

Hydrogenase and Nitrogenase: Key Catalysts in Biohydrogen Production



(This article belongs to the Section Green Chemistry)
Abstract
:1. Introduction
2. Biohydrogen Production (BHP) Systems
2.1. Biophotolysis
2.2. Fermentation
3. Key Enzymes Involved in Biohydrogen Production and Their Structural Characteristics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Species | Protein Components | Cofactor | Fe–S Clusters | Active Site | Reference |
|---|---|---|---|---|---|---|
| [NiFe] hydrogenase | Desulfovibrio gigas | Two subunits | [NiFe] cluster | Two [4Fe–4S] and one [3Fe–4S] | [NiFe] cluster | [14] |
| [FeFe] hydrogenase | Clostridium pasteurianum | Single subunit | H-cluster | Three [4Fe–4S] and one [2Fe–2S] | H-cluster | [36] |
| [Fe] hydrogenase | Methanothermobacter marburgenis | Homodimer | FeGP | None | Fe (II) site of FeGP | [37,38] |
| Mo-nitrogenase | Rhodopseudomonas palustris | Dimer of heterodimer | M-cluster (FeMoco) | P-cluster and one [4Fe–4S] | FeMoco | [15] |
| V-nitrogenase | Azotobacter chroococcum/Azotobacter vinelandii | Dimer of heterodimer | V-cluster (FeVco) | P-cluster and one [4Fe–4S] | FeVco | [39] |
| Fe-nitrogenase | Rhodobacter capsulatus | Dimer of heterodimer | Fe-cluster (FeFeco) | P-cluster and one [4Fe–4S] | FeFeco (proposed) | [40] |
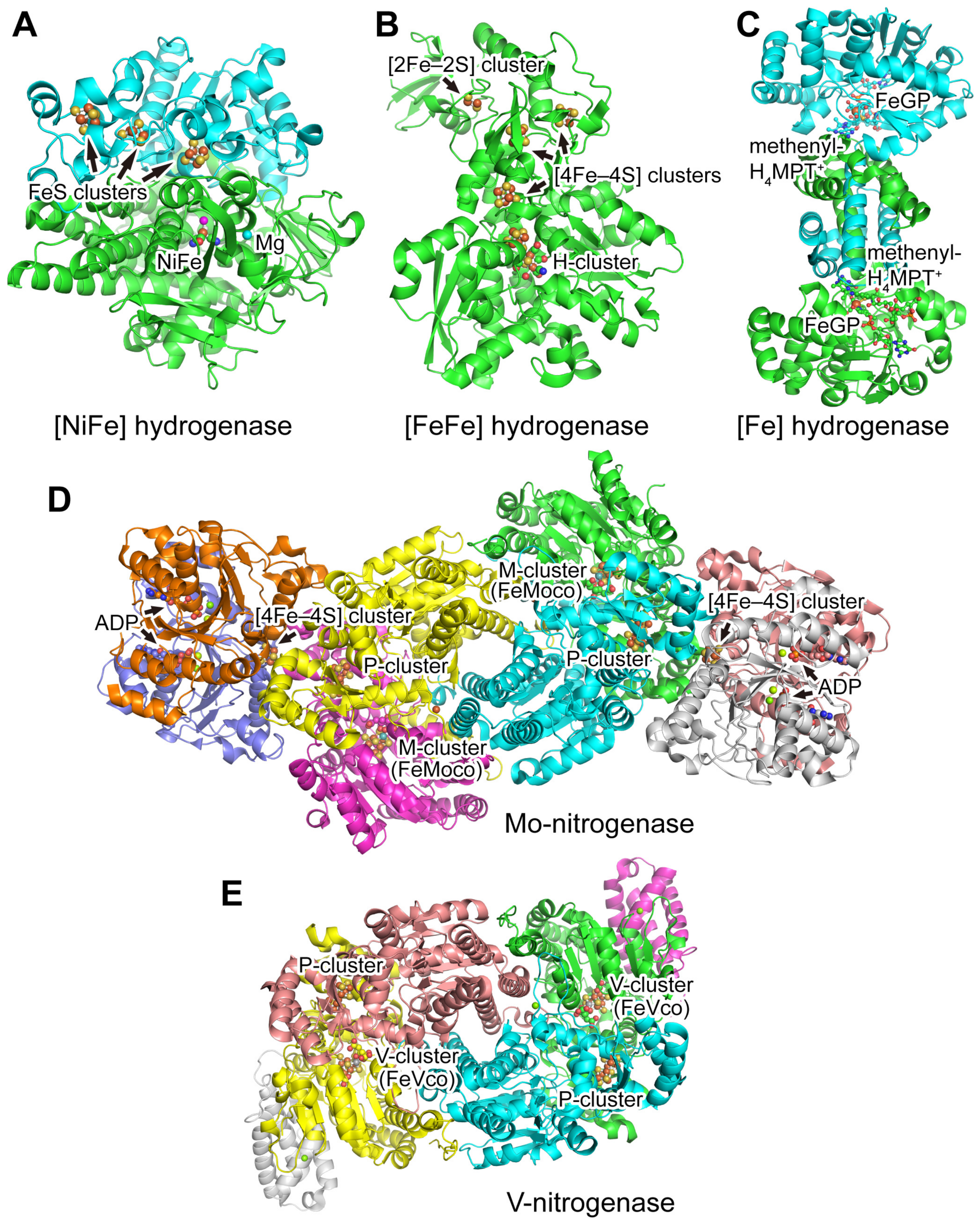
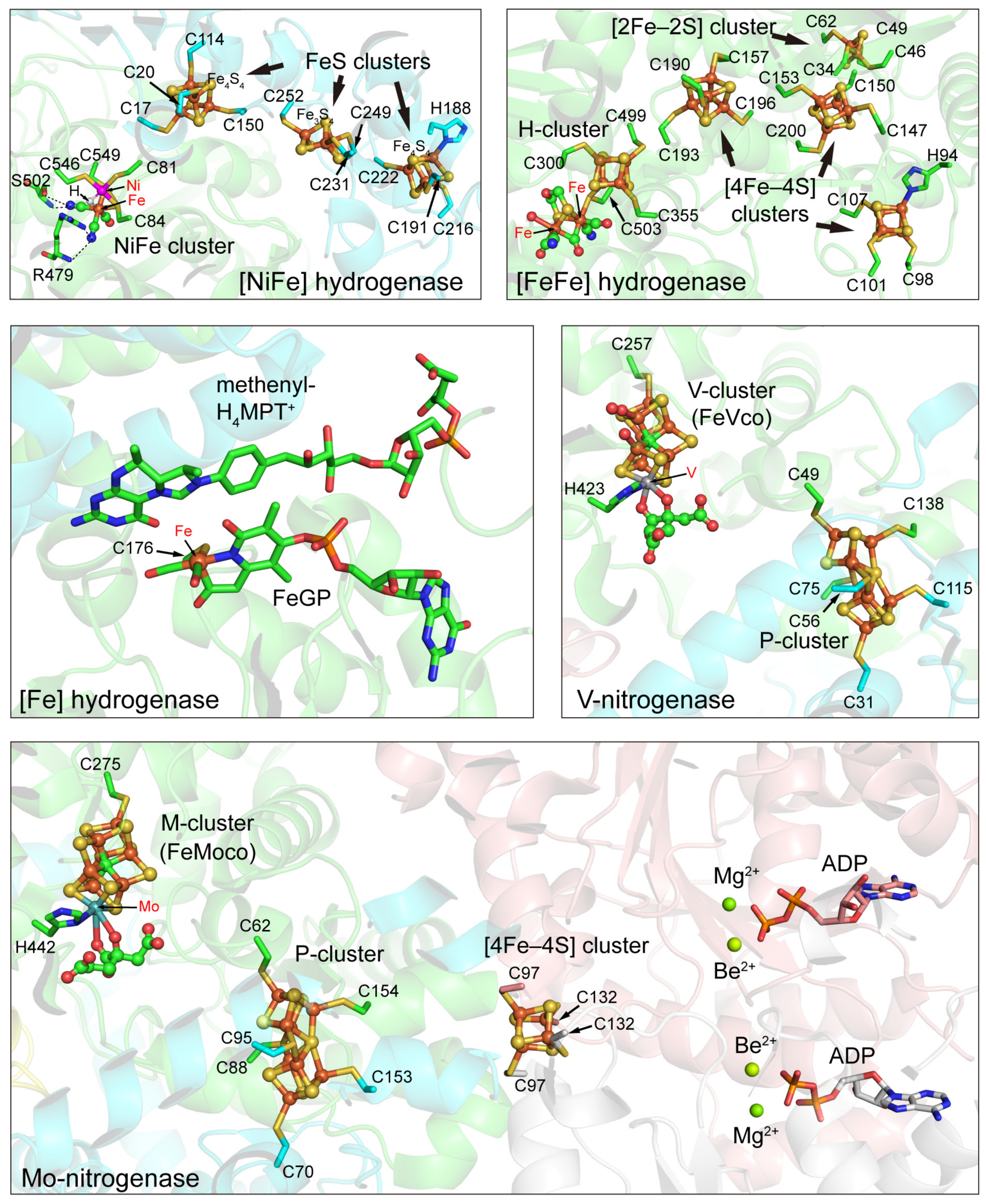
3.1. Hydrogenases
3.1.1. [NiFe] Hydrogenases
3.1.2. [FeFe] Hydrogenases
3.1.3. [Fe] Hydrogenases
3.2. Nitrogenases
4. Bioengineering Approaches for Hydrogen Production
4.1. Improvement of O2 Tolerance
4.2. Immobilization Technology
4.3. Modification of Nitrogenase Substrate Selectivity
4.4. Enzyme Compartmentalization
4.5. Metabolic Engineering
4.6. Artificial Hydrogenases
5. Summary and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wong, Y.M.; Wu, T.Y.; Juan, J.C. A review of sustainable hydrogen production using seed sludge via dark fermentation. Renew. Sustain. Energy Rev. 2014, 34, 471–482. [Google Scholar] [CrossRef]
- Hosseini, S.E.; Wahid, M.A. Hydrogen production from renewable and sustainable energy resources: Promising green energy carrier for clean development. Renew. Sustain. Energy Rev. 2016, 57, 850–866. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, M.S. Hydrogenases for biological hydrogen production. Bioresour. Technol. 2011, 102, 8423–8431. [Google Scholar] [CrossRef]
- IEA. The Future of Hydrogen; IEA: Paris, France, 2019. [Google Scholar]
- Ngoh, S.K.; Njomo, D. An overview of hydrogen gas production from solar energy. Renew. Sustain. Energy Rev. 2012, 16, 6782–6792. [Google Scholar] [CrossRef]
- Smolinka, T. Fuels—Hydrogen production | Water electrolysis. In Encyclopedia of Electrochemical Power Sources; Garche, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 394–413. [Google Scholar] [CrossRef]
- Toledo-Alarcón, J.; Capson-Tojo, G.; Marone, A.; Paillet, F.; Júnior, A.D.N.F.; Chatellard, L.; Bernet, N.; Trably, E. Basics of bio-hydrogen production by dark fermentation. In Bioreactors for Microbial Biomass and Energy Conversion; Liao, Q., Chang, J.-S., Herrmann, C., Xia, A., Eds.; Springer: Singapore, 2018; pp. 199–220. [Google Scholar] [CrossRef]
- Kothari, R.; Singh, D.P.; Tyagi, V.V.; Tyagi, S.K. Fermentative hydrogen production—An alternative clean energy source. Renew. Sustain. Energy Rev. 2012, 16, 2337–2346. [Google Scholar] [CrossRef]
- Trchounian, A. Mechanisms for hydrogen production by different bacteria during mixed-acid and photo-fermentation and perspectives of hydrogen production biotechnology. Crit. Rev. Biotechnol. 2015, 35, 103–113. [Google Scholar] [CrossRef]
- Khetkorn, W.; Rastogi, R.P.; Incharoensakdi, A.; Lindblad, P.; Madamwar, D.; Pandey, A.; Larroche, C. Microalgal hydrogen production—A review. Bioresour. Technol. 2017, 243, 1194–1206. [Google Scholar] [CrossRef]
- Santos-Merino, M.; Singh, A.K.; Ducat, D.C. New applications of synthetic biology tools for cyanobacterial metabolic engineering. Front. Bioeng. Biotechnol. 2019, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Goyal, Y.; Kumar, M.; Gayen, K. Metabolic engineering for enhanced hydrogen production: A review. Can. J. Microbiol. 2013, 59, 59–78. [Google Scholar] [CrossRef]
- Yang, D.W.; Syn, J.W.; Hsieh, C.H.; Huang, C.C.; Chien, L.F. Genetically engineered hydrogenases promote biophotocatalysis-mediated H2 production in the green alga Chlorella sp. DT. Int. J. Hydrog. Energy 2019, 44, 2533–2545. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Rudiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef]
- Einsle, O.; Rees, D.C. Structural enzymology of nitrogenase enzymes. Chem. Rev. 2020, 120, 4969–5004. [Google Scholar] [CrossRef]
- Dubini, A.; Ghirardi, M.L. Engineering photosynthetic organisms for the production of biohydrogen. Photosynth. Res. 2015, 123, 241–253. [Google Scholar] [CrossRef]
- Ramprakash, B.; Lindblad, P.; Eaton-Rye, J.J.; Incharoensakdi, A. Current strategies and future perspectives in biological hydrogen production: A review. Renew. Sustain. Energy Rev. 2022, 168, 112773. [Google Scholar] [CrossRef]
- McKinlay, J.B.; Harwood, C.S. Photobiological production of hydrogen gas as a biofuel. Curr. Opin. Biotechnol. 2010, 21, 244–251. [Google Scholar] [CrossRef]
- Turner, J.; Sverdrup, G.; Mann, M.K.; Maness, P.C.; Kroposki, B.; Ghirardi, M.; Evans, R.J.; Blake, D. Renewable hydrogen production. Int. J. Energy Res. 2008, 32, 379–407. [Google Scholar] [CrossRef]
- Sakurai, H.; Masukawa, H. Promoting R & D in photobiological hydrogen production utilizing mariculture-raised cyanobacteria. Mar. Biotechnol. 2007, 9, 128–145. [Google Scholar] [CrossRef]
- Melis, A. Photosynthetic H2 metabolism in Chlamydomonas reinhardtii (unicellular green algae). Planta 2007, 226, 1075–1086. [Google Scholar] [CrossRef]
- Oncel, S.; Vardar-Sukan, F. Photo-bioproduction of hydrogen by Chlamydomonas reinhardtii using a semi-continuous process regime. Int. J. Hydrog. Energy 2009, 34, 7592–7602. [Google Scholar] [CrossRef]
- Koku, H.; Eroglu, I.; Gunduz, U.; Yucel, M.; Turker, L. Aspects of the metabolism of hydrogen production by Rhodobacter sphaeroides. Int. J. Hydrog. Energy 2002, 27, 1315–1329. [Google Scholar] [CrossRef]
- Honda, R.; Fukushi, K.; Yamamoto, K. Optimization of wastewater feeding for single-cell protein production in an anaerobic wastewater treatment process utilizing purple non-sulfur bacteria in mixed culture condition. J. Biotechnol. 2006, 125, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Ren, N.Q.; Guo, W.Q.; Liu, B.F.; Cao, G.L.; Ding, J. Biological hydrogen production by dark fermentation: Challenges and prospects towards scaled-up production. Curr. Opin. Biotechnol. 2011, 22, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Hung, C.H.; Lin, C.N.; Chen, H.W.; Lee, A.S.; Chang, J.S. Fermentative hydrogen production and bacterial community structure in high-rate anaerobic bioreactors containing silicone-immobilized and self-flocculated sludge. Biotechnol. Bioeng. 2006, 93, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Kapdan, I.K.; Kargi, F. Bio-hydrogen production from waste materials. Enzyme Microb. Technol. 2006, 38, 569–582. [Google Scholar] [CrossRef]
- Ren, N.Q.; Wang, A.J.; Cao, G.L.; Xu, J.F.; Gao, L.F. Bioconversion of lignocellulosic biomass to hydrogen: Potential and challenges. Biotechnol. Adv. 2009, 27, 1051–1060. [Google Scholar] [CrossRef]
- Thauer, R.K.; Jungermann, K.; Decker, K. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 1977, 41, 100–180. [Google Scholar] [CrossRef]
- Jayachandran, V.; Basak, N.; De Philippis, R.; Adessi, A. Novel strategies towards efficient molecular biohydrogen production by dark fermentative mechanism: Present progress and future perspective. Bioprocess Biosyst. Eng. 2022, 45, 1595–1624. [Google Scholar] [CrossRef]
- Oh, Y.K.; Raj, S.M.; Jung, G.Y.; Park, S. Current status of the metabolic engineering of microorganisms for biohydrogen production. Bioresour. Technol. 2011, 102, 8357–8367. [Google Scholar] [CrossRef]
- Kumar, G.; Mudhoo, A.; Sivagurunathan, P.; Nagarajan, D.; Ghimire, A.; Lay, C.H.; Lin, C.Y.; Lee, D.J.; Chang, J.S. Recent insights into the cell immobilization technology applied for dark fermentative hydrogen production. Bioresour. Technol. 2016, 219, 725–737. [Google Scholar] [CrossRef]
- Singh, R.; White, D.; Demirel, Y.; Kelly, R.; Noll, K.; Blum, P. Uncoupling fermentative synthesis of molecular hydrogen from biomass formation in Thermotoga maritima. Appl. Environ. Microbiol. 2018, 84, e00998-18. [Google Scholar] [CrossRef]
- Ergal, İ.; Gräf, O.; Hasibar, B.; Steiner, M.; Vukotić, S.; Bochmann, G.; Fuchs, W.; Rittmann, S.K.M.R. Biohydrogen production beyond the Thauer limit by precision design of artificial microbial consortia. Commun. Biol. 2020, 3, 443. [Google Scholar] [CrossRef]
- Jasniewski, A.J.; Lee, C.C.; Ribbe, M.W.; Hu, Y. Reactivity, mechanism, and assembly of the alternative nitrogenases. Chem. Rev. 2020, 120, 5107–5157. [Google Scholar] [CrossRef]
- Kleinhaus, J.T.; Wittkamp, F.; Yadav, S.; Siegmund, D.; Apfel, U.P. [FeFe]-Hydrogenases: Maturation and reactivity of enzymatic systems and overview of biomimetic models. Chem. Soc. Rev. 2021, 50, 1668–1784. [Google Scholar] [CrossRef]
- Hu, B.W.; Chen, D.F.; Hu, X.L. Reversible dimerization of mononuclear models of [Fe]-hydrogenase. Chem. Eur. J. 2013, 19, 6221–6224. [Google Scholar] [CrossRef]
- Shima, S.; Pilak, O.; Vogt, S.; Schick, M.; Stagni, M.S.; Meyer-Klaucke, W.; Warkentin, E.; Thauer, R.K.; Ermler, U. The crystal structure of [Fe]-hydrogenase reveals the geometry of the active site. Science 2008, 321, 572–575. [Google Scholar] [CrossRef]
- Sippel, D.; Einsle, O. The structure of vanadium nitrogenase reveals an unusual bridging ligand. Nat. Chem. Biol. 2017, 13, 956–960. [Google Scholar] [CrossRef]
- Harris, D.F.; Lukoyanov, D.A.; Shaw, S.; Compton, P.; Tokmina-Lukaszewska, M.; Bothner, B.; Kelleher, N.; Dean, D.R.; Hoffman, B.M.; Seefeldt, L.C. Mechanism of N2 reduction catalyzed by Fe-nitrogenase involves reductive elimination of H2. Biochemistry 2018, 57, 701–710. [Google Scholar] [CrossRef]
- Ogata, H.; Nishikawa, K.; Lubitz, W. Hydrogens detected by subatomic resolution protein crystallography in a [NiFe] hydrogenase. Nature 2015, 520, 571–574. [Google Scholar] [CrossRef]
- Pandey, A.S.; Harris, T.V.; Giles, L.J.; Peters, J.W.; Szilagyi, R.K. Dithiomethylether as a ligand in the hydrogenase H-cluster. J. Am. Chem. Soc. 2008, 130, 4533–4540. [Google Scholar] [CrossRef]
- Schindelin, H.; Kisker, C.; Schlessman, J.L.; Howard, J.B.; Rees, D.C. Structure of ADP·AIF4−-stabilized nitrogenase complex and its implications for signal transduction. Nature 1997, 387, 370–376. [Google Scholar] [CrossRef]
- Rutledge, H.L.; Cook, B.D.; Nguyen, H.P.M.; Herzik, M.A.; Tezcan, F.A. Structures of the nitrogenase complex prepared under catalytic turnover conditions. Science 2022, 377, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, M.; Stickland, L.H. Hydrogenase: A bacterial enzyme activating molecular hydrogen: The properties of the enzyme. Biochem. J. 1931, 25, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Vignais, P.M.; Billoud, B. Occurrence, classification, and biological function of hydrogenases: An overview. Chem. Rev. 2007, 107, 4206–4272. [Google Scholar] [CrossRef] [PubMed]
- Esmieu, C.; Raleiras, P.; Berggren, G. From protein engineering to artificial enzymes—Biological and biomimetic approaches towards sustainable hydrogen production. Sustain. Energy Fuels 2018, 2, 724–750. [Google Scholar] [CrossRef]
- Schafer, C.; Friedrich, B.; Lenz, O. Novel, oxygen-insensitive group 5 [NiFe]-hydrogenase in Ralstonia eutropha. Appl. Environ. Microbiol. 2013, 79, 5137–5145. [Google Scholar] [CrossRef]
- Silva, P.J.; van den Ban, E.C.D.; Wassink, H.; Haaker, H.; de Castro, B.; Robb, F.T.; Hagen, W.R. Enzymes of hydrogen metabolism in Pyrococcus furiosus. Eur. J. Biochem. 2000, 267, 6541–6551. [Google Scholar] [CrossRef]
- Schut, G.J.; Nixon, W.J.; Lipscomb, G.L.; Scott, R.A.; Adams, M.W. Mutational analyses of the enzymes involved in the metabolism of hydrogen by the hyperthermophilic archaeon Pyrococcus furiosus. Front. Microbiol. 2012, 3, 163. [Google Scholar] [CrossRef]
- Straub, C.T.; Counts, J.A.; Nguyen, D.M.N.; Wu, C.-H.; Zeldes, B.M.; Crosby, J.R.; Conway, J.M.; Otten, J.K.; Lipscomb, G.L.; Schut, G.J.; et al. Biotechnology of extremely thermophilic archaea. FEMS Microbiol. Rev. 2018, 42, 543–578. [Google Scholar] [CrossRef] [Green Version]
- Volbeda, A.; Charon, M.H.; Piras, C.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373, 580–587. [Google Scholar] [CrossRef]
- Lubitz, W.; Reijerse, E.; van Gastel, M. [NiFe] and [FeFe] hydrogenases studied by advanced magnetic resonance techniques. Chem. Rev. 2007, 107, 4331–4365. [Google Scholar] [CrossRef]
- Ogata, H.; Lubitz, W.; Higuchi, Y. Structure and function of [NiFe] hydrogenases. J. Biochem. 2016, 160, 251–258. [Google Scholar] [CrossRef]
- Higuchi, Y.; Ogata, H.; Miki, K.; Yasuoka, N.; Yagi, T. Removal of the bridging ligand atom at the Ni-Fe active site of [NiFe] hydrogenase upon reduction with H2, as revealed by X-ray structure analysis at 1.4 Å resolution. Structure 1999, 7, 549–556. [Google Scholar] [CrossRef]
- Sommer, C.; Adamska-Venkatesh, A.; Pawlak, K.; Birrell, J.A.; Rüdiger, O.; Reijerse, E.J.; Lubitz, W. Proton coupled electronic rearrangement within the H-cluster as an essential step in the catalytic cycle of [FeFe] hydrogenases. J. Am. Chem. Soc. 2017, 139, 1440–1443. [Google Scholar] [CrossRef]
- Huang, G.; Wagner, T.; Wodrich, M.D.; Ataka, K.; Bill, E.; Ermler, U.; Hu, X.; Shima, S. The atomic-resolution crystal structure of activated [Fe]-hydrogenase. Nat. Catal. 2019, 2, 537–543. [Google Scholar] [CrossRef]
- Dementin, S.; Burlat, B.; Fourmond, V.; Leroux, F.; Liebgott, P.P.; Abou Hamdan, A.; Leger, C.; Rousset, M.; Guigliarelli, B.; Bertrand, P. Rates of intra- and intermolecular electron transfers in hydrogenase deduced from steady-state activity measurements. J. Am. Chem. Soc. 2011, 133, 10211–10221. [Google Scholar] [CrossRef]
- De Lacey, A.L.; Gutiérrez-Sánchez, C.; Fernández, V.M.; Pacheco, I.; Pereira, I.A.C. FTIR spectroelectrochemical characterization of the Ni–Fe–Se hydrogenase from Desulfovibrio vulgaris Hildenborough. J. Biol. Inorg. Chem. 2008, 13, 1315–1320. [Google Scholar] [CrossRef]
- Vignais, P.M.; Billoud, B.; Meyer, J. Classification and phylogeny of hydrogenases. FEMS Microbiol. Rev. 2001, 25, 455–501. [Google Scholar] [CrossRef]
- Schut, G.J.; Adams, M.W.W. The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: A new perspective on anaerobic hydrogen production. J. Bacteriol. 2009, 191, 4451–4457. [Google Scholar] [CrossRef] [Green Version]
- Wittkamp, F.; Senger, M.; Stripp, S.T.; Apfel, U.P. [FeFe]-Hydrogenases: Recent developments and future perspectives. Chem. Commun. 2018, 54, 5934–5942. [Google Scholar] [CrossRef]
- Zirngibl, C.; Hedderich, R.; Thauer, R.K. N5,N10-Methylenetetrahydromethanopterin dehydrogenase from Methanobacterium thermoautotrophicum has hydrogenase activity. FEBS Lett. 1990, 261, 112–116. [Google Scholar] [CrossRef]
- Zirngibl, C.; Van Dongen, W.; Schworer, B.; Von Bunau, R.; Richter, M.; Klein, A.; Thauer, R.K. H2-forming methylenetetrahydromethanopterin dehydrogenase, a novel type of hydrogenase without iron-sulfur clusters in methanogenic archaea. Eur. J. Biochem. 1992, 208, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Shima, S.; Chen, D.F.; Xu, T.; Wodrich, M.D.; Fujishiro, T.; Schultz, K.M.; Kahnt, J.; Ataka, K.; Hu, X.L. Reconstitution of [Fe]-hydrogenase using model complexes. Nat. Chem. 2015, 7, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Pilak, O.; Mamat, B.; Vogt, S.; Hagemeier, C.H.; Thauer, R.K.; Shima, S.; Vonrhein, C.; Warkentin, E.; Ermler, U. The crystal structure of the apoenzyme of the iron-sulphur cluster-free hydrogenase. J. Mol. Biol. 2006, 358, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Eady, R.R. Structure-function relationships of alternative nitrogenases. Chem. Rev. 1996, 96, 3013–3030. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Rees, D.C. Crystallographic structure and functional implications of the nitrogenase molybdenum-iron protein from Azotobacter vinelandii. Nature 1992, 360, 553–560. [Google Scholar]
- Kim, J.; Rees, D.C. Structural models for the metal centers in the nitrogenase molybdenum-iron protein. Science 1992, 257, 1677–1682. [Google Scholar] [CrossRef]
- Weare, W.W.; Dai, X.; Byrnes, M.J.; Chin, J.M.; Schrock, R.R.; Müller, P. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Proc. Natl. Acad. Sci. USA 2006, 103, 17099–17106. [Google Scholar] [CrossRef]
- Jimenez-Vicente, E.; Yang, Z.Y.; Ray, W.K.; Echavarri-Erasun, C.; Cash, V.L.; Rubio, L.M.; Seefeldt, L.C.; Dean, D.R. Sequential and differential interaction of assembly factors during nitrogenase MoFe protein maturation. J. Biol. Chem. 2018, 293, 9812–9823. [Google Scholar] [CrossRef]
- Cao, L.L.; Borner, M.C.; Bergmann, J.; Caldararu, O.; Ryde, U. Geometry and electronic structure of the P-cluster in nitrogenase studied by combined quantum mechanical and molecular mechanical calculations and quantum refinement. Inorg. Chem. 2019, 58, 9672–9690. [Google Scholar] [CrossRef] [Green Version]
- Leipe, D.D.; Wolf, Y.I.; Koonin, E.V.; Aravind, L. Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 2002, 317, 41–72. [Google Scholar] [CrossRef]
- Georgiadis, M.M.; Komiya, H.; Chakrabarti, P.; Woo, D.; Kornuc, J.J.; Rees, D.C. Crystallographic structure of the nitrogenase iron protein from Azotobacter vinelandii. Science 1992, 257, 1653–1659. [Google Scholar] [CrossRef]
- Sickerman, N.S.; Hu, Y.; Ribbe, M.W. Nitrogenases. In Metalloproteins: Methods and Protocols; Hu, Y., Ed.; Springer: New York, NY, USA, 2019; pp. 3–24. [Google Scholar] [CrossRef]
- Golden, J.W.; Yoon, H.S. Heterocyst development in Anabaena. Curr. Opin. Microbiol. 2003, 6, 557–563. [Google Scholar] [CrossRef]
- Morra, S.; Arizzi, M.; Valetti, F.; Gilardi, G. Oxygen stability in the new [FeFe]-hydrogenase from Clostridium beijerinckii SM10 (CbA5H). Biochemistry 2016, 55, 5897–5900. [Google Scholar] [CrossRef]
- Burgdorf, T.; Lenz, O.; Buhrke, T.; van der Linden, E.; Jones, A.K.; Albracht, S.P.J.; Friedrich, B. [NiFe]-hydrogenases of Ralstonia eutropha H16: Modular enzymes for oxygen-tolerant biological hydrogen oxidation. J. Mol. Microbiol. Biotechnol. 2005, 10, 181–196. [Google Scholar] [CrossRef]
- Lukey, M.J.; Parkin, A.; Roessler, M.M.; Murphy, B.J.; Harmer, J.; Palmer, T.; Sargent, F.; Armstrong, F.A. How Escherichia coli is equipped to oxidize hydrogen under different redox conditions. J. Biol. Chem. 2010, 285, 3928–3938. [Google Scholar] [CrossRef]
- Topin, J.; Diharce, J.; Fiorucci, S.; Antonczak, S.; Golebiowski, J. O2 migration rates in [NiFe] hydrogenases. A joint approach combining free-energy calculations and kinetic modeling. J. Phys. Chem. B 2014, 118, 676–681. [Google Scholar] [CrossRef]
- Wu, X.; Liang, Y.; Li, Q.; Zhou, J.; Long, M. Characterization and cloning of oxygen-tolerant hydrogenase from Klebsiella oxytoca HP1. Res. Microbiol. 2011, 162, 330–336. [Google Scholar] [CrossRef]
- Liebgott, P.P.; Dementin, S.; Leger, C.; Rousset, M. Towards engineering O2-tolerance in [Ni-Fe] hydrogenases. Energy Environ. Sci. 2011, 4, 33–41. [Google Scholar] [CrossRef]
- Wulff, P.; Thomas, C.; Sargent, F.; Armstrong, F.A. How the oxygen tolerance of a [NiFe]-hydrogenase depends on quaternary structure. J. Biol. Inorg. Chem. 2016, 21, 121–134. [Google Scholar] [CrossRef]
- Volbeda, A.; Montet, Y.; Vernede, X.; Hatchikian, E.C.; Fontecilla-Camps, J.C. High-resolution crystallographic analysis of Desulfovibrio fructiosovorans [NiFe] hydrogenase. Int. J. Hydrog. Energy 2002, 27, 1449–1461. [Google Scholar] [CrossRef]
- Bingham, A.S.; Smith, P.R.; Swartz, J.R. Evolution of an [FeFe] hydrogenase with decreased oxygen sensitivity. Int. J. Hydrog. Energy 2012, 37, 2965–2976. [Google Scholar] [CrossRef]
- Koo, J.; Swartz, J.R. System analysis and improved [FeFe] hydrogenase O2 tolerance suggest feasibility for photosynthetic H2 production. Metab. Eng. 2018, 49, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Elman, T.; Schweitzer, S.; Shahar, N.; Swartz, J.; Yacoby, I. Engineered clostridial [FeFe]-hydrogenase shows improved O2 tolerance in Chlamydomonas reinhardtii. Int. J. Hydrog. Energy 2020, 45, 30201–30210. [Google Scholar] [CrossRef]
- Rudiger, O.; Gutierrez-Sanchez, C.; Olea, D.; Pereira, I.A.C.; Velez, M.; Fernandez, V.M.; De Lacey, A.L. Enzymatic anodes for hydrogen fuel cells based on covalent attachment of Ni-Fe hydrogenases and direct electron transfer to SAM-modified gold electrodes. Electroanalysis 2010, 22, 776–783. [Google Scholar] [CrossRef]
- Liu, J.; Wu, W.J.; Fang, F.; Zorin, N.A.; Chen, M.; Qian, D.J. Immobilization of hydrogenase on carbon nanotube polyelectrolytes as heterogeneous catalysts for electrocatalytic interconversion of protons and hydrogen. J. Nanopart. Res. 2016, 18, 220. [Google Scholar] [CrossRef]
- Reddy, K.R.; Hassan, M.; Gomes, V.G. Hybrid nanostructures based on titanium dioxide for enhanced photocatalysis. Appl. Catal. A 2015, 489, 1–16. [Google Scholar] [CrossRef]
- Liu, X.; Risbakk, S.; Carvalho, P.A.; Yang, M.Y.; Backe, P.H.; Bjoras, M.; Norby, T.; Chatzitakis, A. Immobilization of FeFe-hydrogenase on black TiO2 nanotubes as biocathodes for the hydrogen evolution reaction. Electrochem. Commun. 2022, 135, 107221. [Google Scholar] [CrossRef]
- Wang, Y.M.; Song, Y.H.; Ma, C.L.; Xia, H.Q.; Wu, R.R.; Zhu, Z.G. Electrochemical characterization of a truncated hydrogenase from Pyrococcus furiosus. Electrochim. Acta 2021, 387, 138502. [Google Scholar] [CrossRef]
- Hageman, R.V.; Burris, R.H. Kinetic studies on electron transfer and interaction between nitrogenase components from Azotobacter vinelandii. Biochemistry 1978, 17, 4117–4124. [Google Scholar] [CrossRef]
- Benemann, J.R.; Weare, N.M. Hydrogen evolution by nitrogen-fixing Anabaena cylindrica cultures. Science 1974, 184, 174–175. [Google Scholar] [CrossRef]
- Stripp, S.T.; Duffus, B.R.; Fourmond, V.; Leger, C.; Leimkushler, S.; Hirota, S.; Hu, Y.L.; Jasniewski, A.; Ogata, H.; Ribbe, M.W. Second and outer coordination sphere effects in nitrogenase, hydrogenase, formate dehydrogenase, and CO dehydrogenase. Chem. Rev. 2022, 122, 11900–11973. [Google Scholar] [CrossRef]
- Mayer, S.M.; Niehaus, W.G.; Dean, D.R. Reduction of short chain alkynes by a nitrogenase α-70Ala-substituted MoFe protein. J. Chem. Soc. Dalton Trans. 2002, 4, 802–807. [Google Scholar] [CrossRef]
- Barney, B.M.; Igarashi, R.Y.; Dos Santos, P.C.; Dean, D.R.; Seefeldt, L.C. Substrate interaction at an iron-sulfur face of the FeMo-cofactor during nitrogenase catalysis. J. Biol. Chem. 2004, 279, 53621–53624. [Google Scholar] [CrossRef]
- Igarashi, R.Y.; Seefeldt, L.C. Nitrogen fixation: The mechanism of the Mo-dependent nitrogenase. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 351–384. [Google Scholar] [CrossRef]
- Weyman, P.D.; Pratte, B.; Thiel, T. Hydrogen production in nitrogenase mutants in Anabaena variabilis. FEMS Microbiol. Lett. 2010, 304, 55–61. [Google Scholar] [CrossRef]
- Kerfeld, C.A.; Aussignargues, C.; Zarzycki, J.; Cai, F.; Sutter, M. Bacterial microcompartments. Nat. Rev. Microbiol. 2018, 16, 277–290. [Google Scholar] [CrossRef]
- Frank, S.; Lawrence, A.D.; Prentice, M.B.; Warren, M.J. Bacterial microcompartments moving into a synthetic biological world. J. Biotechnol. 2013, 163, 273–279. [Google Scholar] [CrossRef]
- Tanaka, S.; Kerfeld, C.A.; Sawaya, M.R.; Cai, F.; Heinhorst, S.; Cannon, G.C.; Yeates, T.O. Atomic-level models of the bacterial carboxysome shell. Science 2008, 319, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- Li, T.P.; Jiang, Q.Y.; Huang, J.F.; Aitchison, C.M.; Huang, F.; Yang, M.R.; Dykes, G.F.; He, H.L.; Wang, Q.; Sprick, R.S.; et al. Reprogramming bacterial protein organelles as a nanoreactor for hydrogen production. Nat. Commun. 2020, 11, 5448. [Google Scholar] [CrossRef]
- Khetkorn, W.; Baebprasert, W.; Lindblad, P.; Incharoensakdi, A. Redirecting the electron flow towards the nitrogenase and bidirectional Hox-hydrogenase by using specific inhibitors results in enhanced H2 production in the cyanobacterium Anabaena siamensis TISTR 8012. Bioresour. Technol. 2012, 118, 265–271. [Google Scholar] [CrossRef]
- Nyberg, M.; Heidorn, T.; Lindblad, P. Hydrogen production by the engineered cyanobacterial strain Nostoc PCC 7120 ΔhupW examined in a flat panel photobioreactor system. J. Biotechnol. 2015, 215, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Seol, E.; Oh, Y.K.; Wang, G.Y.; Park, S. Hydrogen production and metabolic flux analysis of metabolically engineered Escherichia coli strains. Int. J. Hydrog. Energy 2009, 34, 7417–7427. [Google Scholar] [CrossRef]
- Maeda, T.; Sanchez-Torres, V.; Wood, T.K. Metabolic engineering to enhance bacterial hydrogen production. Microb. Biotechnol. 2008, 1, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Kanai, T.; Simons, J.R.; Tsukamoto, R.; Nakajima, A.; Omori, Y.; Matsuoka, R.; Beppu, H.; Imanaka, T.; Atomi, H. Overproduction of the membrane-bound [NiFe]-hydrogenase in Thermococcus kodakarensis and its effect on hydrogen production. Front. Microbiol. 2015, 6, 847. [Google Scholar] [CrossRef]
- Maeda, T.; Vardar, G.; Self, W.T.; Wood, T.K. Inhibition of hydrogen uptake in Escherichia coli by expressing the hydrogenase from the cyanobacterium Synechocystis sp. PCC 6803. BMC Biotechnol. 2007, 7, 25. [Google Scholar] [CrossRef]
- Raleiras, P.; Khanna, N.; Miranda, H.; Meszaros, L.S.; Krassen, H.; Ho, F.; Battchikova, N.; Aro, E.M.; Magnuson, A.; Lindblad, P.; et al. Turning around the electron flow in an uptake hydrogenase. EPR spectroscopy and in vivo activity of a designed mutant in HupSL from Nostoc punctiforme. Energy Environ. Sci. 2016, 9, 581–594. [Google Scholar] [CrossRef]
- Kars, G.; Gündüz, U.; Rakhely, G.; Yücel, M.; Eroğlu, İ.; Kovacs, K.L. Improved hydrogen production by uptake hydrogenase deficient mutant strain of Rhodobacter sphaeroides O.U.001. Int. J. Hydrog. Energy 2008, 33, 3056–3060. [Google Scholar] [CrossRef]
- Klein, M.; Ansorge-Schumacher, M.B.; Fritsch, M.; Hartmeier, W. Influence of hydrogenase overexpression on hydrogen production of Clostridium acetobutylicum DSM 792. Enzyme Microb. Technol. 2010, 46, 384–390. [Google Scholar] [CrossRef]
- Morimoto, K.; Kimura, T.; Sakka, K.; Ohmiya, K. Overexpression of a hydrogenase gene in Clostridium paraputrificum to enhance hydrogen gas production. FEMS Microbiol. Lett. 2005, 246, 229–234. [Google Scholar] [CrossRef]
- Sybirna, K.; Ezanno, P.; Baffert, C.; Leger, C.; Bottin, H. Arginine171 of Chlamydomonas reinhardtii [Fe-Fe] hydrogenase HydA1 plays a crucial role in electron transfer to its catalytic center. Int. J. Hydrog. Energy 2013, 38, 2998–3002. [Google Scholar] [CrossRef]
- Ryu, M.-H.; Hull, N.C.; Gomelsky, M. Metabolic engineering of Rhodobacter sphaeroides for improved hydrogen production. Int. J. Hydrog. Energy 2014, 39, 6384–6390. [Google Scholar] [CrossRef]
- Ma, H.; Yang, H.; Zheng, X.; Lie, T.; Yan, W. Promoting photo-fermentative hydrogen production performance by substituting the rnf promoter in Rhodobacter capsulatus. Int. J. Hydrog. Energy 2021, 46, 3742–3752. [Google Scholar] [CrossRef]
- Ma, H.; Zheng, X.; Yang, H. Enhancement on hydrogen production performance of Rhodobacter sphaeroides HY01 by overexpressing fdxN. Int. J. Hydrog. Energy 2018, 43, 17082–17090. [Google Scholar] [CrossRef]
- Wu, X.-M.; Zhu, L.-Y.; Zhu, L.-Y.; Wu, L. Improved ammonium tolerance and hydrogen production in nifA mutant strains of Rhodopseudomonas palustris. Int. J. Hydrog. Energy 2016, 41, 22824–22830. [Google Scholar] [CrossRef]
- Wang, X.; Wu, X.; Hu, J.; Zhang, A.; Chen, D.; Yang, H.; Ma, X.; Guo, L. Isolation of a Rhodobacter sphaeroides mutant with enhanced hydrogen production capacity from transposon mutagenesis by NH4+ nitrogen resource. Int. J. Hydrog. Energy 2018, 43, 13821–13828. [Google Scholar] [CrossRef]
- Wang, X.; Yang, H.; Zhang, Y.; Guo, L. Remarkable enhancement on hydrogen production performance of Rhodobacter sphaeroides by disrupting spbA and hupSL genes. Int. J. Hydrog. Energy 2014, 39, 14633–14641. [Google Scholar] [CrossRef]
- Kim, E.-J.; Kim, M.-S.; Lee, J.K. Hydrogen evolution under photoheterotrophic and dark fermentative conditions by recombinant Rhodobacter sphaeroides containing the genes for fermentative pyruvate metabolism of Rhodospirillum rubrum. Int. J. Hydrog. Energy 2008, 33, 5131–5136. [Google Scholar] [CrossRef]
- Akhtar, M.K.; Jones, P.R. Construction of a synthetic YdbK-dependent pyruvate:H2 pathway in Escherichia coli BL21(DE3). Metab. Eng. 2009, 11, 139–147. [Google Scholar] [CrossRef]
- Öztürk, Y.; Gökçe, A.; Peksel, B.; Gürgan, M.; Özgür, E.; Gündüz, U.; Eroğlu, İ.; Yücel, M. Hydrogen production properties of Rhodobacter capsulatus with genetically modified redox balancing pathways. Int. J. Hydrog. Energy 2012, 37, 2014–2020. [Google Scholar] [CrossRef]
- Kim, J.Y.H.; Jo, B.H.; Cha, H.J. Production of biohydrogen by heterologous expression of oxygen-tolerant Hydrogenovibrio marinus [NiFe]-hydrogenase in Escherichia coli. J. Biotechnol. 2011, 155, 312–319. [Google Scholar] [CrossRef]
- Siebel, J.F.; Adamska-Venkatesh, A.; Weber, K.; Rumpel, S.; Reijerse, E.; Lubitz, W. Hybrid [FeFe]-hydrogenases with modified active sites show remarkable residual enzymatic activity. Biochemistry 2015, 54, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.; Richers, C.P.; Lubitz, W.; Rauchfuss, T.B.; Reijerse, E.J. A [RuRu] analogue of an [FeFe]-hydrogenase traps the key hydride intermediate of the catalytic cycle. Angew. Chem. Int. Ed. 2018, 57, 5429–5432. [Google Scholar] [CrossRef] [PubMed]
- Kertess, L.; Wittkamp, F.; Sommer, C.; Esselborn, J.; Rüdiger, O.; Reijerse, E.J.; Hofmann, E.; Lubitz, W.; Winkler, M.; Happe, T.; et al. Chalcogenide substitution in the [2Fe] cluster of [FeFe]-hydrogenases conserves high enzymatic activity. Dalton Trans. 2017, 46, 16947–16958. [Google Scholar] [CrossRef] [PubMed]
- Wittkamp, F.; Boydas, E.B.; Roemelt, M.; Apfel, U.P. New phosphorous-based [FeFe]-hydrogenase models. Catalysts 2020, 10, 522. [Google Scholar] [CrossRef]
- Song, L.-C.; Zhao, P.-H.; Du, Z.-Q.; Tang, M.-Y.; Hu, Q.-M. Unexpected synthesis of tetrahedral Fe/S clusters via highly reactive butterfly intermediates (μ-HS)2Fe2(CO)5[RP(CH2OH)2]. Organometallics 2010, 29, 5751–5753. [Google Scholar] [CrossRef]
- Quentel, F.; Passard, G.; Gloaguen, F. A binuclear iron-thiolate catalyst for electrochemical hydrogen production in aqueous micellar solution. Chem. Eur. J. 2012, 18, 13473–13479. [Google Scholar] [CrossRef]
- Streich, D.; Astuti, Y.; Orlandi, M.; Schwartz, L.; Lomoth, R.; Hammarström, L.; Ott, S. High-turnover photochemical hydrogen production catalyzed by a model complex of the [FeFe]-hydrogenase active site. Chem. Eur. J. 2010, 16, 60–63. [Google Scholar] [CrossRef]
- Durgaprasad, G.; Bolligarla, R.; Das, S.K. Synthesis, structural characterization and electrochemical studies of [Fe2(μ-L)(CO)6] and [Fe2(μ-L)(CO)5(PPh3)] (L = pyrazine-2,3-dithiolate, quinoxaline-2,3-dithiolate and pyrido[2,3-b]pyrazine-2,3-dithiolate): Towards modeling the active site of [FeFe]–Hydrogenase. J. Organomet. Chem. 2011, 696, 3097–3105. [Google Scholar] [CrossRef]
- Hall, G.B.; Chen, J.; Mebi, C.A.; Okumura, N.; Swenson, M.T.; Ossowski, S.E.; Zakai, U.I.; Nichol, G.S.; Lichtenberger, D.L.; Evans, D.H.; et al. Redox chemistry of noninnocent quinones annulated to 2Fe2S cores. Organometallics 2013, 32, 6605–6612. [Google Scholar] [CrossRef]
- Charreteur, K.; Kdider, M.; Capon, J.-F.; Gloaguen, F.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J. Effect of electron-withdrawing dithiolate bridge on the electron-transfer steps in diiron molecules related to [2Fe]H subsite of the [FeFe]-Hydrogenases. Inorg. Chem. 2010, 49, 2496–2501. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Wang, W.-G.; Si, G.; Wang, F.; Tung, C.-H.; Wu, L.-Z. Photocatalytic hydrogen evolution from rhenium(I) complexes to [FeFe] hydrogenase mimics in aqueous SDS micellar systems: A biomimetic pathway. Langmuir 2010, 26, 9766–9771. [Google Scholar] [CrossRef]
- Na, Y.; Wang, M.; Pan, J.; Zhang, P.; Åkermark, B.; Sun, L. Visible light-driven electron transfer and hydrogen generation catalyzed by bioinspired [2Fe2S] complexes. Inorg. Chem. 2008, 47, 2805–2810. [Google Scholar] [CrossRef]
- Brezinski, W.P.; Karayilan, M.; Clary, K.E.; McCleary-Petersen, K.C.; Fu, L.; Matyjaszewski, K.; Evans, D.H.; Lichtenberger, D.L.; Glass, R.S.; Pyun, J. Macromolecular engineering of the outer coordination sphere of [2Fe–2S] metallopolymers to enhance catalytic activity for H2 production. ACS Macro Lett. 2018, 7, 1383–1387. [Google Scholar] [CrossRef]
- Wang, F.; Wang, W.-G.; Wang, X.-J.; Wang, H.-Y.; Tung, C.-H.; Wu, L.-Z. A highly efficient photocatalytic system for hydrogen production by a robust hydrogenase mimic in an aqueous solution. Angew. Chem. Int. Ed. 2011, 50, 3193–3197. [Google Scholar] [CrossRef]
- Slater, J.W.; Shafaat, H.S. Nickel-substituted rubredoxin as a minimal enzyme model for hydrogenase. J. Phys. Chem. Lett. 2015, 6, 3731–3736. [Google Scholar] [CrossRef]
- Gao, S.; Liu, Y.; Shao, Y.D.; Jiang, D.Y.; Duan, Q. Iron carbonyl compounds with aromatic dithiolate bridges as organometallic mimics of [FeFe] hydrogenases. Coord. Chem. Rev. 2020, 402, 213081. [Google Scholar] [CrossRef]
- Land, H.; Senger, M.; Berggren, G.; Stripp, S.T. Current state of [FeFe]-hydrogenase research: Biodiversity and spectroscopic investigations. ACS Catal. 2020, 10, 7069–7086. [Google Scholar] [CrossRef]
- Wang, M.; Chen, L.; Li, X.Q.; Sun, L.C. Approaches to efficient molecular catalyst systems for photochemical H2 production using [FeFe]-hydrogenase active site mimics. Dalton Trans. 2011, 40, 12793–12800. [Google Scholar] [CrossRef]
- Zamader, A.; Reuillard, B.; Pecaut, J.; Billon, L.; Bousquet, A.; Berggren, G.; Artero, V. Non-covalent integration of a [FeFe]-hydrogenase mimic to multiwalled carbon nanotubes for electrocatalytic hydrogen evolution. Chem. Eur. J. 2022, 28, e202202260. [Google Scholar] [CrossRef]
- Wright, J.A.; Webster, L.; Jablonskyte, A.; Woi, P.M.; Ibrahim, S.K.; Pickett, C.J. Protonation of [FeFe]-hydrogenase sub-site analogues: Revealing mechanism using FTIR stopped-flow techniques. Faraday Discuss. 2011, 148, 359–371. [Google Scholar] [CrossRef]
- Frederix, P.; Kania, R.; Wright, J.A.; Lamprou, D.A.; Ulijn, R.V.; Pickett, C.J.; Hunt, N.T. Encapsulating [FeFe]-hydrogenase model compounds in peptide hydrogels dramatically modifies stability and photochemistry. Dalton Trans. 2012, 41, 13112–13119. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. Before enzymes and templates: Theory of surface metabolism. Microbiol. Rev. 1988, 52, 452–484. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. On the chemistry and evolution of the pioneer organism. Chem. Biodivers. 2007, 4, 584–602. [Google Scholar] [CrossRef]
- Menzel, K.; Apfel, U.P.; Wolter, N.; Ruger, R.; Alpermann, T.; Steiniger, F.; Gabel, D.; Forster, S.; Weigand, W.; Fahr, A. [FeFe]-Hydrogenase models assembled into vesicular structures. J. Liposome Res. 2014, 24, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Himiyama, T.; Waki, M.; Esquivel, D.; Onoda, A.; Hayashi, T.; Van der Voort, P.; Inagaki, S. A heterogeneous hydrogen-evolution catalyst based on a mesoporous organosilica with a diiron catalytic center modelling [FeFe]-hydrogenase. ChemCatChem 2018, 10, 4908–4913. [Google Scholar] [CrossRef]



| Enzymes | O2 Sensitivity | Hydrogen Production Rate |
|---|---|---|
| Nitrogenases | Significantly sensitive | 3–4 fold lower than [FeFe] hydrogenases |
| [NiFe] hydrogenases | Inactivated reversibly | 100-fold lower than [FeFe] hydrogenases |
| [FeFe] hydrogenases | Highly sensitive and inactivated irreversibly | Highest |
| [Fe] hydrogenases | Resistance to O2 | — |
| Strategies | Metabolic Pathways | Methods and Genes | Effects | Organisms | Reference |
|---|---|---|---|---|---|
| Reducing hydrogen consumption | Central carbon metabolism | Deletion of hya and hyb | Disrupting uptake hydrogenase | E. coli | [106,107] |
| Fermentative hydrogen production | Disruption of hyh and alaAT | Inactivating two hydrogen consumption enzymes | Thermococcus kodakarensis | [108] | |
| Fermentative hydrogen production | Cloning hoxEFUYH from the cyanobacterium | Inhibition of hydrogen uptake activity | E. coli | [109] | |
| Electron transfer step | Mutation of C12P in fusion protein f-HupS | Modification of Fe–S cluster in uptake hydrogenase | Nostoc punctiforme ATCC 29133 | [110] | |
| Consumption of hydrogen by uptake hydrogenase | Site-directed mutagenesis | Disruption of uptake hydrogenase | Rhodobacter sphaeroides O.U.001 | [111] | |
| Improving hydrogen-producing enzymes | Fermentative hydrogen production | Using a stronger constitutive promoter to replace the promoter of membrane-bound [NiFe]-hydrogenase | Overexpressing [NiFe] hydrogenase | Thermococcus kodakarensis | [108] |
| Fermentative hydrogen production | Insertion of hydACa and hydACb | Overexpressing two [FeFe] hydrogenases | Clostridium acetobutylicum DSM 792 | [112] | |
| Anaerobic dark fermentation | Cloning multiple copies of hydA | Overexpressing [Fe] hydrogenase | Clostridium paraputrificum | [113] | |
| Electron transfer step | Mutation of R171D in HydA1 | Enhancing [FeFe] hydrogenase catalytic activity | Chlamydomonas reinhardtii | [114] | |
| Electron flow | Cloning rnf operon | Overexpressing the Rnf complex to increase the supply of reductants | Rhodobacter sphaeroides 2.4.1 | [115] | |
| Electron flow | Cloning rnf operon under different promoters | Overexpressing Rnf complex, enhancing nitrogenase activity | Rhodobacter capsulatus SB 1003 | [116] | |
| Electron transfer flux | Insertion of fdxN | Overexpressing fdxN (electron donor), enhancing nitrogenase activity | Rhodobacter sphaeroides HY01 | [117] | |
| Photo-fermentative hydrogen production | Mutation of nitrogenase-regulating genes | Enhancing nitrogenase activity | Rhodopseudomonas palustris; Rhodobacter sphaeroides HY01; Rhodobacter sphaeroides | [118,119,120] | |
| Gene coexpression | Photoheterotrophic hydrogen production | Cloning fermentative metabolic genes including [Fe] hydrogenase | Expression of FHL, [Fe] hydrogenase, and nitrogenase | Rhodobacter sphaeroides KCTC 12085 | [121] |
| Redirecting metabolic pathways | Dark fermentative hydrogen production | Construction of synthetic pyruvate:H2 pathway | Co-expression of six proteins including [FeFe]-hydrogenase | E. coli BL21 | [122] |
| Redox balancing pathway | Deletion of uptake hydrogenase gene | Inactivation of Calvin–Bensone–Bassham (CBB) pathway | Rhodobacter capsulatus YO | [123] | |
| Electron flow, ammonia tolerance | Mutation of hupSL, phbC, pucBA | Elimination of nonessential reductive pathways | Rhodobacter sphaeroides 2.4.1 | [115] | |
| Reducing gas tolerance | Aerobic fermentative hydrogen production | Cloning hydS and hydL from Hydrogenovibrio marinus | Heterologous expression of O2-tolerant [NiFe]-hydrogenase | E. coli | [124] |
| Ammonia tolerance | Mutation of nifA | Expression of ammonia-tolerant NifA | Rhodobacter sphaeroides 2.4.1 | [115] | |
| photo-fermentative hydrogen production | Mutation of nitrogenase-regulating genes | Ammonium tolerance improvement | Rhodopseudomonas palustris; Rhodobacter sphaeroides HY01; Rhodobacter sphaeroides | [118,119,120] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xuan, J.; He, L.; Wen, W.; Feng, Y. Hydrogenase and Nitrogenase: Key Catalysts in Biohydrogen Production. Molecules 2023, 28, 1392. https://doi.org/10.3390/molecules28031392
Xuan J, He L, Wen W, Feng Y. Hydrogenase and Nitrogenase: Key Catalysts in Biohydrogen Production. Molecules. 2023; 28(3):1392. https://doi.org/10.3390/molecules28031392
Chicago/Turabian StyleXuan, Jinsong, Lingling He, Wen Wen, and Yingang Feng. 2023. "Hydrogenase and Nitrogenase: Key Catalysts in Biohydrogen Production" Molecules 28, no. 3: 1392. https://doi.org/10.3390/molecules28031392
APA StyleXuan, J., He, L., Wen, W., & Feng, Y. (2023). Hydrogenase and Nitrogenase: Key Catalysts in Biohydrogen Production. Molecules, 28(3), 1392. https://doi.org/10.3390/molecules28031392