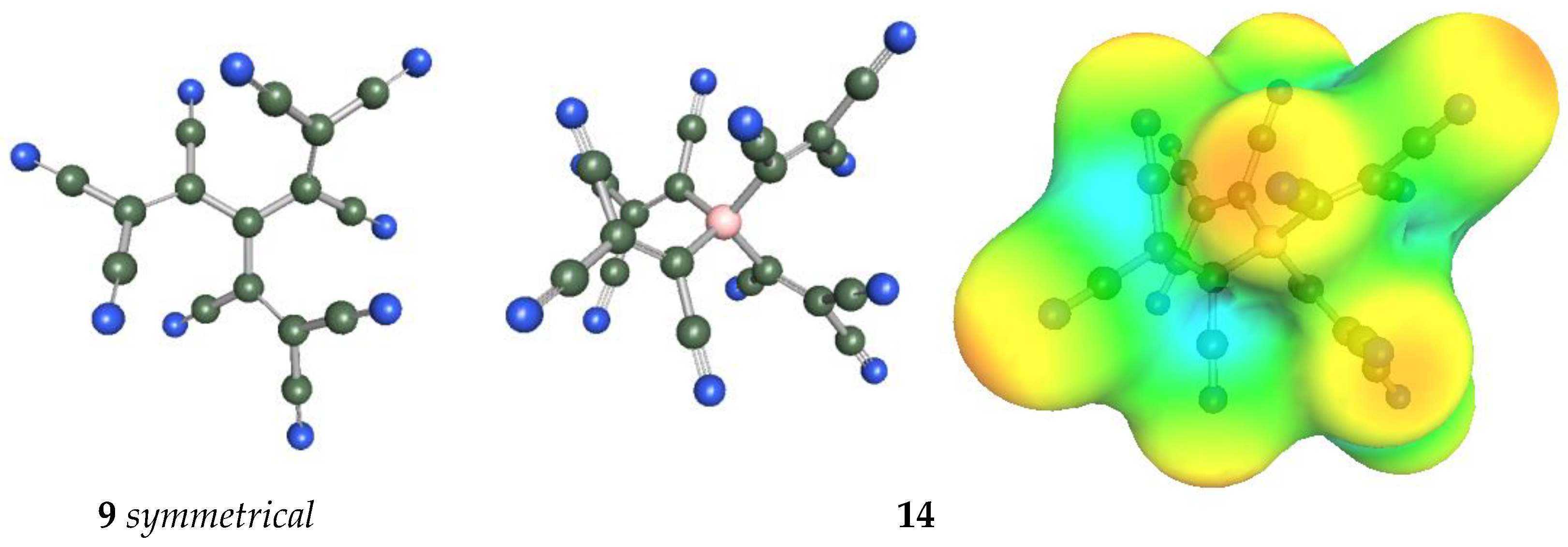{kind=link}
| No. | Compd | PA [kcal/mol] | GA [kcal/mol] | pKa(MeCN) | pKa(DMSO) | pKa(DCE) | |
|---|---|---|---|---|---|---|---|
| 1H | [CN-O]H | T2 | 341.0 [341.2 a] | 334.6 [334.7 a] | 24.2 | 12.4 | 60.6 |
| 2H | [O-TCNV]H | T1 | 289.4 | 282.1 | 5.1 [4.39 e (3.1) f] | −7.0 | 38.6 [36.3 i] |
| 3H | [(CN)2N]H | T2 | 311.7 | 304.5 | 12.5 | 0.5 | 47.1 |
| 4H | [CN-N-TCNV]H | T2 | 290.4 | 283.5 | 5.7 | −6.3 | 39.3 |
| 5H | [N-TCNV2]H | T1 | 276.4 [(278.0) b] | 269.1 | 1.2 [(0.3) f] | −11.0 | 34.1 [33.0 i] |
| 6H | [(CN)3C]H | T2 | 293.0 [(293.7) b] | 286.0 [294.8 c] | 4.9 [5.0 f (5.1) f] | −7.2 | 38.4 [38.6 i] |
| 7H | [(CN)2-C-TCNV]H | T2 | 274.4 [(274.8) b] | 266.6 | −2.4 [(−2.8) f] | −14.7 | 29.9 [29.7 i] |
| 8H | [CN-C-TCNV2]H | T2 | 265.7 [(266.0) b] | 258.6 | −5.2 | −17.5 | 26.7 |
| 9H | [C-TCNV3]H | T4 | 257.7 | 252.3 | −7.6 | −20.0 | 23.9 |
| 10H | [(CN)4B]H | T2 | 265.6 | 258.7 | −3.5 [(−1.0) f] | −15.8 | 28.7 [31.7 i] |
| 11H | [(CN)3-B-TCNV]H | T2 | 260.7 | 253.9 | −4.1 | −16.4 | 28.0 |
| 12H | [(CN)2-B-TCNV2]H | T2 | 256.5 | 249.3 | −4.9 | −17.2 | 27.0 |
| 13H | [CN-B-TCNV3]H | T2 | 253.3 | 246.0 | −5.9 | −18.2 | 25.9 |
| 14H | [B-TCNV4]H | T6 | 236.2 | 229.3 | −12.9 | −25.3 | 17.9 |
| 15H | HCN | 349.3 [350.5 a] | 341.9 [343.2 a] | 28.4 [(23.40) g] | 16.8 [12.9 h] | 65.4 | |
| 16H | C(CN)2=CH(CN) | 330.7 | 323.2 | 29.7 | 18.1 | 66.9 | |
| 17H | [B12(CN)12]H- | 295.1 | 288.7 | −8.1 | −20.4 | 23.4 | |
| 18H | [B12(CN)12]H2 | 243.9 | 237.2 | −11.4 | −23.8 | 19.6 | |
| 19H | [CB11(CN)12]H | 231.6 | 224.9 [(225.0) d] | −14.2 | −26.7 | 16.3 |


| Compound | Turbomole | Gaussian | |||||||
|---|---|---|---|---|---|---|---|---|---|
| BP86/ def-TZVP | BP86/ def2-TZVPP | BP86/ def2-TZVPPD | B3LYP /def2-TZVPPD | B3LYP/ 6-311 + G(d,p) | G4(MP2) | W1RO | CBS-APNO | ||
| [CNO]H | 1H | 338.3 | 340.3 | 338.1 | 342.8 | 334.6 | 335.2 | 335.1 | 335.4 |
| [O-TCNV]H | 2H | 282.5 | 284.2 | 283.5 | 284.2 | 282.1 | 287.1 | 286.5 | 285.3 |
| [(CN)2N]H | 3H | 307.1 | 308.6 | 307.7 | 306.7 | 304.5 | 303.8 | 303.6 | 303.7 |
| [CN-N-TCNV]H | 4H | 285.0 | 286.4 | 285.9 | 285.5 | 283.5 | 283.9 | 283.5 | 282.9 |
| [N-TCNV2]H | 5H | 269.8 | 270.1 | 270.3 | 270.7 | 269.1 | 274.6 | 271.5 | |
| [(CN)3C]H | 6H | 287.9 | 289.3 | 288.7 | 287.5 | 286.0 | 286.7 | 286.1 | 286.0 |
| [(CN)2-C-TCNV]H | 7H | 268.1 | 269.5 | 268.7 | 268.0 | 266.6 | 267.8 | 265.9 | |
| [CN-C-TCNV2]H | 8H | 259.6 | 260.9 | 259.4 | 260.9 | 258.6 | 260.2 | 256.9 | |
| [C-TCNV3]H | 9H | 250.6 | 250.4 | 248.4 | 250.1 | 252.3 | 259.5 | ||
| [(CN)4B]H | 10H | 261.1 | 262.2 | 261.7 | 260.4 | 258.7 | 258.8 | 258.7 | 258.9 |
| [(CN)3-B-TCNV]H | 11H | 256.2 | 257.4 | 257.4 | 255.7 | 253.9 | 254.1 | 253.5 | |
| [(CN)2-sB-TCNV2]H | 12H | 251.9 | 253.0 | 252.6 | 251.1 | 249.3 | 249.8 | ||
| [CN-B-TCNV3]H | 13H | 248.5 | 249.7 | 249.3 | 248.3 | 246.0 | 247.0 | ||
| [B-TCNV4]H | 14H | 233.1 | 234.4 | 232.3 | 229.3 | ||||
| HCN | 15H | 341.9 | 340.2 | 341.9 | |||||
| C(CN)2=CH(CN) | 16H | 323.6 | 323.8 | 323.2 | |||||
| [B12(CN)12]H- | 17H | 291.5 | 292.3 | 288.7 | |||||
| [B12(CN)12]H2 | 18H | 239.8 | 241.3 | 237.2 | |||||
| [CB11(CN)12]H | 19H | 228.0 | 229.1 | 224.9 | |||||
2.2. Solution Phase Acidities
r2 = 0.966; S = 0.416; n = 15
2.3. Structural Details
3. Materials and Methods
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; Wiley-VCH: Weinheim, Germany, 2011; ISBN 978-3-527-32473-6. [Google Scholar]
- Kütt, A.; Selberg, S.; Kaljurand, I.; Tshepelevitsh, S.; Heering, A.; Darnell, A.; Kaupmees, K.; Piirsalu, M.; Leito, I. pKa Values in Organic Chemistry—Making Maximum Use of the Available Data. Tetrahedron Lett. 2018, 59, 3738–3748. [Google Scholar] [CrossRef]
- Kütt, A.; Rodima, T.; Saame, J.; Raamat, E.; Mäemets, V.; Kaljurand, I.; Koppel, I.A.; Garlyauskayte, R.Y.; Yagupolskii, Y.L.; Yagupolskii, L.M.; et al. Equilibrium Acidities of Superacids. J. Org. Chem. 2011, 76, 391–395. [Google Scholar] [CrossRef] [PubMed]
- McCallum, C.; Pethybridge, A.D. Conductance of Acids in Dimethyl-Sulphoxide—II. Conductance of Some Strong Acids in DMSO at 25 °C. Electrochim. Acta 1975, 20, 815–818. [Google Scholar] [CrossRef]
- Webster, O.W. Diazotetracyanocyclopentadiene1. J. Am. Chem. Soc. 1966, 88, 4055–4060. [Google Scholar] [CrossRef]
- Middleton, W.J.; Little, E.L.; Coffman, D.D.; Engelhardt, V.A. Cyanocarbon Chemistry. V.1 Cyanocarbon Acids and Their Salts. J. Am. Chem. Soc. 1958, 80, 2795–2806. [Google Scholar] [CrossRef]
- Webster, O.W. Hexacyanobutadiene. J. Am. Chem. Soc. 1964, 86, 2898–2902. [Google Scholar] [CrossRef]
- Vianello, R.; Maksić, Z.B. Polycyano Derivatives of Some Organic Tri- and Hexacyclic Molecules Are Powerful Super- and Hyperacids in the Gas Phase and DMSO: Computational Study by DFT Approach. J. Org. Chem. 2010, 75, 7670–7681. [Google Scholar] [CrossRef]
- Vianello, R.; Maksić, Z.B. The Engineering of Powerful Non-Ionic Superacids in Silico—A DFT-B3LYP Study of Open Chain Polycyanopolyenes. New J. Chem. 2009, 33, 739–748. [Google Scholar] [CrossRef]
- Vianello, R.; Maksić, Z.B. Rees Polycyanated Hydrocarbons and Related Compounds Are Extremely Powerful Brønsted Superacids in the Gas-Phase and DMSO—A Density Functional B3LYP Study. New J. Chem. 2008, 32, 413–427. [Google Scholar] [CrossRef]
- Maksić, Z.B.; Vianello, R. Extending the Acidity Ladder of Neutral Organic Superacids—A DFT-B3LYP Study of Deprotonation of Nonacyanofluorene. Tetrahedron Lett. 2004, 45, 8663–8666. [Google Scholar] [CrossRef]
- Vianello, R.; Maksić, Z.B. Strong Acidity of Some Polycyclic Aromatic Compounds Annulated to a Cyclopentadiene Moiety and Their Cyano Derivatives—A Density Functional B3LYP Study. Eur. J. Org. Chem. 2005, 2005, 3571–3580. [Google Scholar] [CrossRef]
- Leito, I.; Kütt, A.; Rõõm, E.-I.; Koppel, I. Anions N[C(CN)2]3− and P[C(CN)2]3− and the Superacidic Properties of Their Conjugate Acids. THEOCHEM-J. Mol. Struc. 2007, 815, 41–43. [Google Scholar] [CrossRef]
- Kütt, A.; Koppel, I.; Koppel, I.A.; Leito, I. Boratabenzene Anions C5B(CN)6− and C5B(CF3)6− and the Superacidic Properties of Their Conjugate Acids. ChemPhysChem 2009, 10, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Valadbeigi, Y.; Gal, J.-F. Organometallic Superacids and Hyperacids: Acidity Enhancement by Internal Bonding with a Strong Electron-Pair Acceptor Group BX2. Chem. Phys. Lett. 2021, 763, 138207. [Google Scholar] [CrossRef]
- Lipping, L.; Leito, I.; Koppel, I.; Krossing, I.; Himmel, D.; Koppel, I.A. Superacidity of Closo-Dodecaborate-Based Brønsted Acids: A DFT Study. J. Phys. Chem. A 2015, 119, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Reed, C.A. Exploration of the Pentacyano-Cyclo-Pentadienide Ion, C5(CN)5−, as a Weakly Coordinating Anion and Potential Superacid Conjugate Base. Silylation and Protonation. Chem. Commun. 2004, 706–707. [Google Scholar] [CrossRef] [PubMed]
- Soltner, T.; Häusler, J.; Kornath, A.J. The Existence of Tricyanomethane. Angew. Chem. Int. Ed. 2015, 54, 13775–13776. [Google Scholar] [CrossRef]
- Bernhardt, E.; Henkel, G.; Willner, H. Die Tetracyanoborate M[B(CN)4], M = [Bu4N]+, Ag+, K+. Z. Anorg. Allg. Chem. 2000, 626, 560–568. [Google Scholar] [CrossRef]
- Küppers, T.; Bernhardt, E.; Willner, H.; Rohm, H.W.; Köckerling, M. Tetracyanoborate Salts M[B(CN)4] with M = Singly Charged Cations: Properties and Structures. Inorg. Chem. 2005, 44, 1015–1022. [Google Scholar] [CrossRef]
- Scheers, J.; Johansson, P.; Jacobsson, P. Anions for Lithium Battery Electrolytes: A Spectroscopic and Theoretical Study of the B (CN)4− Anion of the Ionic Liquid C2mim [B(CN)4]. J. Electrochem. Soc. 2008, 155, A628. [Google Scholar] [CrossRef]
- Angenendt, K.; Johansson, P. Ionic Liquid Based Lithium Battery Electrolytes: Charge Carriers and Interactions Derived by Density Functional Theory Calculations. J. Phys. Chem. B 2011, 115, 7808–7813. [Google Scholar] [CrossRef] [PubMed]
- Martins, V.L.; Rennie, A.J.R.; Sanchez-Ramirez, N.; Torresi, R.M.; Hall, P.J. Improved Performance of Ionic Liquid Supercapacitors by Using Tetracyanoborate Anions. ChemElectroChem 2018, 5, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Ershov, O.V.; Eremkin, A.V.; Kayukov, Y.S.; Lyshchikov, A.N.; Nasakin, O.E.; Tafeenko, V.A. Three-Component Synthesis of 2-(4-Amino-2,5-Dihydro-1H-Imidazol-5-Ylidene)Malononitriles. Russ. J. Org. Chem. 2008, 44, 570–576. [Google Scholar] [CrossRef]
- Gardner, H.C.; Kochi, J.K. Alkylation of Tetracyanoethylene. Reaction with Grignard Reagents. J. Am. Chem. Soc. 1976, 98, 558–566. [Google Scholar] [CrossRef]
- Kütt, A. Step-by-Step Replacement of Cyano Groups by Tricyanovinyls—The Influence on the Acidity. 2023. Available online: https://datadoi.ee/handle/33/575 (accessed on 24 November 2023).
- Koppel, I.A.; Burk, P.; Koppel, I.; Leito, I. Generalized Principle of Designing Neutral Superstrong Brønsted Acids. J. Am. Chem. Soc. 2002, 124, 5594–5600. [Google Scholar] [CrossRef] [PubMed]
- Toomsalu, E.; Koppel, I.A.; Burk, P. Critical Test of Some Computational Chemistry Methods for Prediction of Gas-Phase Acidities and Basicities. J. Chem. Theory Comput. 2013, 9, 3947–3958. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 Theory Using Reduced Order Perturbation Theory. J. Chem. Phys. 2007, 127, 124105. [Google Scholar] [CrossRef]
- Hunter, E.P.; Lias, S.G. Proton Affinity Evaluation. In NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2016; p. 20899. [Google Scholar] [CrossRef]
- Raamat, E.; Kaupmees, K.; Ovsjannikov, G.; Trummal, A.; Kütt, A.; Saame, J.; Koppel, I.; Kaljurand, I.; Lipping, L.; Rodima, T.; et al. Acidities of Strong Neutral Brønsted Acids in Different Media. J. Phys. Org. Chem. 2013, 26, 162–170. [Google Scholar] [CrossRef]
- Lipping, L.; Leito, I.; Koppel, I.; Koppel, I.A. Gas-Phase Brønsted Superacidity of Some Derivatives of Monocarba-Closo-Borates: A Computational Study. J. Phys. Chem. A 2009, 113, 12972–12978. [Google Scholar] [CrossRef]
- Kütt, A.; Tshepelevitsh, S.; Saame, J.; Lõkov, M.; Kaljurand, I.; Selberg, S.; Leito, I. Strengths of Acids in Acetonitrile. Eur. J. Org. Chem. 2021, 2021, 1407–1419. [Google Scholar] [CrossRef]
- Ding, F.; Smith, J.M.; Wang, H. First-Principles Calculation of pKa Values for Organic Acids in Nonaqueous Solution. J. Org. Chem. 2009, 74, 2679–2691. [Google Scholar] [CrossRef] [PubMed]
- Bordwell, F.G. Equilibrium Acidities in Dimethyl Sulfoxide Solution. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar] [CrossRef]
- Paenurk, E.; Kaupmees, K.; Himmel, D.; Kütt, A.; Kaljurand, I.; Koppel, I.A.; Krossing, I.; Leito, I. A Unified View to Brønsted Acidity Scales: Do We Need Solvated Protons? Chem. Sci. 2017, 8, 6964–6973. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A. Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Klamt, A.; Jonas, V.; Bürger, T.; Lohrenz, J.C.W. Refinement and Parametrization of COSMO-RS. J. Phys. Chem. A 1998, 102, 5074–5085. [Google Scholar] [CrossRef]
- Eckert, F.; Klamt, A. Fast Solvent Screening via Quantum Chemistry: COSMO-RS Approach. AIChE J. 2002, 48, 369–385. [Google Scholar] [CrossRef]
- Klamt, A.; Eckert, F.; Arlt, W. COSMO-RS: An Alternative to Simulation for Calculating Thermodynamic Properties of Liquid Mixtures. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 101–122. [Google Scholar] [CrossRef]
- Eckert, F.; Leito, I.; Kaljurand, I.; Kütt, A.; Klamt, A.; Diedenhofen, M. Prediction of Acidity in Acetonitrile Solution with COSMO-RS. J. Comput. Chem. 2009, 30, 799–810. [Google Scholar] [CrossRef]
- Kütt, A.; Leito, I.; Kaljurand, I.; Sooväli, L.; Vlasov, V.M.; Yagupolskii, L.M.; Koppel, I.A. A Comprehensive Self-Consistent Spectrophotometric Acidity Scale of Neutral Brønsted Acids in Acetonitrile. J. Org. Chem. 2006, 71, 2829–2838. [Google Scholar] [CrossRef]
- TURBOMOLE V7.2, 2017, A Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 24 November 2023).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- BIOVIA COSMOconfX 2021, 2021, Version 21.0. BIOVIA COSMOthermX19, Release 2023. Dassault Systèmes. Available online: http://www.3ds.com (accessed on 24 November 2023).
- University of Tartu, “UT Rocket”. Share.neic.no. Available online: https://hpc.ut.ee/science/referencing (accessed on 24 November 2023).







| Initial Compound | Step 1 | Step 2 | Step 3 | Step 4 |
|---|---|---|---|---|
 |  | |||
| 1 | 2 | |||
 |  |  | ||
| 3 | 4 | 5 | ||
 |  |  |  | |
| 6 | 7 | 8 | 9 | |
 |  |  |  |  |
| 10 | 11 | 12 | 13 | 14 |
 |  | [B12(CN)12]2– | [B12(CN)12]H– | [CB12(CN)12]– |
|---|---|---|---|---|
| 15 | 16 | 17 | 18 | 19 |
| Compound | LITERATURE pKa(MeCN) | COSMO-RS pKa(MeCN) | |
|---|---|---|---|
| NH2-TCNP | 3.30 | 4.9 | |
| 3,4-(MeO)2-C6H3-TCNP | 1.08 | 3.2 | |
| 4-MeO-C6H4-TCNP | 1.03 | 3.2 | |
| Ph-TCNP | 0.28 | 2.5 | |
| 3-CF3-C6H4-TCNP | −0.50 | 1.5 | |
| H-TCNP | −1.34 | 1.3 | |
| Br-TCNP | −1.90 | 0.4 | |
| 3,5-(CF3)2-C6H3-TCNP | −1.84 | 0.4 | |
| Cl-TCNP | −1.87 | 0.1 | |
| CN-CH2-TCNP | −2.19 | −0.1 | |
| CF3-TCNP | −2.90 | −0.5 | |
| Me-TCNP | 0.39 | 3.3 | |
| [N-TCNV2]H | 5H | −0.96 | 0.3 |
| [(CN)3C]H | 6H | 2.76 | 5.1 |
| [(CN)2C-TCNV]H | 7H | −4.57 | −2.8 |
| [O-TCNV]H | 2H | 3.1 | 2.93 |
| [(CN)4B]H | 10H | −1.0 | −5.61 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kütt, A. Step-by-Step Replacement of Cyano Groups by Tricyanovinyls—The Influence on the Acidity. Molecules 2023, 28, 8157. https://doi.org/10.3390/molecules28248157
Kütt A. Step-by-Step Replacement of Cyano Groups by Tricyanovinyls—The Influence on the Acidity. Molecules. 2023; 28(24):8157. https://doi.org/10.3390/molecules28248157
Chicago/Turabian StyleKütt, Agnes. 2023. "Step-by-Step Replacement of Cyano Groups by Tricyanovinyls—The Influence on the Acidity" Molecules 28, no. 24: 8157. https://doi.org/10.3390/molecules28248157
APA StyleKütt, A. (2023). Step-by-Step Replacement of Cyano Groups by Tricyanovinyls—The Influence on the Acidity. Molecules, 28(24), 8157. https://doi.org/10.3390/molecules28248157