Abstract
Novel polyhydroxylated ammonium, imidazolium, and pyridinium salt organocatalysts were prepared through N-alkylation sequences using glycidol as the key precursor. The most active pyridinium iodide catalyst effectively promoted the carbonation of a set of terminal epoxides (80 to >95% yields) at a low catalyst loading (5 mol%), ambient pressure of CO2, and moderate temperature (75 °C) in batch operations, also demonstrating high recyclability and simple downstream separation from the reaction mixture. Moving from batch to segmented flow conditions with the operation of thermostated (75 °C) and pressurized (8.5 atm) home-made reactors significantly reduced the process time (from hours to seconds), increasing the process productivity up to 20.1 mmol(product) h−1 mmol(cat)−1, a value ~17 times higher than that in batch mode.
1. Introduction
Over the past two decades, there has been a dramatic increase in the amount of CO2 being emitted into the atmosphere, resulting in global warming and subsequent environmental harm [1]. Thus, the balance between anthropogenic emissions and removals from the atmosphere is, today, actively pursued to achieve carbon neutrality in the near future [2]. Accordingly, many kinds of liquid and solid sorbents are being investigated for the development of efficient carbon capture and storage (CCS) strategies [3]. In this scenario, reusing CO2 as a renewable C1 building block to produce added-value chemicals and fuels is becoming a crucial goal for competitiveness, as CO2 refinery may compensate the costs and energy consumption associated with its capture and transportation [4]. However, despite the great interest of academia, industry, and policy makers in carbon capture and utilization (CCU) methodologies, important challenges still need to be addressed including the achievement of levels of process efficiency comparable to those of the petrochemical industry [5]. The kinetic and thermodynamic stability of CO2 (intrinsic factor) is the major limitation of CO2 utilization as a chemical feedstock, which can be overcome by reacting high-energy substrates such as epoxides and aziridines in the presence of extremely active catalysts. The chemical efficiency of CO2 fixation, however, should also take into consideration extrinsic energetic, environmental, and economic factors, thus making preferable the application of inexpensive sustainable catalysts (organocatalysts [6,7] and non-noble metals [8]) with high recyclability, and the operation at a moderate temperature and pressure of CO2 (<100 °C, <10 atm) [9,10,11].
Flow chemistry has recently been proven to have great potential as an enabling technology for the process intensification of gas–liquid reactions of CO2 [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31]. In this reaction set-up, millimeter-sized droplets are generated as confined units with increased superficial area determining an improved mass transfer rate across the gas–liquid interface, which is often the rate-limiting step of gaseous CO2 reactions. Aside from enhanced kinetics, additional advantages of flow conditions in CCU strategies are the better heat transfer, safety, and process reliability, easy control of pressure, facile scaling-out by extending the period of product collection, and straightforward scale-up using the numbering-up approach [32,33].
The atom economical insertion of CO2 into epoxides to yield cyclic carbonates is emerging as a strategic transformation for the chemical process industry because it has been estimated that it will consume, together with the dry reforming of methane, up to 25% of waste CO2 produced annually [6]. Cyclic carbonates are used as high boiling point aprotic solvents [34,35] and electrolytes in secondary batteries [36,37]; moreover, they are valuable monomers for the production of polycarbonates and polyurethanes [38,39,40,41,42], and intermediates for the synthesis of fine chemicals and pharmaceuticals [43,44]. A plethora of homogeneous and heterogeneous catalytic systems have been proposed for this transformation including metal complexes, metal oxides, organocatalysts, and simple alkaline salts [6,7,8,45,46,47,48,49]; however, only a restricted number of these catalysts can be applied without high temperature and pressure requirements, and be recycled using economical downstream purification steps [9,10,11]. In light of this, various metal-based ionic liquids (ILs) have been recently introduced in the literature, [50,51,52,53] showing good to excellent yields and selectivity, typically by the application of high CO2 pressures (up to 50 bar). Additionally, ILs based on quaternary ammonium, imidazolium, and pyridinium salts, eventually immobilized on inorganic [54] and polymeric [55] solid supports, or prepared as hybrid materials, [56] have received considerable attention, showing advantageous features such as uninflammability, low volatility, thermal stability, and flexible structure-tailorability [57,58,59,60,61,62,63,64]. In particular, it has been demonstrated that the presence of hydroxyl groups on the IL moiety significantly increases the catalyst activity as a result of the synergistic effect of hydrogen bonding with the oxygen atom of epoxides, which effectively contributes to the ring-opening process promoted by the halide nucleophilic attack (Figure 1) [65,66,67,68,69,70,71]. On the basis of the same mechanistic rationale, we propose herein a set of novel polyhydroxylated ionic liquids, namely ammonium, imidazolium, and pyridinium organocatalysts, whose activity and recyclability has been initially tested in the carbonation of terminal epoxides under conventional batch conditions. Afterwards, following our interest in the development of efficient technology platforms for the intensification of gas–liquid reactions (aerosol reactors), [72,73] we further developed our study by moving from batch to segmented flow processes with the detection of significant improvements in terms of reaction time and productivity.
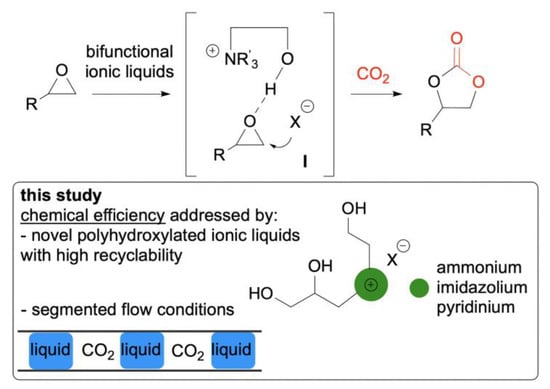
Figure 1.
Design of the organocatalytic conversion of CO2 into cyclic carbonates under segmented flow conditions.
2. Results and Discussion
The synthesis of the polyhydroxylated ammonium iodide salt 3 was first addressed by coupling dibutyl amine with glycidol 1 refluxing under acidic conditions to give the intermediate amine 2 in satisfactory yield (72%; Scheme 1). Afterwards, following standard procedures for the N-alkylation of tertiary amines, the diol 2 was refluxed in alcoholic solvent (EtOH) with iodoethanol, affording an inseparable mixture of the target salt 3 (32%) and the by-product 4 (68%), as detected using 1H NMR spectroscopy and further confirmed with MS analysis (Table 1, entry 1). Lowering the temperature to 60 °C improved the reaction selectivity toward 3 at the expense, however, of the efficiency conversion (21%; entry 2). Similar unsatisfactory results were also registered using different polar aprotic solvents (DMF, THF) and temperatures (entries 4–5). The unexpected reaction outcome was explained by hydriodic acid elimination from iodoethanol promoted by 2 yielding 4 and acetaldehyde (Scheme 1). Fortunately, we observed that neat conditions (75 °C, 48 h) could suppress the side-reaction path, affording the desired polyhydroxylated ammonium salt 3 as the sole product in quantitative yield and 95% purity (1H NMR analysis; entry 8).
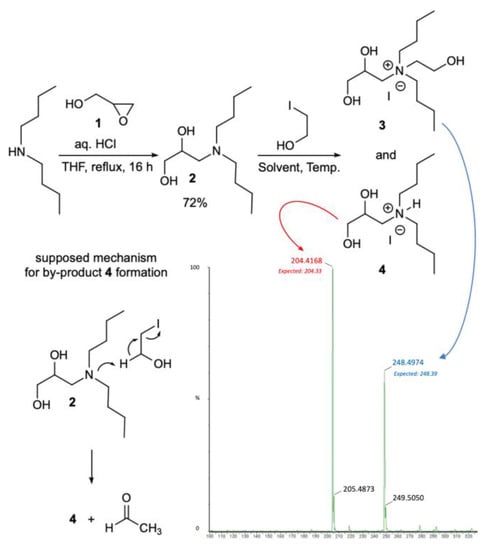
Scheme 1.
Synthesis of polyhydroxylated ammonium iodide salt 3 and side-reaction path. Solvent and temperature variations are reported in Table 1.

Table 1.
Optimization of ammonium salt 3 synthesis.
Next, with the aim of verifying the effect of halide variation on the efficiency of epoxide ring-opening (vide infra), the optimized double N-alkylation sequence was applied to the synthesis of the polyhydroxylated ammonium bromide salt 5 using bromoethanol for quaternarization (Scheme 2). The same protocol was also employed with little modification to produce the bifunctional organocatalyst 8 featuring the designed polyhydroxylated imidazolium moiety.

Scheme 2.
Synthesis of polyhydroxylated (a) ammonium- and (b) imidazolium-based organocatalysts 5 and 8.
The access to the target pyridinium salts 14 and 16 (Scheme 3) initially required the set-up of a practical procedure for the synthesis of the alkylating agent 9 starting from glycidol, which was identified in this study as the common precursor for introducing the 1,2-propanediol group in the final organocatalysts. The iodide 9 is a known compound and it is typically produced in three steps from glycerol [74]; on the basis of previous observations [75], the straightforward and regioselective conversion of glycidol into the vicinal halohydrin 9 was achieved in satisfactory yield (75%) using lithium iodide in combination with acetic acid under mild reaction conditions. Our synthetic plan proceeded with the synthesis of the intermediate N-alkyl-4-amine pyridines 13 and 15 featuring the hydroxyalkyl chain with a different degree of free rotation. Gratifyingly, microwave-heating (130 °C, 3 h) of the mixtures of 4-chloropyridine 10 with excess of either methylamino-ethanol 11 or prolinol 12 gave pyridines 13 and 15, respectively, in almost quantitative yields after simple evaporation of unreacted 11/12. Finally, the completion of the synthetic sequence from 13/15 was straightforward, affording the polyhydroxylated pyridinium salts 14 and 16 by N-alkylation with iodide 9 under the previously optimized neat conditions.
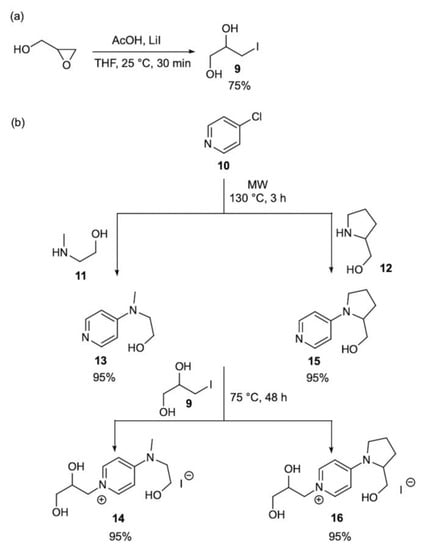
Scheme 3.
Synthesis of: (a) 3-iodopropane-1,2-diol 9 and (b) polyhydroxylated pyridinium-based organocatalysts 14 and 16.
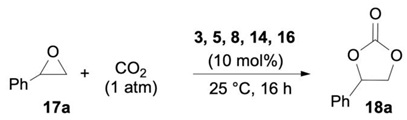
The catalytic activity of the novel bifunctional organocatalysts 3, 5, 8, 14, and 16 (10 mol%) was tested at ambient temperature and pressure in the model conversion of styrene oxide 17a into styrene carbonate 18a (Table 2). In agreement with the order of nucleophilicity of halide anions and their coordination ability of an acidic hydrogen (intermediate I, Figure 1) [6], iodide ammonium salt 3 outperformed the bromide counterpart 5 affording 18a in 30% yield with complete selectivity (entries 1–2). Among the iodide salts 3, 8, 14, and 16, the polyhydroxylated pyridinium organocatalyst 16 emerged as the most effective promoter (18a: 44%; entry 5), somehow substantiating the importance of some rigidity in the hydroxyalkyl chain for transition state stabilization (16 vs. 14).
Table 2.
Screening of organocatalyst 3, 5, 8, 14, and 16 activity.
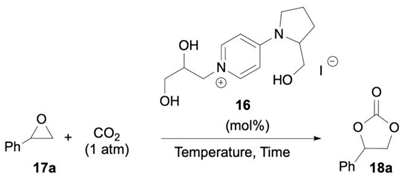
Different conditions were then screened with the selected organocatalyst 16 to improve the process productivity (Table 3). Increasing the temperature up to 75 °C allowed the full conversion of 17a with complete selectivity towards 18a (16: 10 mol%; reaction time: 16 h; entries 1–3). Satisfyingly, the same reaction outcome was reproduced by halving the catalyst loading to 5 mol% (entry 4), while a further decrease in the 16 amount (2 mol%) or a shorter reaction time (12 h) resulted in significantly lower conversions (entries 5–6). Based on previous findings [23,70], DMF and H2O were tested as additives, detecting, however, a marked drop in reaction efficiency (entries 7–8). It is important to emphasize that under the optimized conditions of entry 4, the reaction mixture at the initial time is heterogeneous, becoming completely homogeneous as the reaction progresses. Therefore, keeping in mind the ultimate goal of process intensification by the application of flow conditions, EtOH (50 mol%) was utilized to obtain full solubilization of catalyst 16 (entry 9); opportunely, only a minimal decrease in the yield of cyclic carbonate 18a (92%) was observed. Overall, the resultant productivity (P, which also corresponds to TOF, Equation (1)) of the optimal batch process (entry 4) was 1.2 mmol(18a) h−1 mmol(16)−1.
Table 3.
Optimization study with catalyst 16 under batch conditions.
The recyclability of the polyhydroxylated pyridinium 16 was investigated over six runs (Figure 2). Upon reaction completion, catalyst recovery consisted of the simple addition of EtOAc. Operating in this way, the catalyst precipitated and the product was collected upon centrifugation. Gratifyingly, only a moderate conversion decrease (~3%) was observed after the fifth recycle, mainly because of the partial loss of catalyst during the recovery and washing steps.
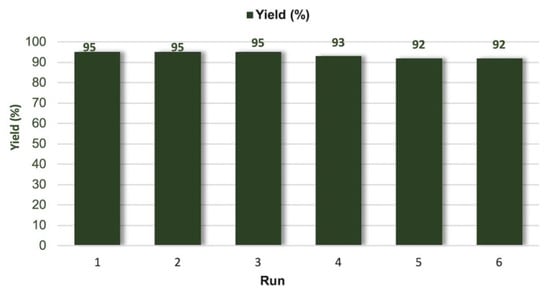
Figure 2.
Recycling experiments (optimized conditions of entry 4, Table 3).
The generality and efficacy of the method was tested through a brief substrate scope study, which was conducted with terminal epoxides 17a–g at atmospheric pressure and mild temperature (Scheme 4). In addition to the styrene oxide derivates 17a,b, the epoxides displaying an alkyl chain 17c–g could also be converted into the corresponding cyclic carbonates 18a–g in good to excellent yields (80% to >95%).

Scheme 4.
Substrate scope (batch procedure).
At this stage of the study, driven by our interest in process intensification by the application of flow techniques [76,77,78,79,80,81,82,83,84,85,86,87,88], we next investigated the transition of the model carbonation of styrene oxide from batch to segmented flow conditions [89,90]. The in-house assembled flow apparatus consisted of a 4.42 mL spiral capillary reactor (FEP tubing; 0.75 mm ID) placed inside a thermostated bath (75 °C). The coil was connected to an HPLC pump and a CO2 cylinder by means of a standard T junction, where the gas and liquid streams were mixed. The exact CO2 volume was delivered into the reactor by a mass flow controller (MFC), while a back-pressure regulator (BPR) maintained a constant pressure of CO2 (8.5 atm) throughout the system (Scheme 5 and Supplementary Material Figure S1). Initial experiments were performed to identify a stable segmented flow regime by variation of the liquid and gas flow rates, always keeping a molar excess of CO2 over styrene oxide. Approximately, each segment length was found to be in a range of 0.5 to 1.0 mm. The residence time (tr) was calculated as the ratio of reactor volume over the total gas and liquid flow rate. After some experimentation, two set of conditions (A and B) were optimized with a constant CO2 flow rate of 3.00 mL min−1 and liquid flow rates of 0.10 and 0.07 mL min−1 corresponding to CO2/17a molar ratios of 1.02 and 1.45, respectively. Significantly, after the attainment of the steady-state regime (ca. 2 min), condition A provided 18a with an instant conversion of 81% (1H NMR of the outlet stream) in only 85 s of residence time, thus resulting in a process productivity of 16.3 mmol(18a) h−1 mmol(16)−1. In accordance with our expectations, the reduction in the liquid flow rate to 0.07 mL min−1 (condition B) further increased the reaction conversion (>95%) with almost the same residence time (86 s), affording 18a with a productivity of 20.1 mmol(18a) h−1 mmol(16)−1. This value is about 17-fold higher than that measured in batch-mode and it was explained by the improved mass transfer of CO2 at the gas–liquid interphase due to the increased pressure and segmented flow regime.
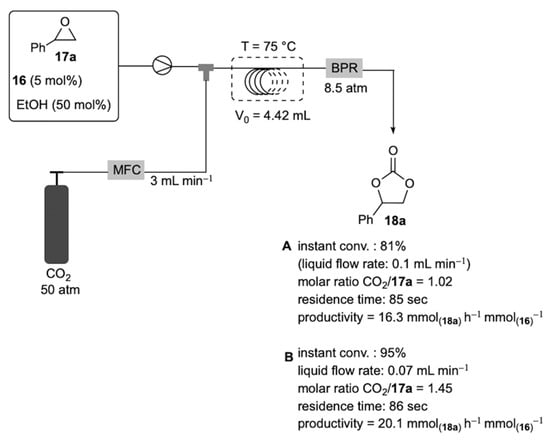
Scheme 5.
Production of styrene carbonate 18a under segmented flow conditions.
The continuous production of cyclic carbonates 18b–g was finally examined under the optimized flow conditions, affording remarkable conversion efficiencies (>85%) and productivities in the range of 17.2–20.1 mmol(18) h−1 mmol(16)−1 (Table 4).
Table 4.
Production of cyclic carbonates 18 under segmented flow conditions.
3. Materials and Methods
Commercially available reagents were purchased from commercial sources and used without any subsequent purification. The solvents used for reactions were distilled from appropriate drying agents and stored over 3 Å molecular sieves. 1H-NMR and 13C-NMR spectra were recorded on Varian Mercury Plus 300 (Varian Inc., Palo Alto, CA, USA) and Varian Mercury Plus 400 (Varian Inc., Palo Alto, CA, USA) spectrometers in CDCl3, DMSO-d6, and D2O at room temperature. 13C{1H} NMR spectra were recorded in 1H broad-band decoupled mode, and chemical shifts (δ) are reported in parts per million relative to the residual solvent peak. Flash column chromatography was performed on silica gel 60 (230−400 mesh). High-resolution mass spectra (HRMS) were recorded in positive ion mode with an Agilent 6520 HPLC-Chip Q/TF-MS nanospray instrument (Agilent Technologies, Santa Clara, CA, USA) using a time-of-flight, a quadrupole, or a hexapole unit to produce spectra.
3.1. Procedures for the Synthesis of Intermediates 2, 6, 9, 13, 15 and Organocatalysts 3, 5, 8, 14, 16
3-(Dibutylamino)propane-1,2-diol(2). Glycidol (15.0 mmol) in THF (10 mL), HCl 37% (1 mL), and dibutyl amine (5 mmol) was added to a round-bottom flask with a refrigerator on top. The mixture was stirred and refluxed overnight. Once the dibutyl amine was completely reacted, the product was purified by acid/base extraction with DCM. The reaction crude was solubilized in DCM and then extracted with a solution of HCl 1M; the organic layer was removed and the aqueous phase was basified with NaOH 1M solution and re-extracted with DCM. The organic layer was treated with anhydrous sodium sulphate and filtered, and the solvent was removed using a rotary evaporator and high-vacuum pump. By following this procedure, 3-(dibutylamino)propane-1,2-diol 2 was obtained as a yellow viscous oil (3.60 mmol, 72% yield). 1H NMR (500 MHz, CDCl3) δ 5.73–5.59 (m, 3H), 5.45 (dd, J = 11.1, 4.5 Hz, 1H), 4.59–4.42 (m, 3H), 4.42–4.31 (m, 3H), 3.47–3.33 (m, 4H), 3.33–3.18 (m, 4H), 2.87 (t, J = 7.4 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3): δ 67.3, 65.0, 57.0, 54.3, 29.2, 20.7, 14.1. HRMS (ESI) m/z: [M + H]+ calcd for C11H26NO2+ 204.1958, found 204.1952.
N-Butyl-N-(2,3-dihydroxypropyl)-N-(3-hydroxypropyl)butan-1-aminium iodide (3). Compound 2 (2.50 mmol) and 2-iodoethanol (2.50 mmol) were added to a 10 mL vial equipped with a small magnetic bar. The vial was sealed, and an argon atmosphere inside the reaction vial was created by three cycles of vacuum and argon pumping. The mixture was vigorously stirred for 48 h at 75 °C to obtain the desired product. No additional purification step is needed. By following this procedure, N-butyl-N-(2,3-dihydropropyl)-N-(3-hydroxypropyl)butan-1-aminium iodide (3) was obtained as a very viscous brown liquid (2.50 mmol, quant.) with a purity of ca. 95% (1H NMR analysis). 1H NMR (300 MHz, DMSO-d6) δ 5.31 (d, J = 5.3 Hz, 1H), 5.20 (t, J = 5.3 Hz, 1H), 5.04 (t, J = 5.3 Hz, 1H), 4.03–3.88 (m, 1H), 3.87–3.71 (m, 2H), 3.63–3.55 (m, 1H), 3.50–3.35 (m, 5H), 3.32–3.11 (m, 3H), 3.01 (broad s, 1H), 1.78–1.46 (m, 4H), 1.36–1.17 (m, 4H), 0.95–0.83 (m, 6H). 13C{1H} NMR (101 MHz, DMSO-d6) δ 66.0, 64.1, 62.8, 61.8, 60.6, 59.8, 59.8, 56.5, 55.0, 23.6, 19.6, 19.0, 13.9. HRMS (ESI) m/z: [M]+ calcd for C13H30NO3+ 248.2226 found 248.2232.
N-Butyl-N-(2,3-dihydroxypropyl)-N-(3-hydroxypropyl)butan-1-aminium bromide (5). Compound 2 (2.50 mmol) and 2-bromoethanol (2.50 mmol) were added to a 10 mL vial equipped with a small magnetic bar. The vial was sealed, and an argon atmosphere inside the reaction vial was created by three cycles of vacuum and argon pumping. The mixture was vigorously stirred for 48 h at 75 °C to obtain the desired product. No purification steps are needed. By following this procedure, N-butyl-N-(2,3-dihydroxypropyl)-N-(3-hydroxypropyl)butan-1-aminium bromide (5) was obtained as a very viscous brown liquid (2.50 mmol, quant.) with a purity of ca. 95% (1H NMR analysis). 1H NMR (300 MHz, DMSO-d6) δ 5.33 (d, J = 5.2 Hz, 1H), 5.21 (t, J = 5.2 Hz, 1H), 5.04 (t, J = 5.2 Hz, 1H), 4.04–3.90 (m, 1H), 3.89–3.73 (m, 2H), 3.73–3.52 (m, 1H), 3.50–3.34 (m, 6H), 3.31–3.12 (m, 2H), 3.03 (s, 1H), 1.81–1.46 (m, 4H), 1.38–1.16 (m, 4H), 0.96–0.81 (m, 6H).13C{1H} NMR (101 MHz, DMSO-d6) δ 66.0, 64.1, 62.7, 61.8, 60.6, 59.8, 59.8, 56.5, 55.0, 23.6, 19.6, 19.0, 13.9. HRMS (ESI) m/z: [M]+ calcd for C13H30NO3+ 248.2226 found 248.2229.
3-(1H-Imidazol-1-yl)propane-1,2-diol (6). Glycidol (15.0 mmol) in acetonitrile (10 mL), DIPEA (10 mmol), and imidazole (10 mmol) were added to a round-bottom flask with a refrigerator on top. The mixture was stirred and refluxed overnight. Once the reaction was complete, the purification took place through flash chromatography using an automatic flash chromatographer CombiFlash. The elution gradient was from 100% A to 100% B in 45 CV, then 100% B in 10 CV (A: AcOEt + 2% NH4OH, B: AcOEt/MeOH = 9/1 + 2% NH4OH). By following this procedure, 3-(1H-imidazol-1-yl)propane-1,2-diol (6) was obtained as a pale yellow viscous oil (5.70 mmol, 57% yield). 1H NMR (500 MHz, D2O) δ 7.68 (s, 1H), 7.18 (s, 1H), 7.03 (s, 1H), 4.19 (dd, J = 13.7, 3.0 Hz, 1H), 4.06–3.98 (m, 2H), 3.60 (dd, J = 11.8, 5.0 Hz, 1H), 3.53 (dd, J = 11.8, 5.0 Hz, 1H).13C{1H} NMR (126 MHz, D2O): δ 138.2, δ 127.2, 120.5, 70.6, δ 62.4, δ 49.0. HRMS (ESI) m/z: [M + H]+ calcd for C6H11N2O2+ 143.0815 found 143.0816.
1-(2,3-Dihydroxypropyl)-3-(2-hydroxyethyl)-1H-imidazol-3-ium iodide (8). Compound 6 (1.40 mmol) and 2-iodoethanol (1.40 mmol) were added to a 10 mL vial equipped with a small magnetic stir bar. The vial was hermetically sealed, and an argon atmosphere inside the reaction vial was created by three cycles of vacuum and argon pumping. The mixture was stirred for 24 h at 75 °C to give the final product. No purification steps are needed. By following this procedure, 1-(2,3-dihydroxypropyl)-3-(2-hydroxyethyl)-1H-imidazol-3-ium iodide (8) was obtained as a very viscous brown liquid (1.40 mmol, quant.) with a purity of ca. 95% (1H NMR analysis). 1H NMR (300 MHz, DMSO-d6) δ 9.05 (s, 1H), 7.69 (d, J = 9.4 Hz, 2H), 5.29 (d, J = 5.0 Hz, 1H), 5.14 (t, J = 5.0 Hz, 1H), 4.93 (t, J = 5.0 Hz, 1H), 4.30 (dd, J = 13.8, 2.7 Hz, 1H), 4.21 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 13.8, 8.2 Hz, 1H), 3.83–3.68 (m, 3H), 3.46–3.36 (m, 1H), 3.26–3.17 (m, 1H). 13C{1H} NMR (101 MHz, DMSO-d6) δ 137.2, 123.5, 122.8, 70.1, 63.2, 59.8, 52.6, 52.0. HRMS (ESI) m/z: [M]+ calcd for C8H15N2O3+ 187.1077 found 187.1071.
3-Iodopropane-1,2-diol (9). LiI (80.0 mmol, 10 g) was added to a solution of glycidol (50.0 mmol) and acetic acid (150 mmol) in anhydrous THF (40 mL), and the solution was kept at room temperature and stirred in argon atmosphere for 40 min. The mixture was diluted with distilled water and extracted with two aliquots of ethyl acetate (2 × 20 mL). The organic layer was treated with anhydrous sodium sulphate and filtered, and the solvent was removed using a rotary evaporator and high-vacuum pump. By following this procedure, 3-iodopropane-1,2-diol (9) was obtained as a yellow amorphous solid (50.0 mmol, quant.) 1H NMR (400 MHz, D2O) δ 3.59–3.42 (m, 3H), 3.25 (dd, J = 10.8, 4.5 Hz, 1H), 3.15 (dd, J = 10.8, 4.5 Hz, 1H). 13C{1H} NMR (101 MHz, D2O) δ 70.6, 64.4, 8.6. HRMS (ESI) m/z: [M + H]+ calcd for C3H8IO2+ 202.9563 found 202.9554 [74].
2-(Methyl(pyridin-4-yl)amino)ethan-1-ol (13). Chloropyridine hydrochloride (5.00 mmol) and N-methylethanolamine (62.5 mmol) were added to a round-bottom flask. The mixture was stirred at 120°C for 24 h. Once the reaction was complete, the excess of unreacted amine was vacuum-evaporated, and then the product was purified by extracting the free amine with DCM from a basic environment (K2CO3). The organic layer was treated with anhydrous sodium sulphate and filtered, and the solvent was removed using a rotary evaporator and high-vacuum pump. By following this procedure, 2-(methyl(pyridin-4-yl)amino)ethan-1-ol (13) was obtained as a white amorphous solid (3.50 mmol, 70% yield). 1H NMR (400 MHz, CDCl3) δ 8.12 (dd, J = 5.1, 1.6 Hz, 2H), 6.52 (dd, J = 5.1, 1.6 Hz, 2H), 3.83 (t, J = 5.7 Hz, 2H), 3.54 (t, J = 5.7 Hz, 2H), 3.04 (s, 3H), 2.70 (broad s, 1H).13C{1H} NMR (126 MHz, CDCl3): δ 149.0, 106.7, 59.6, 53.6, 38.2. HRMS (ESI) m/z: [M + H]+ calcd for C8H13N2O+ 153.1022 found 153.1024.
1-(2,3-Dihydroxypropyl)-4-((2-hydroxyethyl)(methyl)amino)pyridin-1-ium iodide (14). Compound 13 (0.25 mmol) and 3-iodopropane-1,2-diol 9 (0.25 mmol) were added to a 10 mL vial equipped with a small magnetic bar. The vial was hermetically sealed, and an argon atmosphere inside the reaction vial was created by three cycles of vacuum and argon pumping. The mixture was stirred for 24 h at 75 °C to give the final product. No purification steps are needed. By following this procedure, 1-(2,3-dihydroxypropyl)-4-((2-hydroxyethyl)(methyl)amino)pyridin-1-ium iodide (14) was obtained as a very viscous yellow liquid (0.25 mmol, quant.) with a purity of ca. 95% (1H NMR analysis). 1H NMR (300 MHz, DMSO-d6) δ 8.13 (dd, J = 12.5, 7.5 Hz, 2H), 7.12 (d, J = 5.2 Hz, 1H), 6.96 (d, J = 7.5 Hz, 1H), 5.24 (d, J = 5.2 Hz, 1H), 4.92 (dd, J = 12.0, 6.2 Hz, 2H), 4.27 (d, J = 10.6 Hz, 1H), 4.01 (dd, J = 13.6, 8.2 Hz, 1H), 3.73 (broad s, 1H), 3.63 (s, 4H), 3.53–3.40 (m, 2H), 3.15 (s, 3H). 13C{1H} NMR (101 MHz, DMSO-d6) δ 156.6, 143.5, 142.8, 108.0, 107.6, 70.6, 63.1, 60.0, 58.4, 54.2. HRMS (ESI) m/z: [M]+ calcd for C11H19N2O3+ 227.1390 found 227.1380.
(1-(Pyridin-4-yl)pyrrolidin-2-yl)methanol (15). 4-Chloropyridine hydrochloride (0.70 mmol) and prolinol (8.75 mmol) were added to a 5 mL vial equipped with a magnetic stir bar. The vial was hermetically sealed and inserted into a microwave. The reaction mixture was stirred and irradiated for 3 h at 120 °C. After this time, the crude was transferred into a round-bottom flask and attached to a high-vacuum pump in order to remove the unreacted excess of prolinol. After 24 h, the crude was diluted in dichloromethane and washed with a 1M solution of K2CO3. The organic layer was concentrated at the rotary evaporator and the flask was attached to the high-vacuum pump for an additional 24 h. By following this procedure, (1-(pyridine-4-yl)pyrrolidine-2-yl)methanol (15) was obtained as a brown amorphous solid (0.70 mmol, quant.). 1H NMR (300 MHz, CDCl3) δ 8.16 (d, J = 5.0 Hz, 2H), 6.48 (d, J = 5.0 Hz, 2H), 3.98–3.83 (m, 1H), 3.70 (dd, J = 11.0, 4.2 Hz, 1H), 3.60 (dd, J = 11.0, 6.9 Hz, 1H), 3.48–3.43 (m, 1H), 3.28–3.08 (m, 1H), 2.20–1.90 (m, 6H). 13C{1H} NMR (101 MHz, CDCl3) δ 149.6, 107.4, 62.6, 59.6, 48.1, 28.3, 23.2. HRMS (ESI) m/z: [M + H]+ calcd for C10H15N2O+ 179.1179 found 179.1181.
1-(2,3-Dihydroxypropyl)-4-(2-(hydroxymethyl)pyrrolidin-1-yl)pyridin-1-ium iodide (16). Compound 15 (0.25 mmol) and 3-iodopropane-1,2-diol 9 (0.25 mmol) was added to a 10 mL vial equipped with a small magnetic stir bar. The vial was hermetically sealed, and an argon atmosphere inside the reaction vial was created by three cycles of vacuum and argon pumping. The mixture was stirred for 24 h at 75 °C. No purification steps are needed. By following this procedure, 1-(2,3-dihydroxypropyl)-4-(2-(hydroxymethyl)pyrrolidin-1-yl)pyridin-1-ium iodide (16) was obtained as a very viscous brown liquid (0.25 mmol, quant.) with a purity of ca. 95% (1H NMR analysis). 1H NMR (300 MHz, DMSO-d6) δ 8.16 (t, J = 6.3 Hz, 2H), 7.06 (dd, J = 7.8, 2.8 Hz, 1H), 6.86 (dd, J = 7.8, 2.8 Hz, 1H), 5.25 (d, J = 5.4 Hz, 1H), 4.96 (dt, J = 13.1, 5.4 Hz, 2H), 4.29 (d, J = 12.3 Hz, 1H), 4.14 (d, J = 5.1 Hz, 1H), 4.03 (dd, J = 13.6, 7.8 Hz, 1H), 3.75 (broad s, 1H), 3.58 (t, J = 10.8 Hz, 1H), 3.51–3.35 (m, 4H), 3.31–3.21 (m, 1H), 2.23–1.71 (m, 4H). 13C{1H} NMR (101 MHz, DMSO-d6) δ 154.0, 143.5, 142.9, 108.8, 108.4, 70.8, 63.2, 61.3, 61.1, 60.1, 49.2, 28.0, 22.8. HRMS (ESI) m/z: [M]+ calcd for C13H21N2O2+ 253.1547 found 253.1551.
3.2. General Procedure for the Synthesis of Styrene Carbonates 18a–g under Batch Conditions
Epoxide 17 (2.00 mmol) and catalyst 16 (5 mol%) were added to a 10 mL vial equipped with a small magnetic stir bar. A CO2 atmosphere inside the reaction vial was created by three cycles of vacuum and CO2 pumping and maintained by a carbon dioxide balloon connected via a needle. The mixture was stirred for 16 h at room temperature, then diluted with EtOAc to precipitate the catalyst 16, and centrifuged to recover the cyclic carbonate 18 in the solution, which was purified using column chromatography.
4-Phenyl-1,3-dioxolan-2-one (18a). By following the general procedure, 18a (318 mg, 1.94 mmol, >95%) was obtained as a viscous colorless oil after short column chromatography on silica gel (9:1 cyclohexane/EtOAc). 1H NMR (500 MHz, CDCl3) δ 7.48–7.41 (m, 3H), 7.36 (dd, J = 7.8, 1.8 Hz, 2H), 5.67 (t, J = 8.0 Hz, 1H), 4.80 (t, J = 8.0 Hz, 1H), 4.34 (t, J = 8.0 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 154.8, 135.8, 129.7, 129.2, 125.9, 78.0, 71.2. HRMS (ESI) m/z: [M + H]+ calcd for C9H9O3+ 165.0546 found 165.0539 [23].
4-(4-Chlorophenyl)-1,3-dioxolan-2-one (18b). By following the general procedure, 18b (356 mg, 1.70 mmol, 85%) was obtained as a viscous pale yellow oil after column chromatography on silica gel (9:1 cyclohexane/EtOAc). 1H NMR (300 MHz, CDCl3) δ 7.43 (d, J = 8.5 Hz, 2H), 7.31 (d, J = 8.5 Hz, 2H), 5.66 (t, J = 8.0 Hz, 1H), 4.80 (t, J = 8.6 Hz, 1H), 4.31 (dd, J = 8.6, 8.0 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3) δ 154.5, 135.8, 134.2, 129.5, 127.2, 77.0, 71.0. HRMS (ESI) m/z: [M + H]+ calcd for C9H8ClO3+ 199.0156 found 199.0163 [91].
4-(But-3-en-1-yl)-1,3-dioxolan-2-one (18c). By following the general procedure, 18c (276 mg, 1.95 mmol, >95%) was obtained as a viscous colorless oil after short column chromatography on silica gel (9:1 cyclohexane/EtOAc). 1H NMR (400 MHz, CDCl3) δ 5.88–5.70 (m, 1H), 5.15–5.01 (m, 2H), 4.78–4.67 (m, 1H), 4.53 (t, J = 8.2 Hz, 1H), 4.08 (t, J = 8.2 Hz, 1H), 2.34–2.10 (m, 2H), 2.00–1.87 (m, 1H), 1.83–1.71 (m, 1H). 13C{1H} NMR (101 MHz, CDCl3) δ 154.9, 136.0, 116.5, 76.3, 69.3, 33.1, 28.7. HRMS (ESI) m/z: [M + H]+ calcd for C7H11O3+ 143.0703 found 143.0699 [23].
4-Butyl-1,3-dioxolan-2-one (18d). By following the general procedure, 18d (230 mg, 1.60 mmol, 80%) was obtained as a viscous colorless oil after column chromatography on silica gel (9:1 cyclohexane/EtOAc). 1H NMR (400 MHz, CDCl3) δ 4.75–4.65 (m, 1H), 4.52 (t, J = 8.1 Hz, 1H), 4.07 (dd, J = 8.1, 7.1 Hz, 1H), 1.88–1.76 (m, 1H), 1.74–1.64 (m, 1H), 1.48–1.31 (m, 4H), 0.93 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (101 MHz, CDCl3) δ 155.0, 77.0, 69.4, 33.6, 26.4, 22.2, 13.8. HRMS (ESI) m/z: [M + H]+ calcd for C7H13O3+ 145.0859 found 145.0861 [23].
4-(Chloromethyl)-1,3-dioxolan-2-one (18e). By following the general procedure, 18e (232 mg, 1.70 mmol, 85%) was obtained as a viscous colorless oil after column chromatography on silica gel (9:1 cyclohexane/EtOAc). 1H NMR (400 MHz, CDCl3) δ 4.99–4.91 (m, 1H), 4.59 (dd, J = 8.9, 8.2 Hz, 1H), 4.41 (dd, J = 8.9, 5.7 Hz, 1H), 3.84–3.68 (m, 2H). 13C{1H} NMR (101 MHz, CDCl3) δ 154.0, 74.2, 67.0, 43.5. HRMS (ESI) m/z: [M + H]+ calcd for C4H6ClO3+ 137.0000 found 136.9998 [23].
4-(Methoxymethyl)-1,3-dioxolan-2-one (18f). By following the general procedure, 18f (250 mg, 1.90 mmol, 95%) was obtained as a viscous colorless oil after short column chromatography on silica gel (9:1 cyclohexane/EtOAc). 1H NMR (300 MHz, CDCl3) δ 4.85–4.75 (m, 1H), 4.49 (t, J = 8.4 Hz, 1H), 4.38 (dd, J = 8.4, 6.1 Hz, 1H), 3.61 (qd, J = 10.9, 3.9 Hz, 2H), 3.43 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3) δ 154.9, 74.9, 71.4, 66.2, 59.7. HRMS (ESI) m/z: [M + H]+ calcd for C4H9O4+ 133.0495 found 133.0489 [92].
4-(Phenoxymethyl)-1,3-dioxolan-2-one (18g). By following general procedure, 18g (337 mg, 1.74 mmol, 87%) was obtained as a white amorphous solid after trituration in Et2O. 1H NMR (300 MHz, CDCl3) δ 7.31 (dd, J = 8.2, 7.3 Hz, 2H), 7.02 (t, J = 7.3 Hz, 1H), 6.91 (d, J = 8.2 Hz, 2H), 5.09–4.98 (m, 1H), 4.66–4.50 (m, 2H), 4.20 (qd, J = 10.5, 3.9 Hz, 2H). 13C{1H} NMR (101 MHz, CDCl3) δ 157.7, 129.7, 122.0, 114.6, 74.0, 66.8, 66.2. HRMS (ESI) m/z: [M + H]+ calcd for C10H11O4+ 195.0652 found 195.0660 [23].
3.3. General Procedure for the Synthesis of Cyclic Carbonates 18a–g under Segmented Flow Conditions
Epoxide 17, ethanol (50 mol%), and organocatalysts 16 (5 mol%) were mixed in the reservoir and the resulting solution was pumped through the thermostated reactor (75 °C) at 0.07 mL min−1. Simultaneously, a CO2 gas flow of 3.0 mL min−1 was delivered. Collection and analysis using 1H NMR of the outlet stream (minute by minute with durene as internal standard for conversion evaluation) was started 4 min after injection and maintained for an additional 6 min. After this period, the collected reaction mixture was diluted with EtOAc to precipitate the catalyst 16, and centrifuged to recover the cyclic carbonate 18 in the solution, which was purified as described in the batch procedure (Section 3.2).
4. Conclusions
In summary, the chemical efficiency of the carbonation of terminal epoxides with CO2 to produce cyclic carbonates has been investigated by reporting a set of novel polyhydroxylated ionic liquids and operating segmented flow reactors. The selected pyridinium iodide organocatalyst guaranteed high conversions at ambient pressure and moderate temperature (75 °C), showing high reusability and simple downstream separation in batch experiments. Transition to segmented flow conditions determined a ∼17-fold increase in process productivity and a reduction in process time from hours to seconds, as a result of the improved CO2 mass transfer at the gas–liquid interphase due to the moderate increase in pressure (8.5 atm) and the segmented flow regime. Therefore, we believe that the flow methodology herein disclosed might represent a new opportunity for further advancements in the process intensification of CO2 fixation into cyclic carbonates.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28041530/s1. Figure S1: Flow apparatus. Figures S2 and S3: 1H and 13C NMR spectra of intermediates and organocatalysts (2, 3, 5, 6, 8, 9, 13, 14, 15,16); Figure S12–S18: 1H and 13C NMR spectra of cyclic carbonates (18a–18g).
Author Contributions
Conceptualization, A.M. and P.D.; methodology, L.P., C.R. and F.V.; validation, D.R., G.D.C. and A.B.; writing—original draft preparation, A.M. and P.D.; writing—review and editing, all authors; supervision, A.M. and P.D.; project administration, P.D.; funding acquisition, P.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the NATO Science for Peace and Security Programme (NATO SPS, Grant No. MYP G5885, project acronym: TANGO).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Paolo Formaglio for NMR experiments, Tatiana Bernardi for HRMS analyses, Ercolina Bianchini for elemental analyses, and the students Luca Erriquez and Matteo Tolio for the preparation of starting materials.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors by request.
References
- Rogelj, J.; Luderer, G.; Pietzcker, R.C.; Kriegler, E.; Schaeffer, M.; Krey, V.; Riahi, K. Energy System Transformations for Limiting End-of-Century Warming to below 1.5 °C. Nat. Clim. Change 2015, 5, 519–527. [Google Scholar] [CrossRef]
- Rogelj, J.; Schaeffer, M.; Meinshausen, M.; Knutti, R.; Alcamo, J.; Riahi, K.; Hare, W. Zero Emission Targets as Long-Term Global Goals for Climate Protection. Environ. Res. Lett. 2015, 10, 105007. [Google Scholar] [CrossRef]
- Bui, M.; Adjiman, C.S.; Bardow, A.; Anthony, E.J.; Boston, A.; Brown, S.; Fennell, P.S.; Fuss, S.; Galindo, A.; Hackett, L.A.; et al. Carbon Capture and Storage (CCS): The Way Forward. Energy Environ. Sci. 2018, 11, 1062–1176. [Google Scholar] [CrossRef]
- Guo, W.; Gómez, J.E.; Cristòfol, À.; Xie, J.; Kleij, A.W. Catalytic Transformations of Functionalized Cyclic Organic Carbonates. Angew. Chem. Int. Ed. 2018, 57, 13735–13747. [Google Scholar] [CrossRef]
- Gao, W.; Liang, S.; Wang, R.; Jiang, Q.; Zhang, Y.; Zheng, Q.; Xie, B.; Toe, C.Y.; Zhu, X.; Wang, J.; et al. Industrial Carbon Dioxide Capture and Utilization: State of the Art and Future Challenges. Chem. Soc. Rev. 2020, 49, 8584–8686. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lamb, K.J.; North, M. Recent Developments in Organocatalysed Transformations of Epoxides and Carbon Dioxide into Cyclic Carbonates. Green Chem. 2020, 23, 77–118. [Google Scholar] [CrossRef]
- Sreejyothi, P.; Mandal, S.K. From CO2 Activation to Catalytic Reduction: A Metal-Free Approach. Chem. Sci. 2020, 11, 10571–10593. [Google Scholar] [CrossRef]
- Yan, S.-S.; Fu, Q.; Liao, L.-L.; Sun, G.-Q.; Ye, J.-H.; Gong, L.; Bo-Xue, Y.-Z.; Yu, D.-G. Transition Metal-Catalyzed Carboxylation of Unsaturated Substrates with CO2. Coord. Chem. Rev. 2018, 374, 439–463. [Google Scholar] [CrossRef]
- Yang, Y.; Lee, J.-W. Toward Ideal Carbon Dioxide Functionalization. Chem. Sci. 2019, 10, 3905–3926. [Google Scholar] [CrossRef]
- Shaikh, R.R.; Pornpraprom, S.; D’Elia, V. Catalytic Strategies for the Cycloaddition of Pure, Diluted, and Waste CO2 to Epoxides under Ambient Conditions. ACS Catal. 2018, 8, 419–450. [Google Scholar] [CrossRef]
- Dabral, S.; Schaub, T. The Use of Carbon Dioxide (CO2) as a Building Block in Organic Synthesis from an Industrial Perspective. Adv. Synth. Catal. 2019, 361, 223–246. [Google Scholar] [CrossRef]
- Martinez, A.S.; Hauzenberger, C.; Sahoo, A.R.; Csendes, Z.; Hoffmann, H.; Bica, K. Continuous Conversion of Carbon Dioxide to Propylene Carbonate with Supported Ionic Liquids. ACS Sustain. Chem. Eng. 2018, 6, 13131–13139. [Google Scholar] [CrossRef]
- Bui, T.Q.; Konwar, L.J.; Samikannu, A.; Nikjoo, D.; Mikkola, J.-P. Mesoporous Melamine-Formaldehyde Resins as Efficient Heterogeneous Catalysts for Continuous Synthesis of Cyclic Carbonates from Epoxides and Gaseous CO2. ACS Sustain. Chem. Eng. 2020, 8, 12852–12869. [Google Scholar] [CrossRef]
- Zanda, N.; Sobolewska, A.; Alza, E.; Kleij, A.W.; Pericàs, M.A. Organocatalytic and Halide-Free Synthesis of Glycerol Carbonate under Continuous Flow. ACS Sustain. Chem. Eng. 2021, 9, 4391–4397. [Google Scholar] [CrossRef]
- Hou, J.; Ee, A.; Cao, H.; Ong, H.; Xu, J.; Wu, J. Visible-Light-Mediated Metal-Free Difunctionalization of Alkenes with CO2 and Silanes or C(Sp3)-H Alkanes. Angew. Chem. Int. Ed. 2018, 57, 17220–17224. [Google Scholar] [CrossRef]
- Wu, X.; Wang, M.; Xie, Y.; Chen, C.; Li, K.; Yuan, M.; Zhao, X.; Hou, Z. Carboxymethyl Cellulose Supported Ionic Liquid as a Heterogeneous Catalyst for the Cycloaddition of CO2 to Cyclic Carbonate. Appl. Catal. A Gen. 2016, 519, 146–154. [Google Scholar] [CrossRef]
- Sathe, A.A.; Nambiar, A.M.K.; Rioux, R.M. Synthesis of Cyclic Organic Carbonates via Catalytic Oxidative Carboxylation of Olefins in Flow Reactors. Catal. Sci. Technol. 2016, 7, 84–89. [Google Scholar] [CrossRef]
- Rigo, D.; Calmanti, R.; Perosa, A.; Selva, M.; Fiorani, G. Diethylene Glycol/NaBr Catalyzed CO2 Insertion into Terminal Epoxides: From Batch to Continuous Flow. Chemcatchem 2021, 13, 2005–2016. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, A.; Liu, X.; Xu, J.; Wang, Y.; Mumford, K.; Stevens, G.W.; Fei, W. Kinetic Study of Highly Efficient CO2 Fixation into Propylene Carbonate Using a Continuous-Flow Reactor. Chem. Eng. Process.-Process Intensif. 2021, 159, 108235. [Google Scholar] [CrossRef]
- North, M.; Villuendas, P.; Young, C. A Gas-Phase Flow Reactor for Ethylene Carbonate Synthesis from Waste Carbon Dioxide. Chem. Eur. J. 2009, 15, 11454–11457. [Google Scholar] [CrossRef]
- Zhao, Y.; Yao, C.; Chen, G.; Yuan, Q. Highly Efficient Synthesis of Cyclic Carbonate with CO2 Catalyzed by Ionic Liquid in a Microreactor. Green Chem. 2012, 15, 446–452. [Google Scholar] [CrossRef]
- Wu, Y.; Ding, Y.; Xu, J.; Wang, Y.; Mumford, K.; Stevens, G.W.; Fei, W. Efficient Fixation of CO2 into Propylene Carbonate with [BMIM]Br in a Continuous-Flow Microreaction System. Green Energy Environ. 2021, 6, 291–297. [Google Scholar] [CrossRef]
- Kozak, J.A.; Wu, J.; Su, X.; Simeon, F.; Hatton, T.A.; Jamison, T.F. Bromine-Catalyzed Conversion of CO2 and Epoxides to Cyclic Carbonates under Continuous Flow Conditions. J. Am. Chem. Soc. 2013, 135, 18497–18501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Su, D.; Xiao, L.; Wu, W. Immobilized Protic Ionic Liquids: Efficient Catalysts for CO2 Fixation with Epoxides. J. CO2 Util. 2017, 17, 37–42. [Google Scholar] [CrossRef]
- Rehman, A.; Fernández, A.M.L.; Resul, M.F.M.G.; Harvey, A. Kinetic Investigations of Styrene Carbonate Synthesis from Styrene Oxide and CO2 Using a Continuous Flow Tube-in-Tube Gas-Liquid Reactor. J. CO2 Util. 2018, 24, 341–349. [Google Scholar] [CrossRef]
- Seo, H.; Katcher, M.H.; Jamison, T.F. Photoredox Activation of Carbon Dioxide for Amino Acid Synthesis in Continuous Flow. Nat. Chem. 2017, 9, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, W.; Lyu, Y.; Chen, X.; Li, C.; Zhang, Y.; Song, X.; Ding, Y. Highly Recyclable Polymer Supported Ionic Liquids as Efficient Heterogeneous Catalysts for Batch and Flow Conversion of CO2 to Cyclic Carbonates. RSC Adv. 2017, 7, 2836–2841. [Google Scholar] [CrossRef]
- James, B.R.; Boissonnault, J.A.; Wong-Foy, A.G.; Matzger, A.J.; Sanford, M.S. Structure Activity Relationships in Metal–Organic Framework Catalysts for the Continuous Flow Synthesis of Propylene Carbonate from CO2 and Propylene Oxide. RSC Adv. 2018, 8, 2132–2137. [Google Scholar] [CrossRef]
- Zanda, N.; Primitivo, L.; Chaudhari, M.; Kleij, A.W.; Pericàs, M.À. Organocatalytic N -Formylation of Amines by CO2 in Batch and Continuous Flow. Org. Chem. Front. 2022, 10, 375–381. [Google Scholar] [CrossRef]
- Wu, J.; Kozak, J.A.; Simeon, F.; Hatton, T.A.; Jamison, T.F. Mechanism-Guided Design of Flow Systems for Multicomponent Reactions: Conversion of CO2 and Olefins to Cyclic Carbonates. Chem. Sci. 2014, 5, 1227–1231. [Google Scholar] [CrossRef]
- Zanda, N.; Zhou, L.; Alza, E.; Kleij, A.W.; Pericàs, M.À. Continuous Organocatalytic Flow Synthesis of 2-Substituted Oxazolidinones Using Carbon Dioxide. Green Chem. 2022, 24, 4628–4633. [Google Scholar] [CrossRef]
- Seo, H.; Nguyen, L.V.; Jamison, T.F. Using Carbon Dioxide as a Building Block in Continuous Flow Synthesis. Adv. Synth. Catal. 2019, 361, 247–264. [Google Scholar] [CrossRef]
- Rehman, A.; Saleem, F.; Javed, F.; Ikhlaq, A.; Ahmad, S.W.; Harvey, A. Recent Advances in the Synthesis of Cyclic Carbonates via CO2 Cycloaddition to Epoxides. J. Environ. Chem. Eng. 2021, 9, 105113. [Google Scholar] [CrossRef]
- Schäffner, B.; Schäffner, F.; Verevkin, S.P.; Börner, A. Organic Carbonates as Solvents in Synthesis and Catalysis. Chem. Rev. 2010, 110, 4554–4581. [Google Scholar] [CrossRef]
- Beattie, C.; North, M.; Villuendas, P. Proline-Catalysed Amination Reactions in Cyclic Carbonate Solvents. Molecules 2011, 16, 3420–3432. [Google Scholar] [CrossRef] [PubMed]
- Xu, K. Nonaqueous Liquid Electrolytes for Lithium-Based Rechargeable Batteries. Chem. Rev. 2004, 104, 4303–4418. [Google Scholar] [CrossRef]
- Zhang, S.S. A Review on Electrolyte Additives for Lithium-Ion Batteries. J. Power Sources 2006, 162, 1379–1394. [Google Scholar] [CrossRef]
- Besse, V.; Camara, F.; Voirin, C.; Auvergne, R.; Caillol, S.; Boutevin, B. Synthesis and Applications of Unsaturated Cyclocarbonates. Polym. Chem. 2013, 4, 4545–4561. [Google Scholar] [CrossRef]
- Pyo, S.-H.; Persson, P.; Mollaahmad, M.A.; Sörensen, K.; Lundmark, S.; Hatti-Kaul, R. Cyclic Carbonates as Monomers for Phosgene- and Isocyanate-Free Polyurethanes and Polycarbonates. Pure Appl. Chem. 2011, 84, 637–661. [Google Scholar] [CrossRef]
- Pescarmona, P.P.; Taherimehr, M. Challenges in the Catalytic Synthesis of Cyclic and Polymeric Carbonates from Epoxides and CO2. Catal. Sci. Technol. 2012, 2, 2169–2187. [Google Scholar] [CrossRef]
- von der Assen, N.; Sternberg, A.; Kätelhön, A.; Bardow, A. Environmental Potential of Carbon Dioxide Utilization in the Polyurethane Supply Chain. Faraday Discuss. 2015, 183, 291–307. [Google Scholar] [CrossRef]
- Fukuoka, S.; Kawamura, M.; Komiya, K.; Tojo, M.; Hachiya, H.; Hasegawa, K.; Aminaka, M.; Okamoto, H.; Fukawa, I.; Konno, S. A Novel Non-Phosgene Polycarbonate Production Process Using by-Product CO2 as Starting Material. Green Chem. 2003, 5, 497–507. [Google Scholar] [CrossRef]
- Clements, J.H. Reactive Applications of Cyclic Alkylene Carbonates. Ind. Eng. Chem. Res. 2003, 42, 663–674. [Google Scholar] [CrossRef]
- Dondoni, A.; Massi, A. Decoration of Dihydropyrimidine and Dihydropyridine Scaffolds with Sugars via Biginelli and Hantzsch Multicomponent Reactions: An Efficient Entry to a Collection of Artificial Nucleosides. Mol. Divers. 2003, 6, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Büttner, H.; Longwitz, L.; Steinbauer, J.; Wulf, C.; Werner, T. Recent Developments in the Synthesis of Cyclic Carbonates from Epoxides and CO2. Top. Curr. Chem. 2017, 375, 50. [Google Scholar] [CrossRef] [PubMed]
- Cokoja, M.; Wilhelm, M.E.; Anthofer, M.H.; Herrmann, W.A.; Kühn, F.E. Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide by Using Organocatalysts. Chemsuschem 2015, 8, 2436–2454. [Google Scholar] [CrossRef]
- Martín, C.; Fiorani, G.; Kleij, A.W. Recent Advances in the Catalytic Preparation of Cyclic Organic Carbonates. ACS Catal. 2015, 5, 1353–1370. [Google Scholar] [CrossRef]
- Comerford, J.W.; Ingram, I.D.V.; North, M.; Wu, X. Sustainable Metal-Based Catalysts for the Synthesis of Cyclic Carbonates Containing Five-Membered Rings. Green Chem. 2014, 17, 1966–1987. [Google Scholar] [CrossRef]
- Alves, M.; Grignard, B.; Mereau, R.; Jerome, C.; Tassaing, T.; Detrembleur, C. Organocatalyzed Coupling of Carbon Dioxide with Epoxides for the Synthesis of Cyclic Carbonates: Catalyst Design and Mechanistic Studies. Catal. Sci. Technol. 2017, 7, 2651–2684. [Google Scholar] [CrossRef]
- Yang, C.; Chen, Y.; Xu, P.; Yang, L.; Zhang, J.; Sun, J. Facile Synthesis of Zinc Halide-Based Ionic Liquid for Efficient Conversion of Carbon Dioxide to Cyclic Carbonates. Mol. Catal. 2020, 480, 110637. [Google Scholar] [CrossRef]
- Calmanti, R.; Sargentoni, N.; Selva, M.; Perosa, A. One-Pot Tandem Catalytic Epoxidation-CO2 Insertion of Monounsaturated Methyl Oleate to the Corresponding Cyclic Organic Carbonate. Catalysts 2021, 11, 1477. [Google Scholar] [CrossRef]
- Xu, A.; Chen, Z.; Jin, L.; Chu, B.; Lu, J.; He, X.; Yao, Y.; Li, B.; Dong, L.; Fan, M. Quaternary Ammonium Salt Functionalized MIL-101-NH2(Cr) as a Bifunctional Catalyst for the Cycloaddition of CO2 with Epoxides to Produce Cyclic Carbonates. Appl. Catal. A Gen. 2021, 624, 118307. [Google Scholar] [CrossRef]
- Cai, X.; Tolvanen, P.; Virtanen, P.; Eränen, K.; Rahkila, J.; Leveneur, S.; Salmi, T. Kinetic Study of the Carbonation of Epoxidized Fatty Acid Methyl Ester Catalyzed over Heterogeneous Catalyst HBimCl-NbCl5/HCMC. Int. J. Chem. Kinet. 2021, 53, 1203–1219. [Google Scholar] [CrossRef]
- Li, X.; Sun, J.; Xue, M.; Yin, J. Catalytic Conversion of CO2 by Supported Ionic Liquid Prepared with Supercritical Fluid Deposition in a Continuous Fixed-Bed Reactor. J. CO2 Util. 2022, 64, 102168. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Su, Q.; Li, Y.; Deng, L.; Dong, L.; Fu, M.; Liu, S.; Cheng, W. Poly(Ionic Liquid) Materials Tailored by Carboxyl Groups for the Gas Phase-Conversion of Epoxide and CO2 into Cyclic Carbonates. J. CO2 Util. 2022, 60, 101976. [Google Scholar] [CrossRef]
- Comin, E.; Aquino, A.S.; Favero, C.; Mignoni, M.L.; de Souza, R.F.; de Souza, M.O.; Pergher, S.B.C.; Campos, C.X.d.S.; Bernardo-Gusmão, K. Cyclic Carbonate Synthesis via Cycloaddition of CO2 and Epoxides Catalysed by Beta Zeolites Containing Alkyl Imidazolium Ionic Liquids Used as Structure-Directing Agents. Mol. Catal. 2022, 530, 112624. [Google Scholar] [CrossRef]
- Hernández, E.; Santiago, R.; Moya, C.; Navarro, P.; Palomar, J. Understanding the CO2 Valorization to Propylene Carbonate Catalyzed by 1-Butyl-3-Methylimidazolium Amino Acid Ionic Liquids. J. Mol. Liq. 2021, 324, 114782. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, Z.; Zhang, X.; Chen, J.; Wang, Y.; Xu, J. Kinetic Study of CO2 Fixation into Propylene Carbonate with Water as Efficient Medium Using Microreaction System. Chin. J. Chem. Eng. 2022, 50, 247–253. [Google Scholar] [CrossRef]
- Shannon, M.S.; Bara, J.E. Properties of Alkylimidazoles as Solvents for CO2 Capture and Comparisons to Imidazolium-Based Ionic Liquids. Ind. Eng. Chem. Res. 2011, 50, 8665–8677. [Google Scholar] [CrossRef]
- Peng, J.; Deng, Y. Cycloaddition of Carbon Dioxide to Propylene Oxide Catalyzed by Ionic Liquids. New J. Chem. 2001, 25, 639–641. [Google Scholar] [CrossRef]
- Girard, A.-L.; Simon, N.; Zanatta, M.; Marmitt, S.; Gonçalves, P.; Dupont, J. Insights on Recyclable Catalytic System Composed of Task-Specific Ionic Liquids for the Chemical Fixation of Carbon Dioxide. Green Chem. 2014, 16, 2815–2825. [Google Scholar] [CrossRef]
- Li, W.; Cheng, W.; Yang, X.; Su, Q.; Dong, L.; Zhang, P.; Yi, Y.; Li, B.; Zhang, S. Synthesis of Cyclic Carbonate Catalyzed by DBU Derived Basic Ionic Liquids. Chin. J. Chem. 2018, 36, 293–298. [Google Scholar] [CrossRef]
- Meng, X.; Ju, Z.; Zhang, S.; Liang, X.; von Solms, N.; Zhang, X.; Zhang, X. Efficient Transformation of CO2 to Cyclic Carbonates Using Bifunctional Protic Ionic Liquids under Mild Conditions. Green Chem. 2019, 21, 3456–3463. [Google Scholar] [CrossRef]
- Galvan, M.; Selva, M.; Perosa, A.; Noè, M. Toward the Design of Halide- and Metal-Free Ionic-Liquid Catalysts for the Cycloaddition of CO2 to Epoxides. Asian J. Org. Chem. 2014, 3, 504–513. [Google Scholar] [CrossRef]
- Shi, T.-Y.; Wang, J.-Q.; Sun, J.; Wang, M.-H.; Cheng, W.-G.; Zhang, S.-J. Efficient Fixation of CO2 into Cyclic Carbonates Catalyzed by Hydroxyl-Functionalized Poly(Ionic Liquids). RSC Adv. 2013, 3, 3726–3732. [Google Scholar] [CrossRef]
- Sun, J.; Wang, J.; Cheng, W.; Zhang, J.; Li, X.; Zhang, S.; She, Y. Chitosan Functionalized Ionic Liquid as a Recyclable Biopolymer -Supported Catalyst for Cycloaddition of CO2. Green Chem. 2012, 14, 654–660. [Google Scholar] [CrossRef]
- Sun, J.; Han, L.; Cheng, W.; Wang, J.; Zhang, X.; Zhang, S. Efficient Acid–Base Bifunctional Catalysts for the Fixation of CO2 with Epoxides under Metal- and Solvent-Free Conditions. Chemsuschem 2011, 4, 502–507. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, S.; Cheng, W.; Ren, J. Hydroxyl-Functionalized Ionic Liquid: A Novel Efficient Catalyst for Chemical Fixation of CO2 to Cyclic Carbonate. Tetrahedron Lett. 2008, 49, 3588–3591. [Google Scholar] [CrossRef]
- Sun, J.; Cheng, W.; Fan, W.; Wang, Y.; Meng, Z.; Zhang, S. Reusable and Efficient Polymer-Supported Task-Specific Ionic Liquid Catalyst for Cycloaddition of Epoxide with CO2. Catal. Today 2009, 148, 361–367. [Google Scholar] [CrossRef]
- Sun, J.; Ren, J.; Zhang, S.; Cheng, W. Water as an Efficient Medium for the Synthesis of Cyclic Carbonate. Tetrahedron Lett. 2009, 50, 423–426. [Google Scholar] [CrossRef]
- Gou, H.; Ma, X.; Su, Q.; Liu, L.; Ying, T.; Qian, W.; Dong, L.; Cheng, W. Hydrogen Bond Donor Functionalized Poly(Ionic Liquid)s for Efficient Synergistic Conversion of CO2 to Cyclic Carbonates. Phys. Chem. Chem. Phys. 2020, 23, 2005–2014. [Google Scholar] [CrossRef]
- Kayahan, E.; Urbani, D.; Dambruoso, P.; Massi, A.; Braeken, L.; Gerven, T.V.; Leblebici, M.E. Overcoming Mass and Photon Transfer Limitations in a Scalable Reactor: Oxidation in an Aerosol Photoreactor. Chem. Eng. J. 2021, 408, 127357. [Google Scholar] [CrossRef]
- Urbani, D.; Rovegno, C.; Massi, A.; Leblebici, M.E.; Kayahan, E.; Polo, E.; Dambruoso, P. Efficiency in CO2-Utilization Strategies: The Case of Styrene Carbonate Synthesis in Microdroplets Conditions. J. CO2 Util. 2023, 67, 102328. [Google Scholar] [CrossRef]
- Pereira, M.P.; Martins, R.d.S.; de Oliveira, M.A.L.; Bombonato, F.I. Amino Acid Ionic Liquids as Catalysts in a Solvent-Free Morita-Baylis-Hillman Reaction. RSC Adv. 2018, 8, 23903–23913. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, J.S.; Anderson, R.C. A Highly Regioselective Conversion of Epoxides to Halohydrins by Lithium Halides. Tetrahedron Lett. 1991, 32, 3021–3024. [Google Scholar] [CrossRef]
- Massi, A.; Cavazzini, A.; Zoppo, L.D.; Pandoli, O.; Costa, V.; Pasti, L.; Giovannini, P.P. Toward the Optimization of Continuous-Flow Aldol and α-Amination Reactions by Means of Proline-Functionalized Silicon Packed-Bed Microreactors. Tetrahedron Lett. 2011, 52, 619–622. [Google Scholar] [CrossRef]
- Bortolini, O.; Caciolli, L.; Cavazzini, A.; Costa, V.; Greco, R.; Massi, A.; Pasti, L. Silica-Supported 5-(Pyrrolidin-2-Yl)Tetrazole: Development of Organocatalytic Processes from Batch to Continuous-Flow Conditions. Green Chem. 2012, 14, 992–1000. [Google Scholar] [CrossRef]
- Bortolini, O.; Cavazzini, A.; Giovannini, P.P.; Greco, R.; Marchetti, N.; Massi, A.; Pasti, L. A Combined Kinetic and Thermodynamic Approach for the Interpretation of Continuous-Flow Heterogeneous Catalytic Processes. Chem. Eur. J. 2013, 19, 7802–7808. [Google Scholar] [CrossRef]
- Greco, R.; Caciolli, L.; Zaghi, A.; Pandoli, O.; Bortolini, O.; Cavazzini, A.; Risi, C.D.; Massi, A. A Monolithic 5-(Pyrrolidin-2-Yl)Tetrazole Flow Microreactor for the Asymmetric Aldol Reaction in Water-Ethanol Solvent. React. Chem. Eng. 2015, 1, 183–193. [Google Scholar] [CrossRef]
- Ragno, D.; Carmine, G.D.; Brandolese, A.; Bortolini, O.; Giovannini, P.P.; Massi, A. Immobilization of Privileged Triazolium Carbene Catalyst for Batch and Flow Stereoselective Umpolung Processes. ACS Catal. 2017, 7, 6365–6375. [Google Scholar] [CrossRef]
- Warias, R.; Zaghi, A.; Heiland, J.J.; Piendl, S.K.; Gilmore, K.; Seeberger, P.H.; Massi, A.; Belder, D. An Integrated Lab-on-a-chip Approach to Study Heterogeneous Enantioselective Catalysts at the Microscale. Chemcatchem 2018, 10, 5382–5385. [Google Scholar] [CrossRef]
- Warias, R.; Ragno, D.; Massi, A.; Belder, D. A Visible-Light-Powered Polymerization Method for the Immobilization of Enantioselective Organocatalysts into Microreactors. Chem. Eur. J. 2020, 26, 13152–13156. [Google Scholar] [CrossRef]
- Risi, C.D.; Bortolini, O.; Brandolese, A.; Carmine, G.D.; Ragno, D.; Massi, A. Recent Advances in Continuous-Flow Organocatalysis for Process Intensification. React. Chem. Eng. 2020, 5, 1017–1052. [Google Scholar] [CrossRef]
- Ragno, D.; Leonardi, C.; Carmine, G.D.; Bortolini, O.; Brandolese, A.; Risi, C.D.; Massi, A. Regiodivergent Isosorbide Acylation by Oxidative N-Heterocyclic Carbene Catalysis in Batch and Continuous Flow. ACS Sustain. Chem. Eng. 2021, 9, 8295–8305. [Google Scholar] [CrossRef]
- Bortolini, O.; Cavazzini, A.; Dambruoso, P.; Giovannini, P.P.; Caciolli, L.; Massi, A.; Pacifico, S.; Ragno, D. Thiazolium-Functionalized Polystyrene Monolithic Microreactors for Continuous-Flow Umpolung Catalysis. Green Chem. 2013, 15, 2981–2992. [Google Scholar] [CrossRef]
- Leonardi, C.; Brandolese, A.; Preti, L.; Bortolini, O.; Polo, E.; Dambruoso, P.; Ragno, D.; Carmine, G.D.; Massi, A. Expanding the Toolbox of Heterogeneous Asymmetric Organocatalysts: Bifunctional Cyclopropenimine Superbases for Enantioselective Catalysis in Batch and Continuous Flow. Adv. Synth. Catal. 2021, 363, 5473–5485. [Google Scholar] [CrossRef]
- Westphal, H.; Warias, R.; Becker, H.; Spanka, M.; Ragno, D.; Gläser, R.; Schneider, C.; Massi, A.; Belder, D. Unveiling Organocatalysts Action-Investigating Immobilized Catalysts at Steady-State Operation via Lab-on-a-Chip Technology. Chemcatchem 2021, 13, 5089–5096. [Google Scholar] [CrossRef]
- Westphal, H.; Warias, R.; Weise, C.; Ragno, D.; Becker, H.; Spanka, M.; Massi, A.; Gläser, R.; Schneider, C.; Belder, D. An Integrated Resource-Efficient Microfluidic Device for Parallelised Studies of Immobilised Chiral Catalysts in Continuous Flow via Miniaturized LC/MS-Analysis. React. Chem. Eng. 2022, 7, 1936–1944. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, G.; Ye, C.; Yuan, Q. Gas-Liquid Two-Phase Flow in Microchannel at Elevated Pressure. Chem. Eng. Sci. 2013, 87, 122–132. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, G.; Moghtaderi, B.; Doroodchi, E. A Review of Microreactors Based on Slurry Taylor (Segmented) Flow. Chem. Eng. Sci. 2021, 247, 117040. [Google Scholar] [CrossRef]
- Meléndez, J.; North, M.; Villuendas, P. One-component catalysts for cyclic carbonate synthesis. Chem. Commun. 2009, 18, 2577–2579. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Shan, H.; Tian, T.; Wang, Y.; Yuan, D.; You, H.; Yao, Y. Conversion of CO2 into Cyclic Carbonates under Ambient Conditions Catalyzed by Rare-Earth Metal Complexes Bearing Poly(phenolato) Ligand. ACS Sustain. Chem. Eng. 2020, 8, 13185–13194. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).