
HS-BAμE: A New Alternative Approach for VOCs Analysis—Application for Monitoring Biogenic Emissions from Tree Species

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. GC-MS Instrumental Conditions
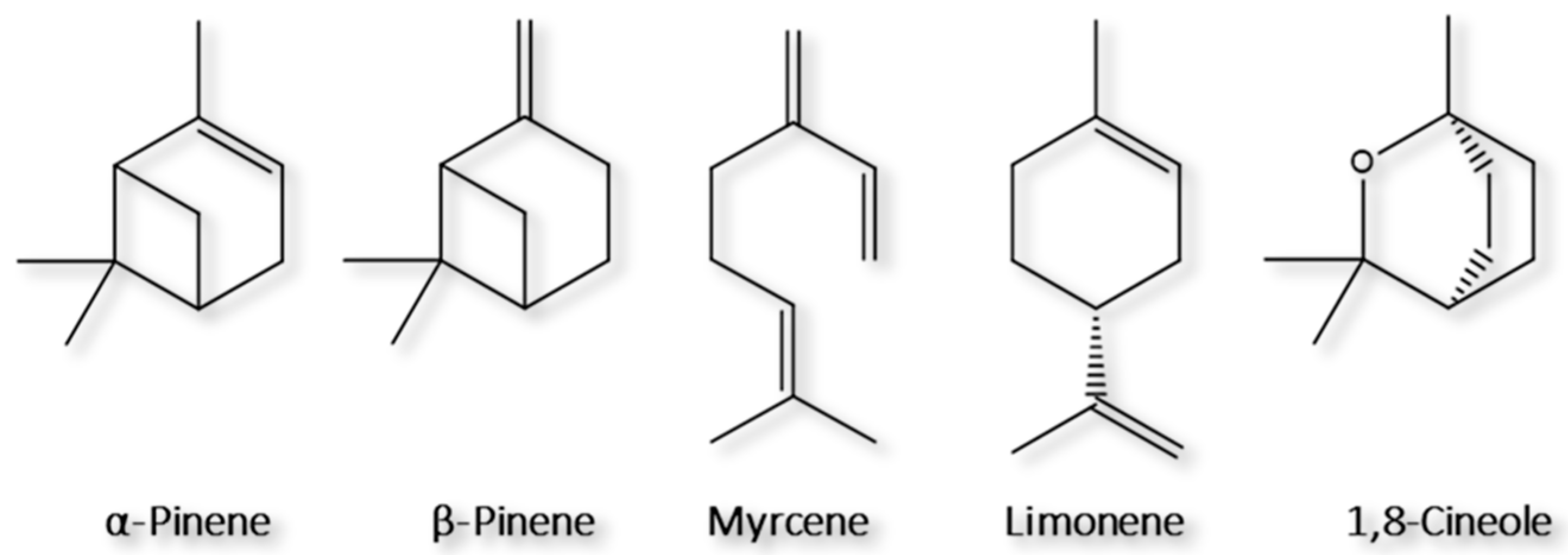
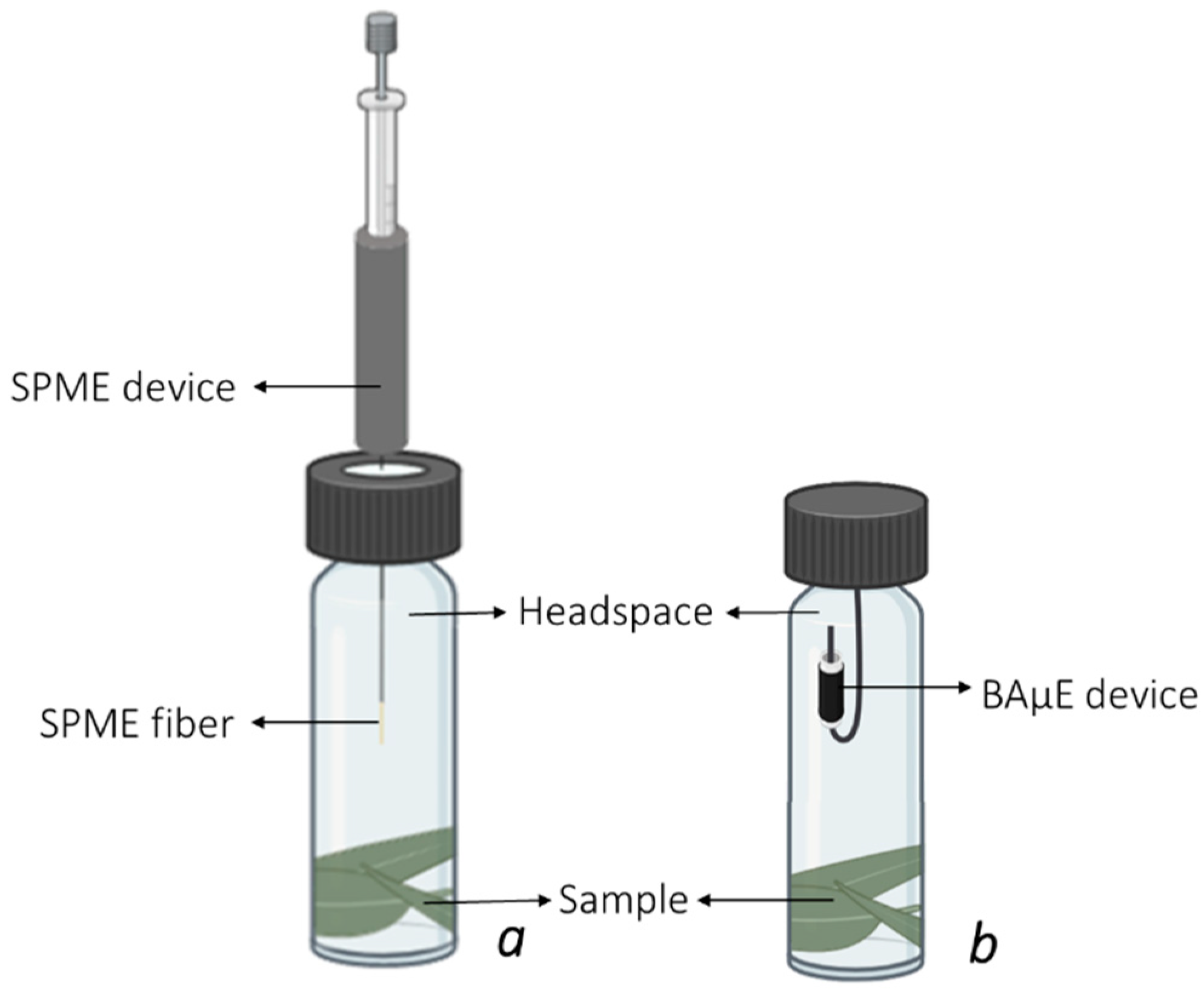
2.2. Development of the Microextraction Methodologies
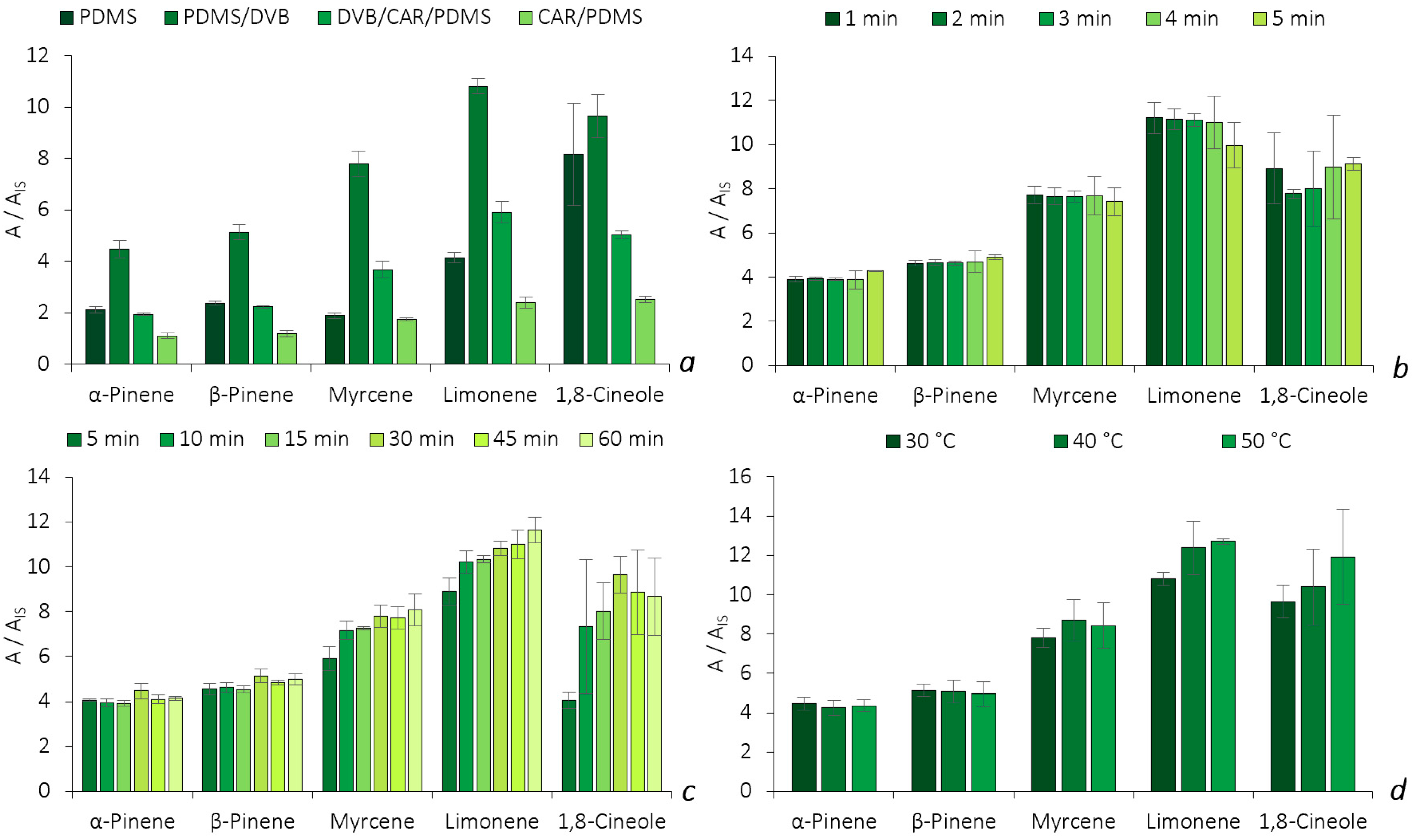
2.2.1. Optimization of the HS-SPME Methodology
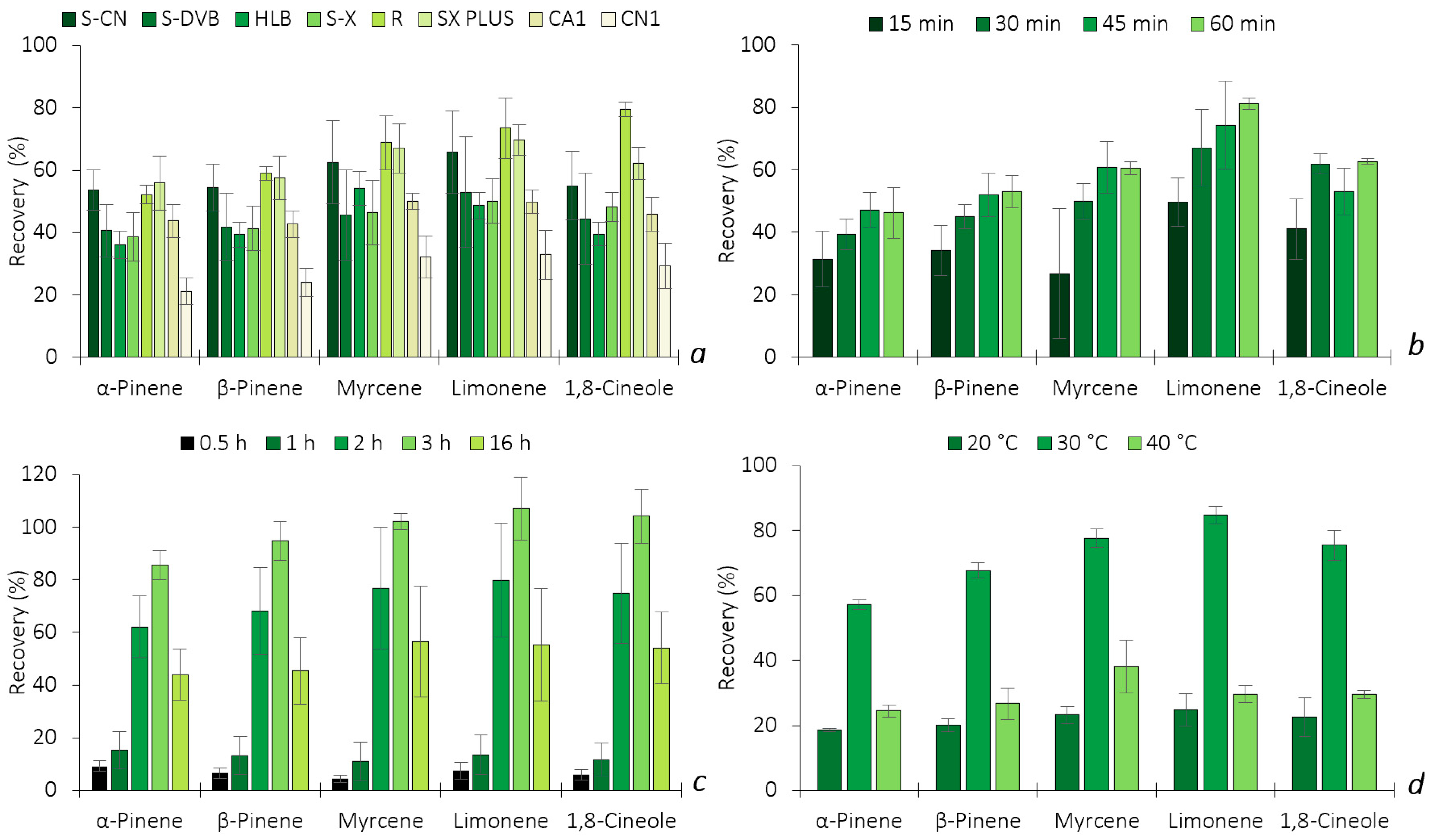
2.2.2. Optimization of the HS-BAμE Methodology
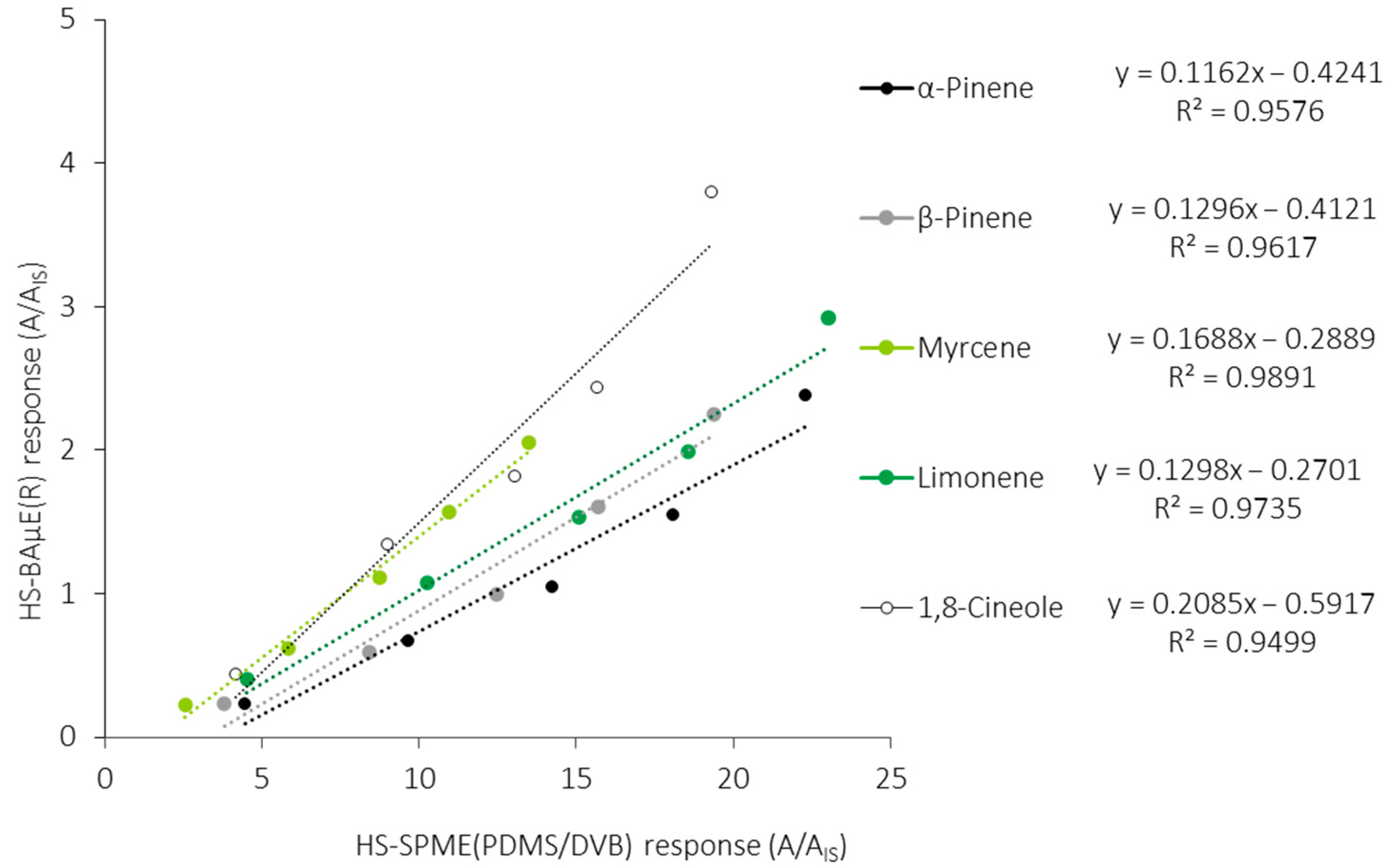
2.3. Validation and Comparison of Both Methodologies
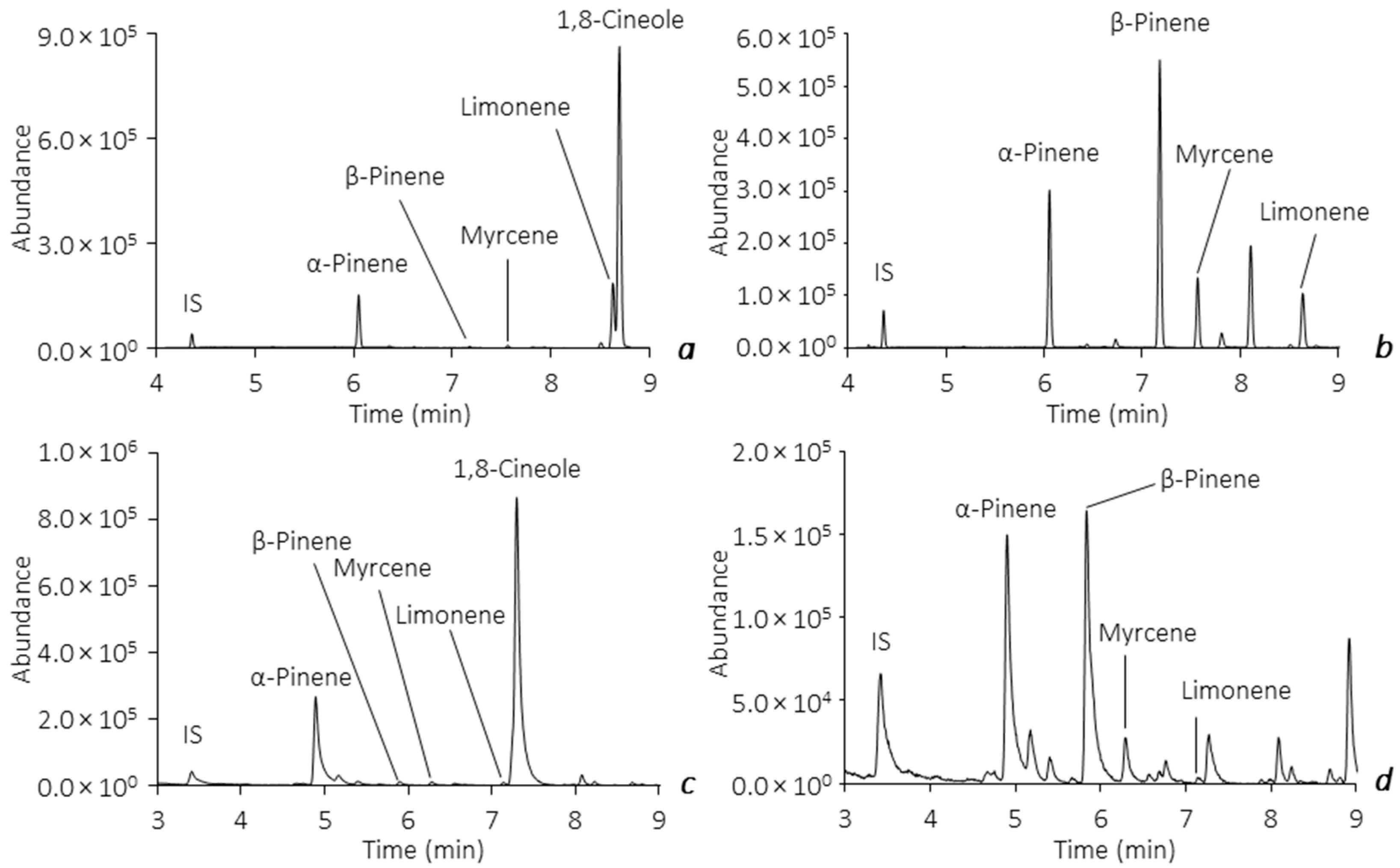
2.4. Application to Real Samples
3. Materials and Methods
3.1. Chemical Standards, Material and Real Samples
3.2. Experimental Set-Up
3.2.1. Preparation and Conditioning of SPME Fibers and BAμE Devices
3.2.2. Experimental Optimization Conditions (HS-SPME and HS-BAμE Assays)
3.2.3. Validation Assays
3.2.4. Real Samples Assays
3.3. Instrumental Set-Up
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chatelon, F.-J.; Sauvagnargues, S.; Dusserre, G.; Balbi, J.-H. Generalized Blaze Flash, a “Flashover” Behavior for Forest Fires—Analysis from the Firefighter’s Point of View. Open J. For. 2014, 4, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Chetehouna, K.; Barboni, T.; Zarguili, I.; Leoni, E.; Simeoni, A.; Fernandez-Pello, A.C. Investigation on the emission of volatile organic compounds from heated vegetation and their potential to cause an accelerating forest fire. Combust. Sci. Technol. 2009, 181, 1273–1288. [Google Scholar] [CrossRef] [Green Version]
- Demeestere, K.; Dewulf, J.; De Witte, B.; Van Langenhove, H. Sample preparation for the analysis of volatile organic compounds in air and water matrices. J. Chromatogr. A 2007, 1153, 130–144. [Google Scholar] [CrossRef]
- Nogueira, J.M.F. Novel sorption-based methodologies for static microextraction analysis: A review on SBSE and related techniques. Anal. Chim. Acta 2012, 757, 1–10. [Google Scholar] [CrossRef]
- Ide, A.H.; Nogueira, J.M.F. New-generation bar adsorptive microextraction (BAμE) devices for a better eco-user-friendly analytical approach–Application for the determination of antidepressant pharmaceuticals in biological fluids. J. Pharm. Biomed. Anal. 2018, 153, 126–134. [Google Scholar] [CrossRef]
- Pawliszyn, J. Handbook of Solid Phase Microextraction; Elsevier: Toronto, ON, Canada, 2012. [Google Scholar]
- Zhu, H.; Zhu, J.; Wang, L.; Li, Z. Development of a SPME-GC-MS method for the determination of volatile compounds in Shanxi aged vinegar and its analytical characterization by aroma wheel. J. Food Sci. Technol. 2016, 53, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Jerković, I.; Marijanović, Z. A short review of headspace extraction and ultrasonic solvent extraction for honey volatiles fingerprinting. Croat. J. Food Sci. Technol. 2009, 1, 28–34. [Google Scholar]
- Rianawati, E.; Balasubramanian, R. Optimization and validation of solid phase micro-extraction (SPME) method for analysis of polycyclic aromatic hydrocarbons in rainwater and stormwater. Phys. Chem. Earth 2009, 34, 857–865. [Google Scholar] [CrossRef]
- Qiu, R.; Trengove, R.; Agarwal, M.; Ren, Y. Optimization of Headspace Solid-Phase Microextraction Conditions for the Identification of Phytophthora cinnamomi Rands. Plant Dis. 2014, 98, 1088–1098. [Google Scholar] [CrossRef] [Green Version]
- Serôdio, P.; Nogueira, J.M.F. Multi-residue screening of endocrine disrupters chemicals in water samples by stir bar sorptive extraction-liquid desorption-capillary gas chromatography–mass spectrometry detection. Anal. Chim. Acta 2004, 517, 21–32. [Google Scholar] [CrossRef]
- Da Silva, C.P.; Emídio, E.S.; De Marchi, M.R.R. Method validation using weighted linear regression models for quantification of UV filters in water samples. Talanta 2015, 131, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.M.; Nogueira, J.M.F. High throughput bar adsorptive microextraction: A novel cost-effective tool for monitoring benzodiazepines in large number of biological samples. Talanta 2019, 199, 195–202. [Google Scholar] [CrossRef]
- Souza Silva, É.A.; Saboia, G.; Jorge, N.C.; Hoffmann, C.; dos Santos Isaias, R.M.; Soares, G.L.G.; Zini, C.A. Development of a HS-SPME-GC/MS protocol assisted by chemometric tools to study herbivore-induced volatiles in Myrcia splendens. Talanta 2017, 175, 9–20. [Google Scholar] [CrossRef]
- Sampedro, L.; Moreira, X.; Llusia, J.; Peñuelas, J.; Zas, R. Genetics, phosphorus availability, and herbivore-derived induction as sources of phenotypic variation of leaf volatile terpenes in a pine species. J. Exp. Bot. 2010, 61, 4437–4447. [Google Scholar] [CrossRef] [Green Version]
- Tavares, C.S.; Martins, A.; Faleiro, M.L.; Miguel, M.G.; Duarte, L.C.; Gameiro, J.A.; Roseiro, L.B.; Figueiredo, A.C. Bioproducts from forest biomass: Essential oils and hydrolates from wastes of Cupressus lusitanica Mill. and Cistus ladanifer L. Ind. Crops Prod. 2020, 144, 112034. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Mestre, A.S.; Neng, N.R.; Ania, C.O.; Carvalho, A.P.; Nogueira, J.M.F. Carbon-Based Sorbent Coatings for the Determination of Pharmaceutical Compounds by Bar Adsorptive Microextraction. ACS Appl. Bio Mater. 2020, 3, 2078–2091. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Calado, B.B.C.; Oliveira, M.N.; Neng, N.R.; Nogueira, J.M.F. Bar adsorptive microextraction coated with carbonbased phase mixtures for performance-enhancement to monitor selected benzotriazoles, benzothiazoles, and benzenesulfonamides in environmental water matrices. Molecules 2020, 25, 2133. [Google Scholar] [CrossRef]
- Almeida, C.; Nogueira, J.M.F. Determination of steroid sex hormones in real matrices by bar adsorptive microextraction (BAμE). Talanta 2015, 136, 145–154. [Google Scholar] [CrossRef]
- George, M.J.; Njobeh, P.B.; Gbashi, S.; Adegoke, G.O.; Dubery, I.A.; Madala, N.E. Rapid Screening of Volatile Organic Compounds from Aframomum danielli Seeds Using Headspace Solid Phase Microextraction Coupled to Gas Chromatography Mass Spectrometry. Int. J. Anal. Chem. 2018, 2018, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Barták, P.; Bednář, P.; Cáp, L.; Ondráková, L.; Stránský, Z. SPME-A valuable tool for investigation of flower scent. J. Sep. Sci. 2003, 26, 715–721. [Google Scholar] [CrossRef]
- Durant, A.A.; Rodríguez, C.; Herrera, L.; Almanza, A.; Santana, A.I.; Spadadora, C.; Gupta, M.P. Anti-malarial activity and HS-SPME-GC-MS chemical profiling of Plinia cerrocampanensis leaf essential oil. Malar. J. 2014, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Almeida, C.; Nogueira, J.M.F. Determination of trace levels of parabens in real matrices by bar adsorptive microextraction using selective sorbent phases. J. Chromatogr. A 2014, 1348, 17–26. [Google Scholar] [CrossRef]
- Neng, N.R.; Nogueira, J.M.F. Development of a bar adsorptive micro-extraction-large-volume injection-gas chromatography-mass spectrometric method for pharmaceuticals and personal care products in environmental water matrices. Anal. Bioanal. Chem. 2012, 402, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.N.; Liang, M.; Yang, Y.; Zheng, F.P.; Wang, X.P.; Yu, A.N. Optimization of a headspace solid-phase microextraction method for the gas chromatography-mass spectrometry analysis aroma compounds of litsea mollis hemsl. Immature fruit. Food Sci. Technol. 2020, 40, 786–793. [Google Scholar] [CrossRef] [Green Version]
- 32004L0042; Directive 2004/42/CE of the European Parliament and of the Council of 21 April 2004 on the Limitation of Emissions of Volatile Organic Compounds Due to the Use of Organic Solvents in Certain Paints and Varnishes and vEhicle Refinishing Products and Amending Directive 1999/13/EC. European Union: Brussels, Belgium, 2004.
- Huang, W.; Ratkowsky, D.A.; Hui, C.; Wang, P.; Su, J.; Shi, P. Leaf fresh weight versus dry weight: Which is better for describing the scaling relationship between leaf biomass and leaf area for broad-leaved plants? Forests 2019, 10, 256. [Google Scholar] [CrossRef] [Green Version]
- Adams, R.P. Identification of Essential Oil Components by Gas Chromatograpy/Mass Spectrometry, 4th ed.; Allured publishing corporation: Carol Stream, IL, USA, 2007. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monoterpenoids | HS-SPME(PDMS/DVB) | HS-BAμE(R) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LOD (ng L−1) | LOQ (ng L−1) | Linear Range (µg L−1) | a | b | r2 | LOD (µg L−1) | LOQ (µg L−1) | Linear Range (mg L−1) | a | b | r2 | |
| α-Pinene | 25.0 | 75.0 | 0.5–17.5 | 26.193 | −0.029 | 0.9987 | 5.0 | 16.6 | 20.0–100.0 | 4.731 | −0.276 | 0.9976 |
| β-Pinene | 50.0 | 175.0 | 32.305 | −0.047 | 0.9984 | 4.734 | −0.296 | 0.9991 | ||||
| Myrcene | 50.0 | 175.0 | 51.874 | −0.480 | 0.9985 | 4.508 | −0.256 | 0.9959 | ||||
| Limonene | 25.0 | 75.0 | 75.885 | −0.324 | 0.9989 | 5.630 | −0.201 | 0.9965 | ||||
| 1,8-Cineole | 25.0 | 175.0 | 54.062 | 0.581 | 0.9966 | 7.184 | −0.362 | 0.9976 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonçalves, O.C.; Cerqueira, J.S.R.F.; Mestre, A.S.; Neng, N.R.; Nogueira, J.M.F. HS-BAμE: A New Alternative Approach for VOCs Analysis—Application for Monitoring Biogenic Emissions from Tree Species. Molecules 2023, 28, 1179. https://doi.org/10.3390/molecules28031179
Gonçalves OC, Cerqueira JSRF, Mestre AS, Neng NR, Nogueira JMF. HS-BAμE: A New Alternative Approach for VOCs Analysis—Application for Monitoring Biogenic Emissions from Tree Species. Molecules. 2023; 28(3):1179. https://doi.org/10.3390/molecules28031179
Chicago/Turabian StyleGonçalves, Oriana C., Jéssica S. R. F. Cerqueira, Ana S. Mestre, Nuno R. Neng, and José M. F. Nogueira. 2023. "HS-BAμE: A New Alternative Approach for VOCs Analysis—Application for Monitoring Biogenic Emissions from Tree Species" Molecules 28, no. 3: 1179. https://doi.org/10.3390/molecules28031179
APA StyleGonçalves, O. C., Cerqueira, J. S. R. F., Mestre, A. S., Neng, N. R., & Nogueira, J. M. F. (2023). HS-BAμE: A New Alternative Approach for VOCs Analysis—Application for Monitoring Biogenic Emissions from Tree Species. Molecules, 28(3), 1179. https://doi.org/10.3390/molecules28031179