

Four Routes to 3-(3-Methoxy-1,3-dioxopropyl)pyrrole, a Core Motif of Rings C and E in Photosynthetic Tetrapyrroles
 , and
, and
Abstract
:
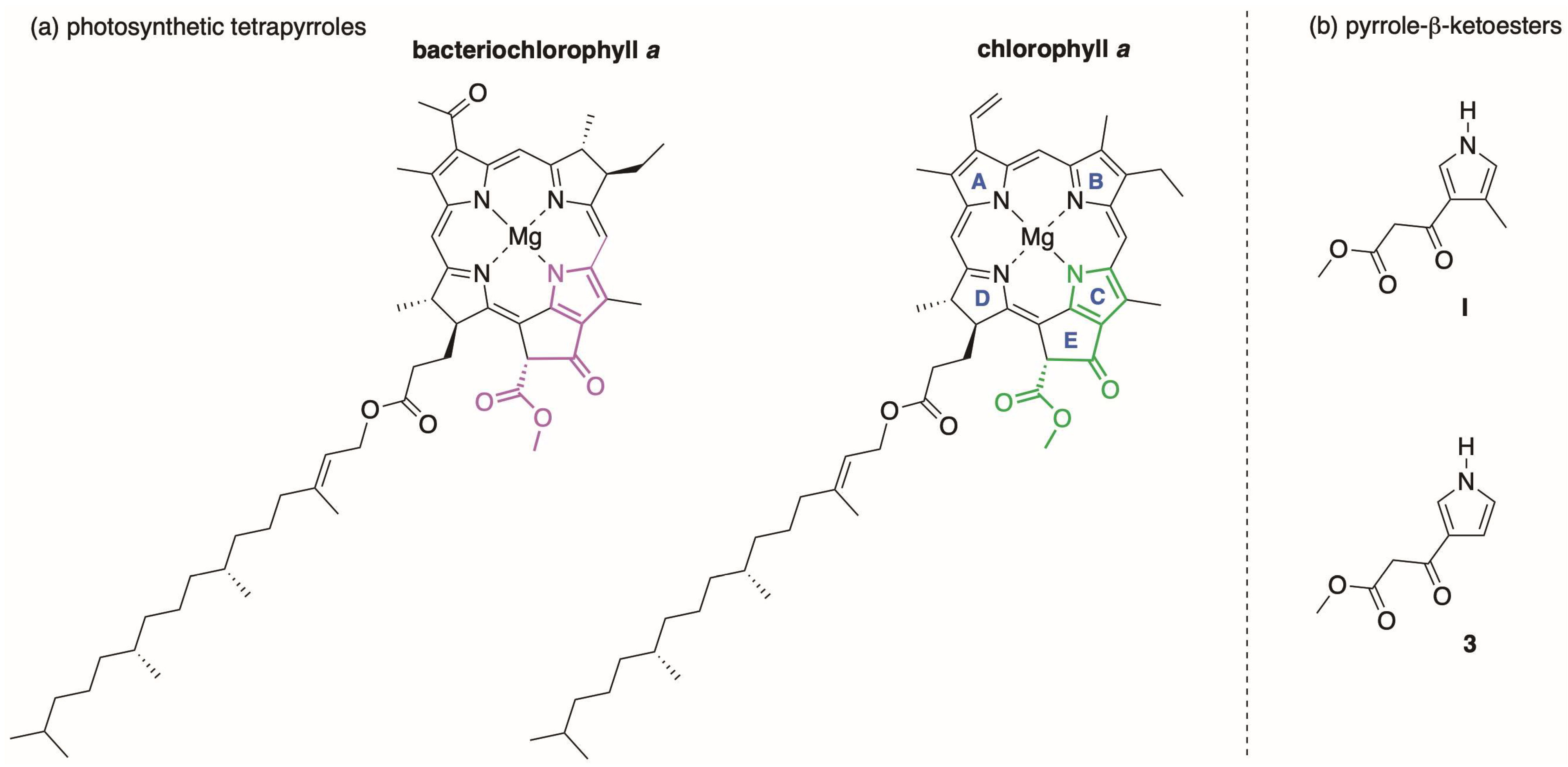
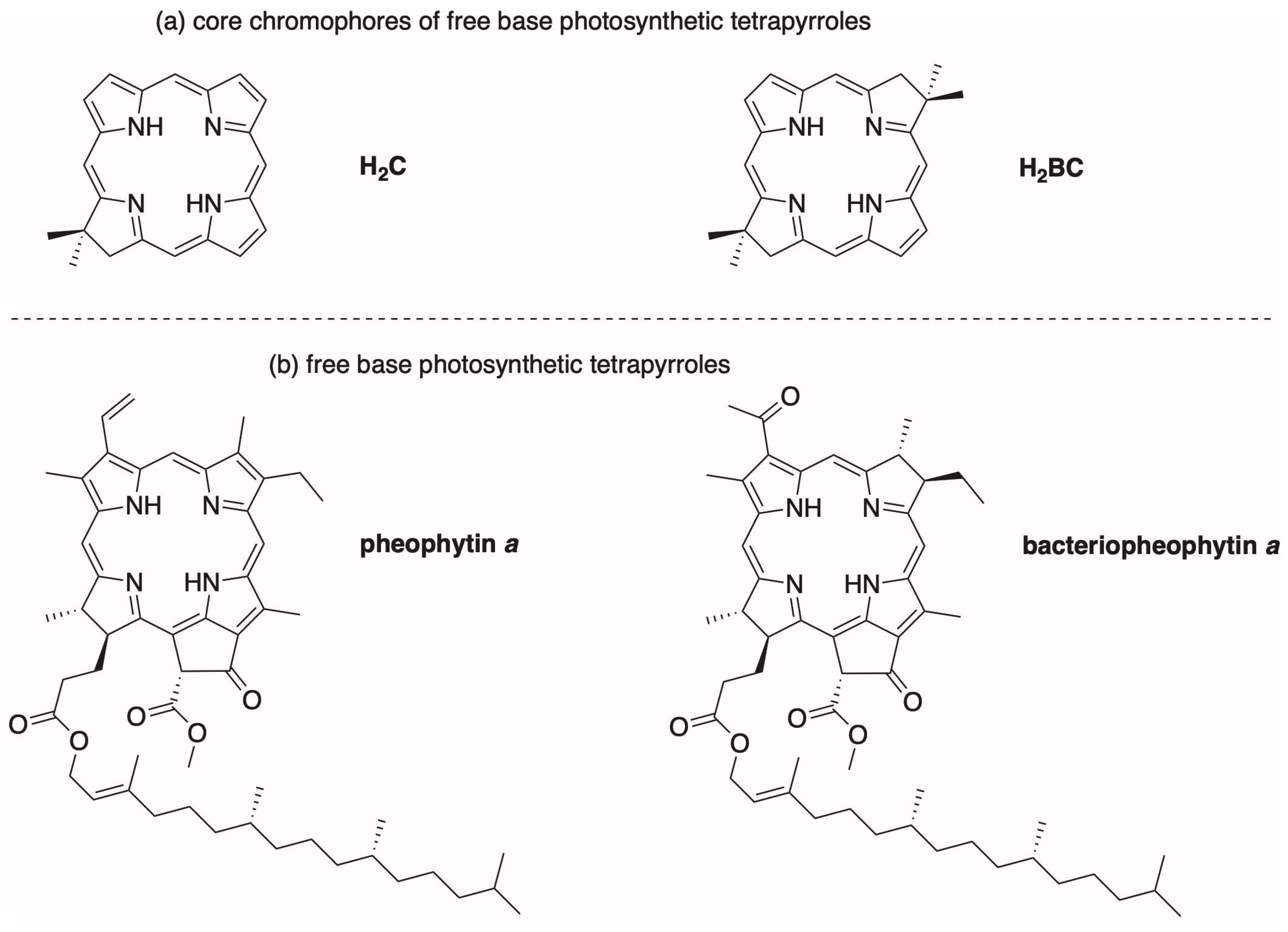
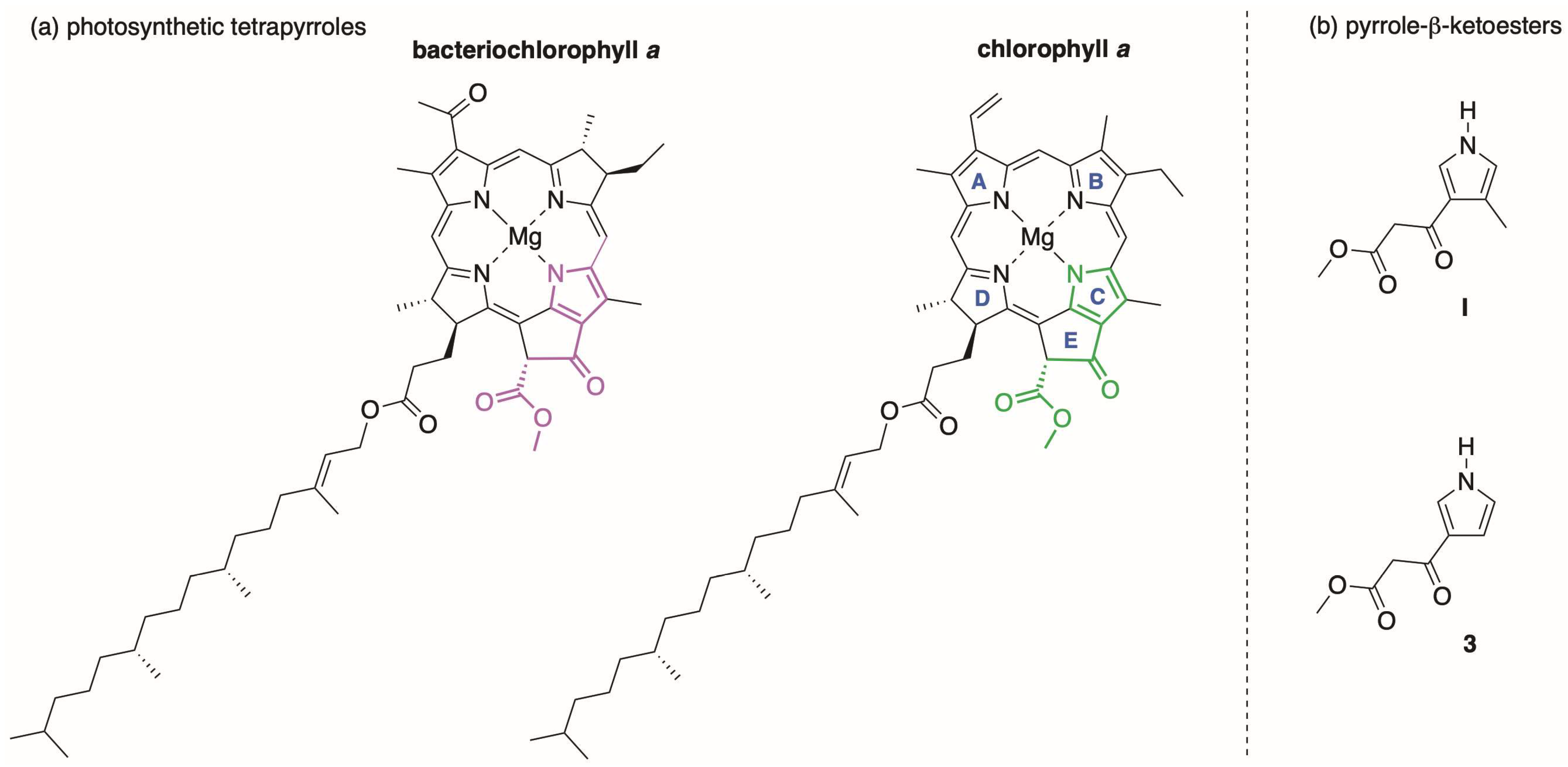
1. Introduction
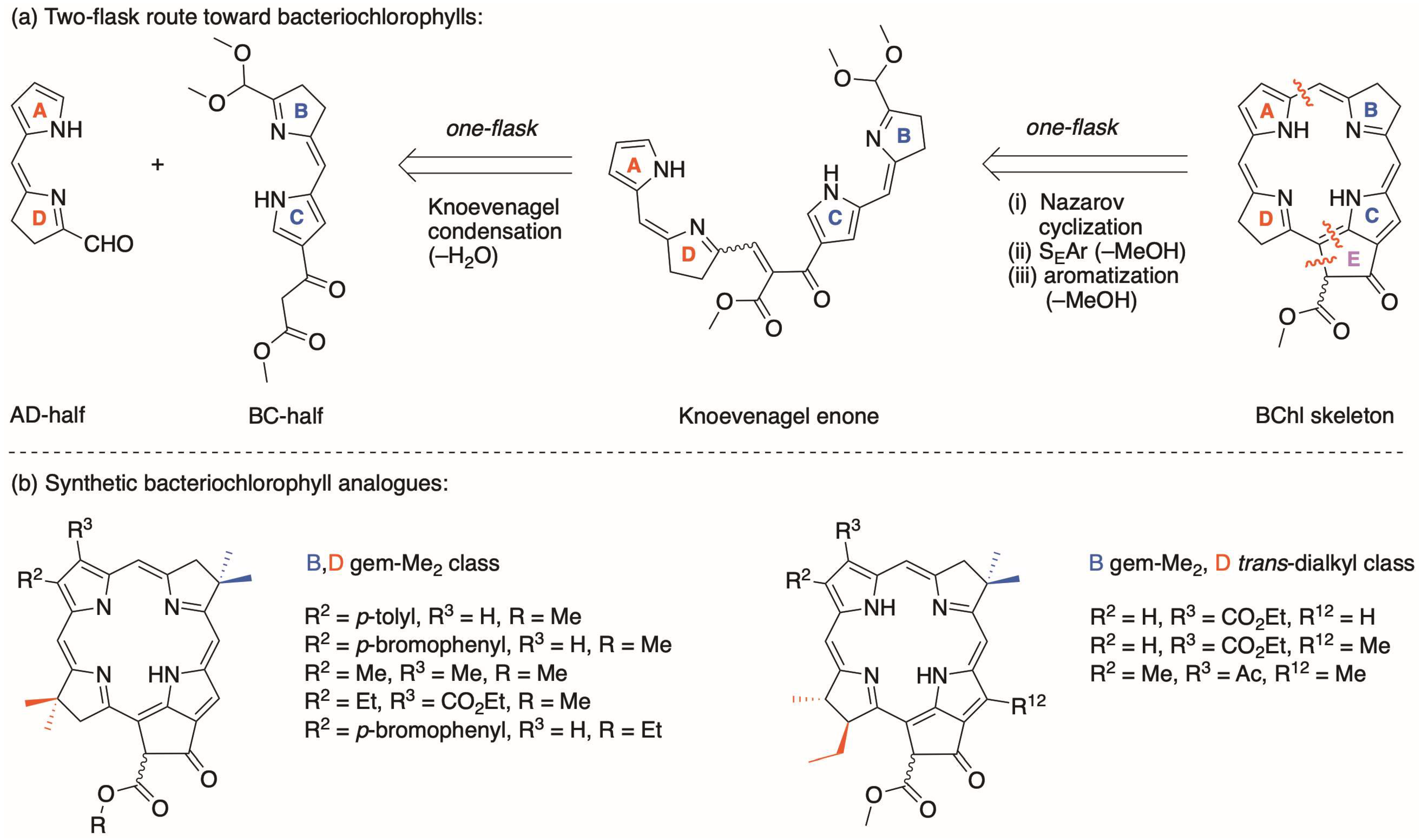
2. Reconnaissance
3. Results
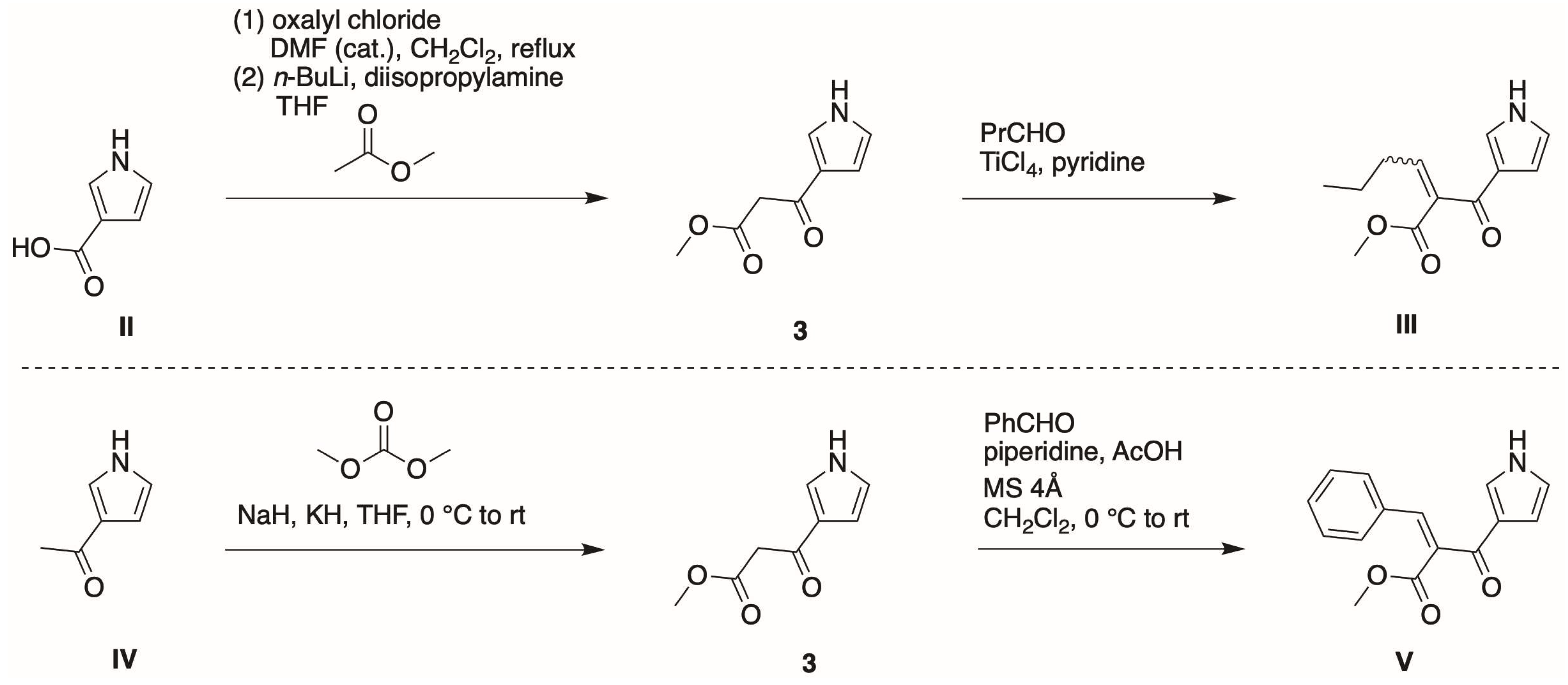
3.1. Synthetic Routes to a Pyrrole-β-Ketoester
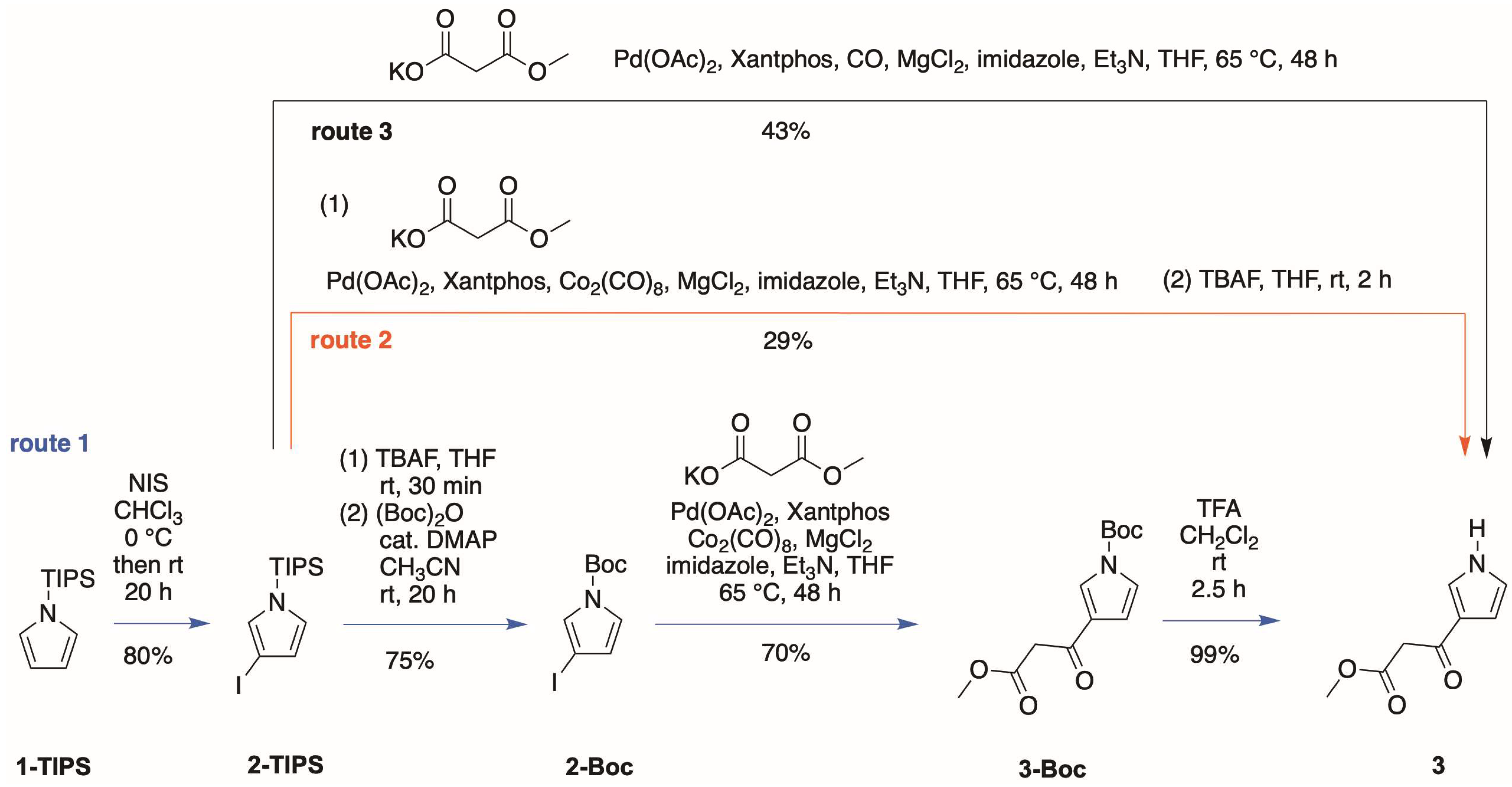
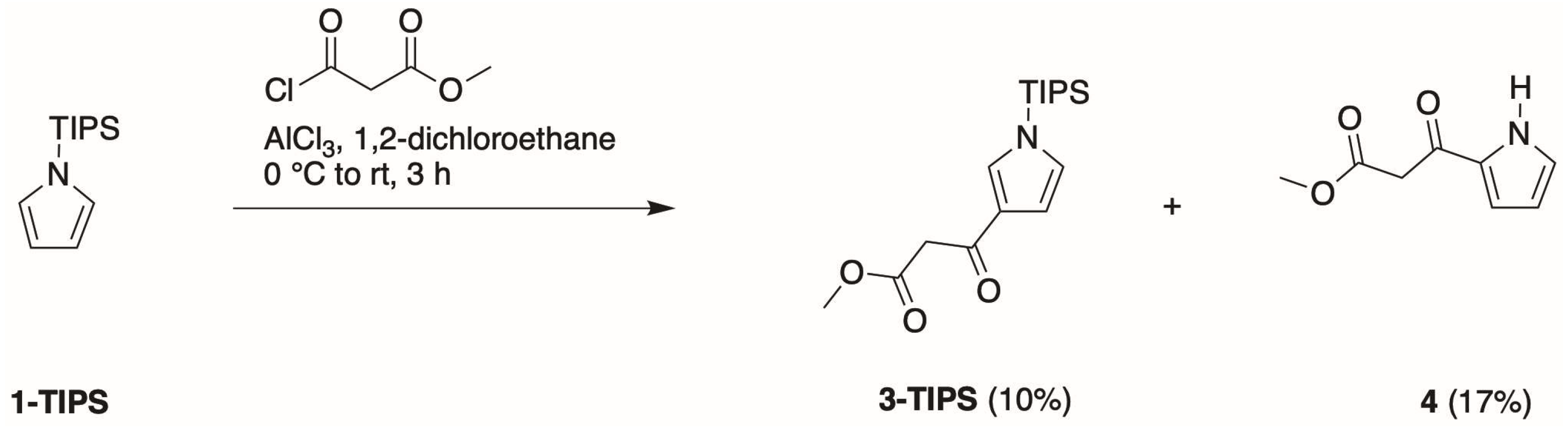
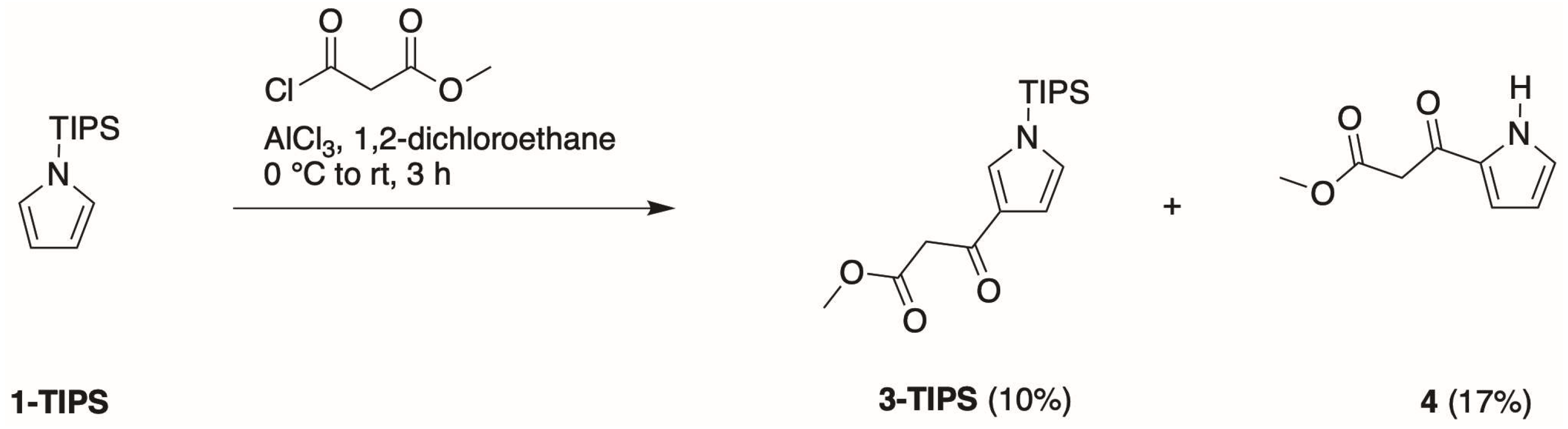
- First, treatment of pyrrole with methyl malonyl chloride in the presence of AlCl3 in 1,2-dichloroethane at 0 °C to room temperature for 3 h gave the 2-substituted isomer (4) in 30% isolated yield.
- Second, Bray and coworkers [21] reported that TIPS-pyrrole would react with potent acyl electrophiles in the absence of a Lewis acid. In our hands, the reaction of 1-TIPS with methyl malonyl chloride in 1,2-dichloroethane containing pyridine at 0 °C to reflux for 24 h gave two unknown products.
- Third, the same reaction of 1-TIPS with methyl malonyl chloride in nitromethane containing one of several Lewis acids also afforded 4 as the only product detectable by 1H-NMR spectroscopy. The Lewis acids were those identified in reactions of heterocycles [29], namely, Ga(OTf)3, Yb(OTf)3, and Hf(OTf)3. The results indicated that the TIPS group had been removed during the course of the reaction, without any treatment with a fluoride reagent, stemming most likely from chloride liberated during the course of the reaction.
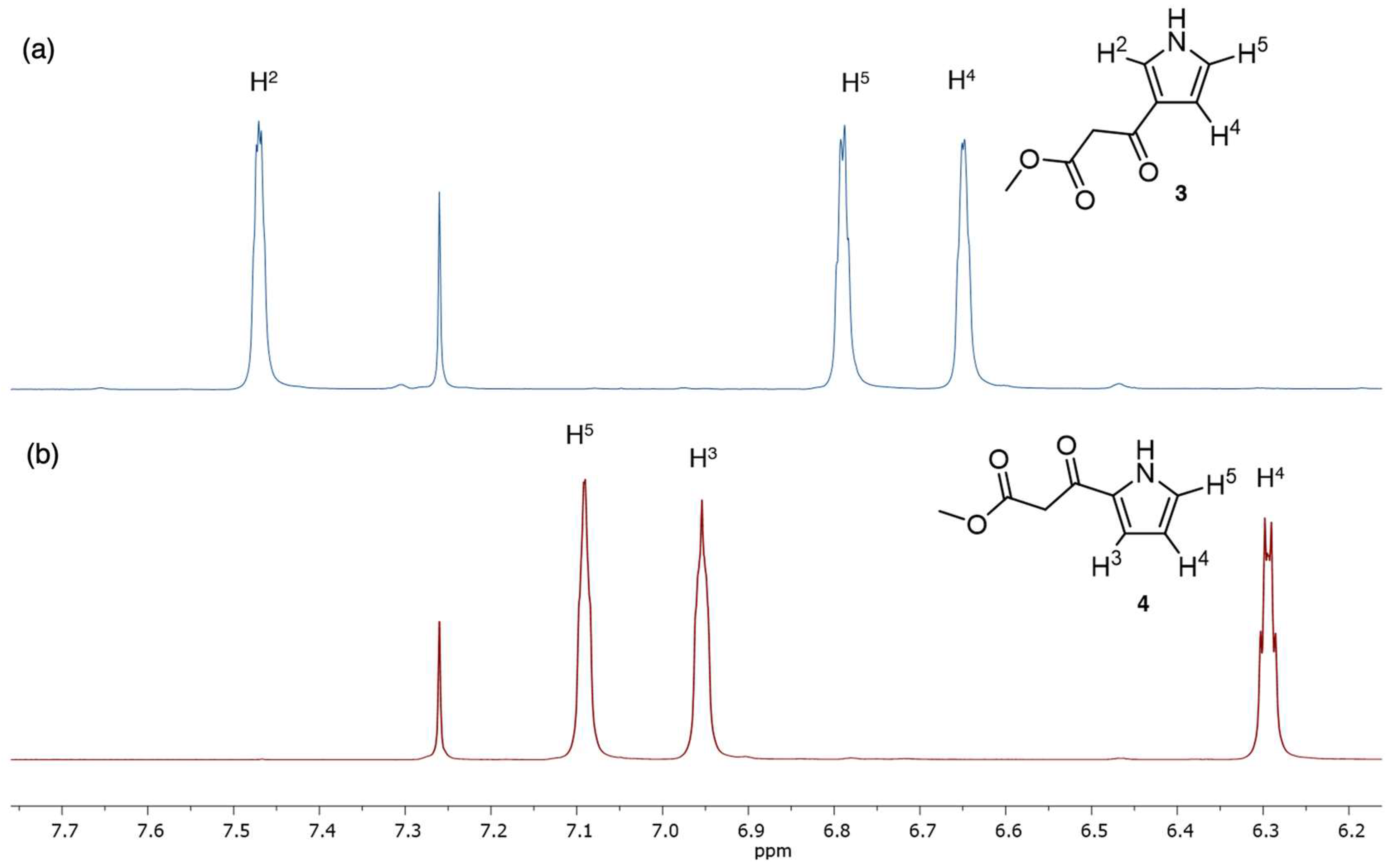
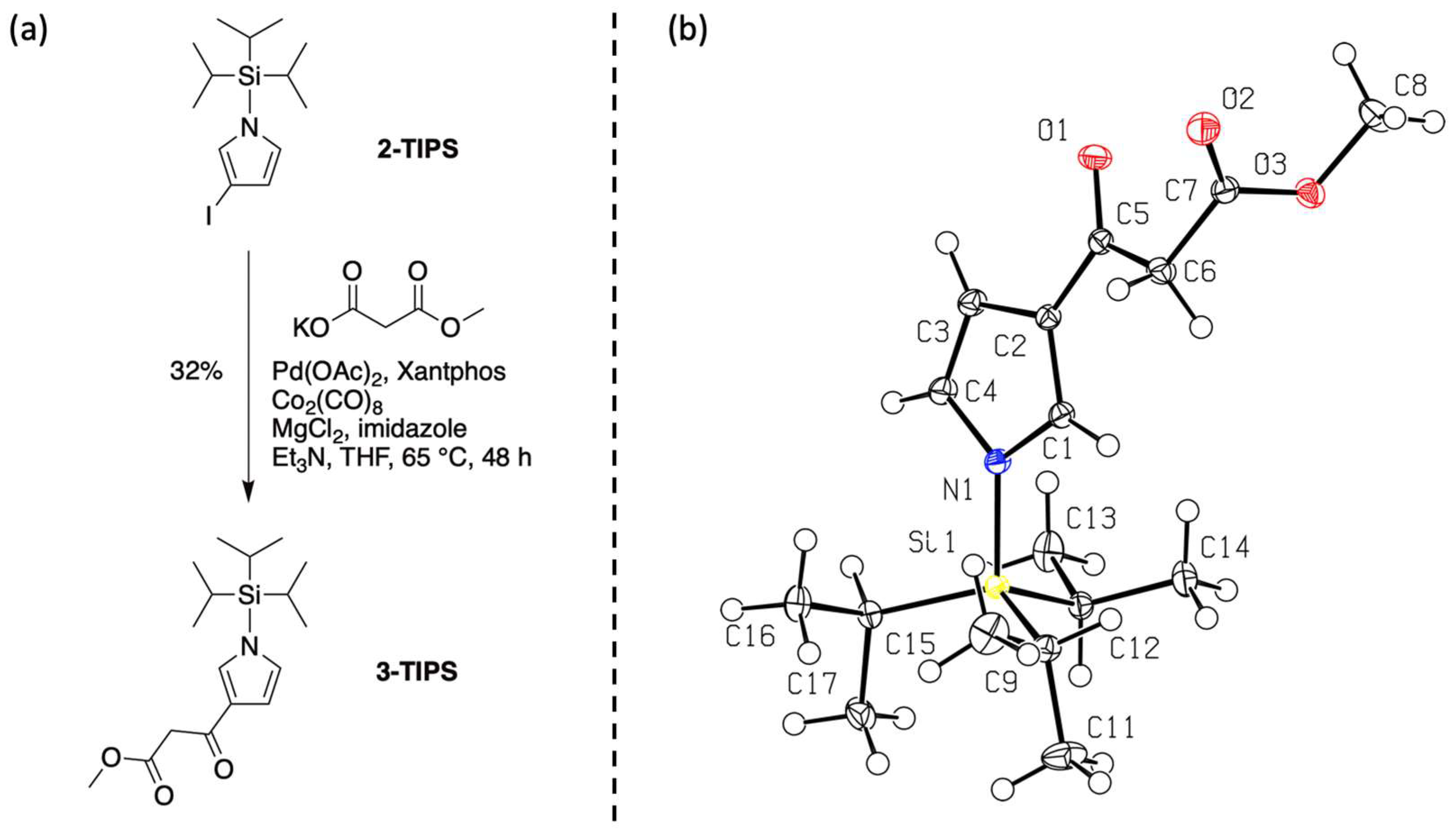
- Fourth, we again treated 1-TIPS with methyl malonyl chloride in 1,2-dichloroethane containing AlCl3 (Scheme 5). The two products isolated were 3-TIPS and 4; thus, 3-acylation occurred with the TIPS group intact, whereas it is most likely that 2-acylation occurred following the adventitious loss of the TIPS group. The subsequent treatment with TBAF, as shown in Scheme 4, then is only required for the conversion of 3-TIPS to the pyrrole 3. To achieve a higher yield in the acylation of 3-TIPS will require identification of reaction conditions wherein the TIPS group remains intact, thereby thwarting 2-acylation. Regardless, route 4 as is remains the most expeditious among the four routes examined. The synthesis of 3-TIPS was achieved by independent means, as described in the next section.
3.2. Structural Studies
4. Discussion
4.1. Comparison of Routes
4.2. Perspective on Design
5. Materials and Methods
5.1. General Methods
5.2. Synthesis and Characterization
5.2.1. Route 1
5.2.2. Route 2
5.2.3. Route 3
5.2.4. Route 4
5.2.5. Exploratory Acylation Studies
5.2.6. Intermediate for XRD Study
5.2.7. XRD Analysis
6. Outlook
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Liu, Y.; Zhang, S.; Lindsey, J.S. Total Synthesis Campaigns Toward Chlorophylls and Related Natural Hydroporphyrins—Diverse Macrocycles, Unrealized Opportunities. Nat. Prod. Rep. 2018, 35, 879–901. [Google Scholar] [CrossRef] [PubMed]
- Scheer, H. An Overview of Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications. In Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications; Grimm, B., Porra, R.J., Rüdiger, W., Scheer, H., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 1–26. [Google Scholar]
- Zhang, S.; Lindsey, J.S. Construction of the Bacteriochlorin Macrocycle with Concomitant Nazarov Cyclization to Form the Annulated Isocyclic Ring: Analogues of Bacteriochlorophyll a. J. Org. Chem. 2017, 82, 2489–2504. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Chau-Nguyen, K.; Lindsey, J.S. Synthesis of the Ring C Pyrrole of Native Chlorophylls and Bacteriochlorophylls. J. Org. Chem. 2019, 84, 11286–11293. [Google Scholar] [CrossRef]
- Wang, P.; Lu, F.; Lindsey, J.S. Use of the Nascent Isocyclic Ring to Anchor Assembly of the Full Skeleton of Model Chlorophylls. J. Org. Chem. 2020, 85, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Chau Nguyen, K.; Wang, P.; Sommer, R.D.; Lindsey, J.S. Asymmetric Synthesis of a Bacteriochlorophyll Model Compound Containing trans-Dialkyl Substituents in Ring D. J. Org. Chem. 2020, 85, 6605–6619. [Google Scholar] [CrossRef]
- Chau Nguyen, K.; Wang, P.; Lindsey, J.S. Study of Conditions for Streamlined Assembly of a Model Bacteriochlorophyll from Two Dihydrodipyrrin Halves. New J. Chem. 2021, 45, 569–581. [Google Scholar] [CrossRef]
- Chung, D.T.M.; Tran, P.V.; Chau Nguyen, K.; Wang, P.; Lindsey, J.S. Synthesis of Model Bacteriochlorophylls Containing Substituents of Native Rings A, C and E. New J. Chem. 2021, 45, 13302–13316. [Google Scholar] [CrossRef]
- List, B. Emil Knoevenagel and the Roots of Aminocatalysis. Angew. Chem. Int. Ed. 2010, 49, 1730–1734. [Google Scholar] [CrossRef]
- Frontier, A.J.; Hernandez, J.J. New Twists in Nazarov Cyclization Chemistry. Acc. Chem. Res. 2020, 53, 1822–1832. [Google Scholar] [CrossRef]
- Lindsey, J.S. De Novo Synthesis of Gem-Dialkyl Chlorophyll Analogues for Probing and Emulating our Green World. Chem. Rev. 2015, 115, 6534–6620. [Google Scholar] [CrossRef]
- Nigst, T.A.; Westermaier, M.; Ofial, A.R.; Mayr, H. Nucleophilic Reactivities of Pyrroles. Eur. J. Org. Chem. 2008, 2008, 2369–2374. [Google Scholar] [CrossRef]
- Mayr, H.; Lakhdar, S.; Maji, B.; Ofial, A.R. A Quantitative Approach to Nucleophilic Organocatalysis. Beilstein J. Org. Chem. 2012, 8, 1458–1478. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; Dogutan, D.K.; Ptaszek, M.; Lindsey, J.S. Synthesis of Hydrodipyrrins Tailored for Reactivity at the 1- and 9-Positions. Tetrahedron 2007, 63, 37–55. [Google Scholar] [CrossRef] [Green Version]
- Malona, J.A.; Colbourne, J.M.; Frontier, A.J. A General Method for the Catalytic Nazarov Cyclization of Heteroaromatic Compounds. Org. Lett. 2006, 8, 5661–5664. [Google Scholar] [CrossRef]
- Fujiwara, M.; Kawatsura, M.; Hayase, S.; Nanjo, M.; Itoh, T. Iron(III) Salt-Catalyzed Nazarov Cyclization/Michael Addition of Pyrrole Derivatives. Adv. Synth. Catal. 2009, 351, 123–128. [Google Scholar] [CrossRef]
- Abbiati, G.; Beccalli, E.M.; Broggini, G.; Zoni, C. Regioselectivity on the Palladium-Catalyzed Intramolecular Cyclization of Indole Derivatives. J. Org. Chem. 2003, 68, 7625–7628. [Google Scholar] [CrossRef]
- Hughes, M.J.; Thomas, E.J. Milbemycin Synthesis: Synthesis of a Macrocyclic Analogue of Non-Aromatic β-Milbemycins. J. Chem. Soc. Perkin Trans. 1 1993, 1493–1505. [Google Scholar] [CrossRef]
- Lehnert, W. Knoevenagel-Kondensationen Mit Titan-tetrachlorid/Base–II Alkyliden- und Arylidenacet- Bzw.—Nitroessigester Bei 0–22 °. Tetrahedron 1972, 28, 663–666. [Google Scholar] [CrossRef]
- Stefan, K.-P.; Schuhmann, W.; Parlar, H.; Korte, F. Synthese neuer 3-substituierter Pyrrole. Chem. Ber. 1989, 122, 169–174. [Google Scholar] [CrossRef]
- Bray, B.L.; Mathies, P.H.; Naef, R.; Solas, D.R.; Tidwell, T.T.; Artis, D.R.; Muchowski, J.M. N-(Triisopropylsilyl)pyrrole. A Progenitor “Par Excellence” of 3-Substituted Pyrroles. J. Org. Chem. 1990, 55, 6317–6328. [Google Scholar] [CrossRef]
- Alvarez, A.; Guzmán, A.; Ruiz, A.; Velarde, E. Synthesis of 3-Arylpyrroles and 3-Pyrrolylacetylenes by Palladium-Catalyzed Coupling Reactions. J. Org. Chem. 1992, 57, 1653–1656. [Google Scholar] [CrossRef]
- Liu, J.-H.; Chan, H.-W.; Xue, F.; Wang, W.-G.; Mak, T.C.W.; Wong, H.N.C. Generation and Trapping Reactions of 1-tert-Butoxycarbonyl-3,4-Didehydro-1H-Pyrrole. J. Org. Chem. 1999, 64, 1630–1634. [Google Scholar] [CrossRef] [PubMed]
- Conrad, J.C.; Kong, J.; Laforteza, B.N.; MacMillan, D.W.C. Enantioselective α-Arylation of Aldehydes via Organo-SOMO Catalysis. An Ortho-Selective Arylation Reaction Based on an Open-Shell Pathway. J. Am. Chem. Soc. 2009, 131, 11640–11641. [Google Scholar] [CrossRef] [Green Version]
- Artico, M. Nitration, Sulfonation, and Halogenation. In Pyrroles, Part One; Jones, R.A., Ed.; The Chemistry of Heterocyclic Compounds; Wiley Interscience: New York, NY, USA, 1990; Volume 48, pp. 329–395. [Google Scholar]
- Baburajan, P.; Elango, K.P. One Pot Direct Synthesis of β-Ketoesters via Carbonylation of Aryl Halides using Cobalt Carbonyl. Tetrahedron Lett. 2014, 55, 3525–3528. [Google Scholar] [CrossRef]
- Anderson, H.J.; Loader, C.E. Alkylation and Acylation. In Pyrroles, Part One; Jones, R.A., Ed.; The Chemistry of Heterocyclic Compounds; Wiley Interscience: New York, NY, USA, 1990; Volume 48, pp. 397–497. [Google Scholar]
- McNab, H.; Montgomery, J.; Parsons, S.; Tredgett, D.G. Pyrrolizine-1,3-dione. Org. Biomol. Chem. 2010, 8, 4383–4387. [Google Scholar] [CrossRef]
- Komoto, I.; Matsuo, J.-I.; Kobayashi, S. Catalytic Friedel-Crafts Acylation of Heteroaromatics. Top. Catal. 2002, 19, 43–47. [Google Scholar] [CrossRef]
- Taniguchi, M.; Ptaszek, M.; McDowell, B.E.; Boyle, P.D.; Lindsey, J.S. Sparsely Substituted Chlorins as Core Constructs in Chlorophyll Analogue Chemistry. Part 3: Spectral and Structural Properties. Tetrahedron 2007, 63, 3850–3863. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, M.; Cramer, D.L.; Bhise, A.D.; Kee, H.L.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Accessing the Near-Infrared Spectral Region with Stable, Synthetic, Wavelength-Tunable Bacteriochlorins. New J. Chem. 2008, 32, 947–958. [Google Scholar] [CrossRef]
- Meyer, M.; Scheer, H.; Breton, J. Probing Native-like Orientation of Pigments in Modified Reaction Centers from Rhodobacter sphaeroides R26 by Linear Dichroism. FEBS Lett. 1996, 393, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, M.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Beyond Green with Synthetic Chlorophylls—Connecting Structural Features with Spectral Properties. J. Photochem. Photobiol. C Photochem. Rev. 2022, 52, 100513. [Google Scholar] [CrossRef]
- Taniguchi, M.; Lindsey, J.S. Database of Absorption and Fluorescence Spectra of >300 Common Compounds for Use in PhotochemCAD. Photochem. Photobiol. 2018, 94, 290–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, M.; Lindsey, J.S. Absorption and Fluorescence Spectral Database of Chlorophylls and Analogues. Photochem. Photobiol. 2021, 97, 136–165. [Google Scholar] [CrossRef] [PubMed]
- Springer, J.W.; Faries, K.M.; Diers, J.R.; Muthiah, C.; Mass, O.; Kee, H.L.; Kirmaier, C.; Lindsey, J.S.; Bocian, D.F.; Holten, D. Effects of Substituents on Synthetic Analogs of Chlorophylls. Part 3: The Distinctive Impact of Auxochromes at the 7- versus 3-Positions. Photochem. Photobiol. 2012, 88, 651–674. [Google Scholar] [CrossRef] [PubMed]
- Tamiaki, H.; Kunieda, M. Photochemistry of Chlorophylls and Their Synthetic Analogs. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing Co., Pte., Ltd.: Singapore, 2011; Volume 11, pp. 223–290. [Google Scholar]
- Vairaprakash, P.; Yang, E.; Sahin, T.; Taniguchi, M.; Krayer, M.; Diers, J.R.; Wang, A.; Niedzwiedzki, D.M.; Kirmaier, C.; Lindsey, J.S.; et al. Extending the Short and Long Wavelength Limits of Bacteriochlorin Near-Infrared Absorption via Dioxo- and Bisimide-Functionalization. J. Phys. Chem. B 2015, 119, 4382–4395. [Google Scholar] [CrossRef]
- Sirohiwal, A.; Berraud-Pache, R.; Neese, F.; Izsák, R.; Pantazis, D.A. Accurate Computation of the Absorption Spectrum of Chlorophyll a with Pair Natural Orbital Coupled Cluster Methods. J. Phys. Chem. B 2020, 124, 8761–8771. [Google Scholar] [CrossRef]
- Silva, P.J.; Osswald-Claro, M.; Mendonça, R.C. How to Tune the Absorption Spectrum of Chlorophylls to Enable Better Use of the Available Solar Spectrum. PeerJ Phys. Chem. 2022, 4, e26. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sorimachi, Y.; Fukayama, D.; Komatsu, H.; Kanjoh, T.; Wada, K.; Kawachi, M.; Miyashita, H.; Ohnishi-Kameyama, M.; Ono, H. Physicochemical Properties of Chlorophylls and Bacteriochlorophylls. In Handbook of Photosynthesis; Pessarakli, M., Ed.; CRC Press: Boca Raton, FL, USA, 2016; pp. 95–147. [Google Scholar]
- Karg, C.A.; Taniguchi, M.; Lindsey, J.S.; Moser, S. Phyllobilins—Bioactive Natural Products Derived from Chlorophyll—Plant Origins, Structures, Absorption Spectra, and Biomedical Properties. Planta Med. 2022. [Google Scholar] [CrossRef]
- Kräutler, B.; Hörtensteiner, S. Chlorophyll Breakdown: Chemistry, Biochemistry, and Biology. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing Co., Pte., Ltd.: Singapore, 2014; Volume 28, pp. 117–185. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
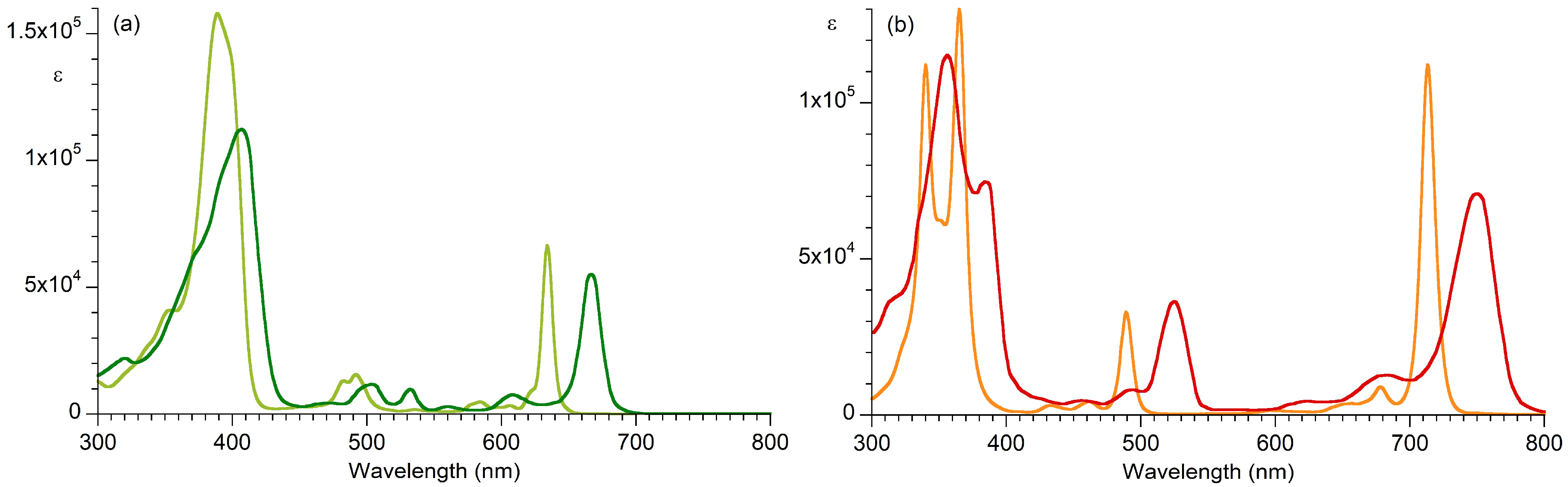
| Absorption | Bx Band | By Band | Qx Band | Qy Band | |
|---|---|---|---|---|---|
| Compound | λabs (fwhm), ε | λabs (fwhm), ε | λabs (fwhm), ε | λabs (fwhm), ε | |
| Bacteriopheophytin a | 356 (46), b,c 115,000 | 384 (23), b,c 74,700 | 525 (24), 36,400 | 750 (37), 71,000 | |
| H2BC | 340 (12), b,d 112,000 | 365 (14), d 130,000 | 489 (10), 33,000 | 713 (12), 112,000 | |
| Pheophytin a | 407 (54), e 112,000 | – f | 667 (17.5), 55,100 | ||
| H2C | 389 (33), e 158,000 | – f | 634 (9), 66,500 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chau Nguyen, K.; Nguyen Tran, A.T.; Wang, P.; Zhang, S.; Wu, Z.; Taniguchi, M.; Lindsey, J.S. Four Routes to 3-(3-Methoxy-1,3-dioxopropyl)pyrrole, a Core Motif of Rings C and E in Photosynthetic Tetrapyrroles. Molecules 2023, 28, 1323. https://doi.org/10.3390/molecules28031323
Chau Nguyen K, Nguyen Tran AT, Wang P, Zhang S, Wu Z, Taniguchi M, Lindsey JS. Four Routes to 3-(3-Methoxy-1,3-dioxopropyl)pyrrole, a Core Motif of Rings C and E in Photosynthetic Tetrapyrroles. Molecules. 2023; 28(3):1323. https://doi.org/10.3390/molecules28031323
Chicago/Turabian StyleChau Nguyen, Khiem, Anh Thu Nguyen Tran, Pengzhi Wang, Shaofei Zhang, Zhiyuan Wu, Masahiko Taniguchi, and Jonathan S. Lindsey. 2023. "Four Routes to 3-(3-Methoxy-1,3-dioxopropyl)pyrrole, a Core Motif of Rings C and E in Photosynthetic Tetrapyrroles" Molecules 28, no. 3: 1323. https://doi.org/10.3390/molecules28031323
APA StyleChau Nguyen, K., Nguyen Tran, A. T., Wang, P., Zhang, S., Wu, Z., Taniguchi, M., & Lindsey, J. S. (2023). Four Routes to 3-(3-Methoxy-1,3-dioxopropyl)pyrrole, a Core Motif of Rings C and E in Photosynthetic Tetrapyrroles. Molecules, 28(3), 1323. https://doi.org/10.3390/molecules28031323