
Structural, Spectroscopic, Electric and Magnetic Properties of New Trigonal K5FeHf(MoO4)6 Orthomolybdate

 , ,
, ,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results and Discussions
2.1. Subsolidus Phase Relations in the K2MoO4–Fe2(MoO4)3–Hf(MoO4)2 System
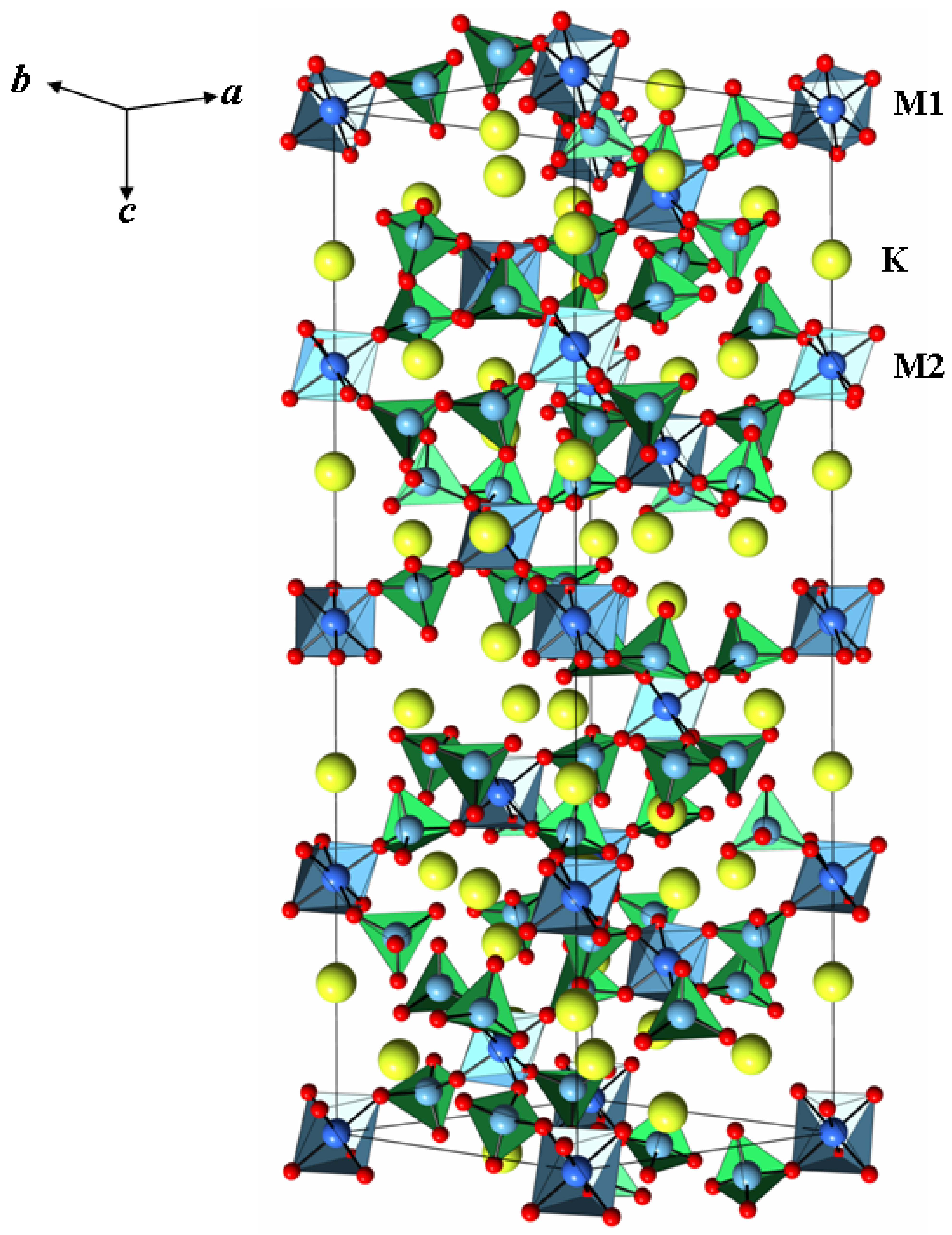
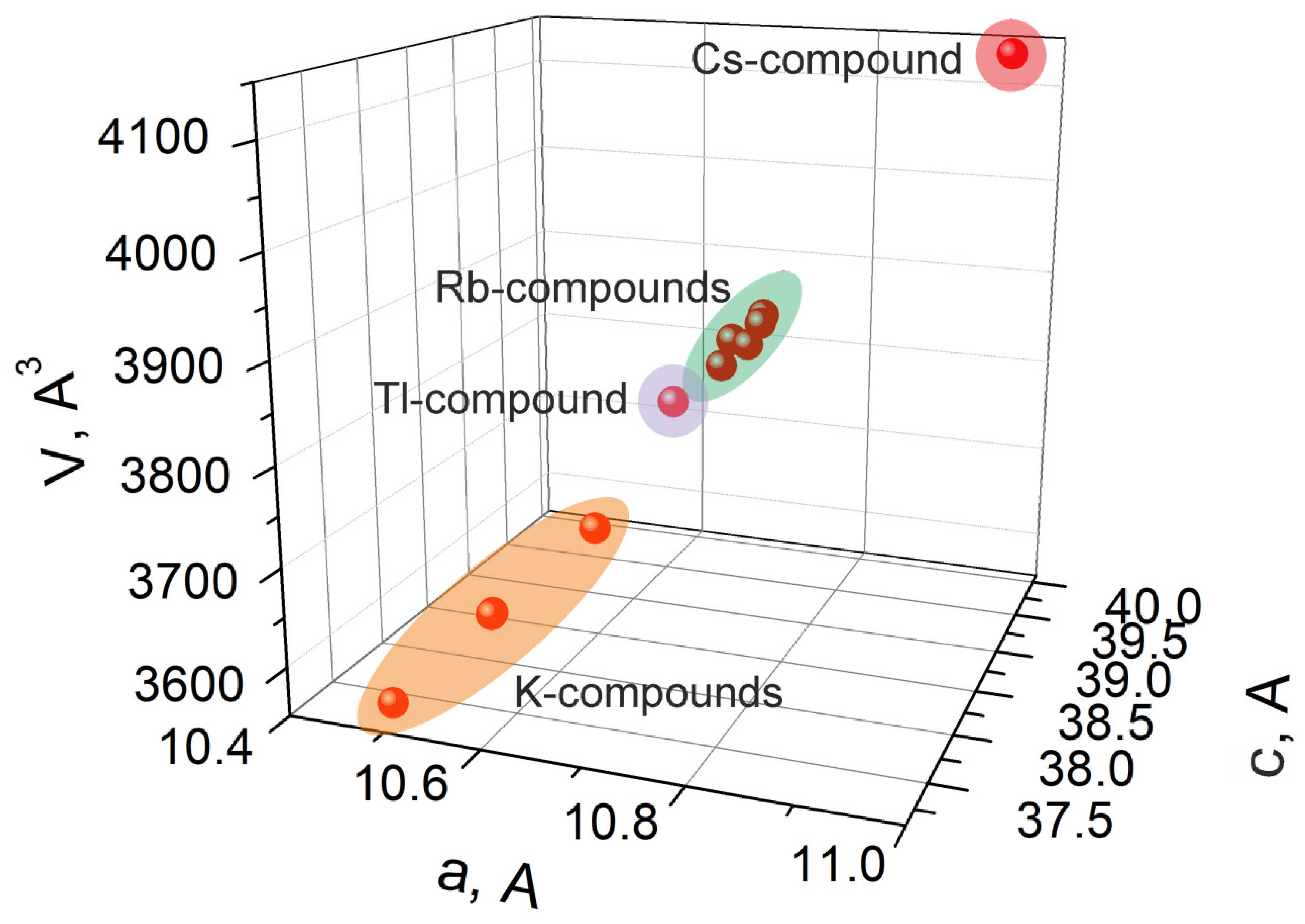
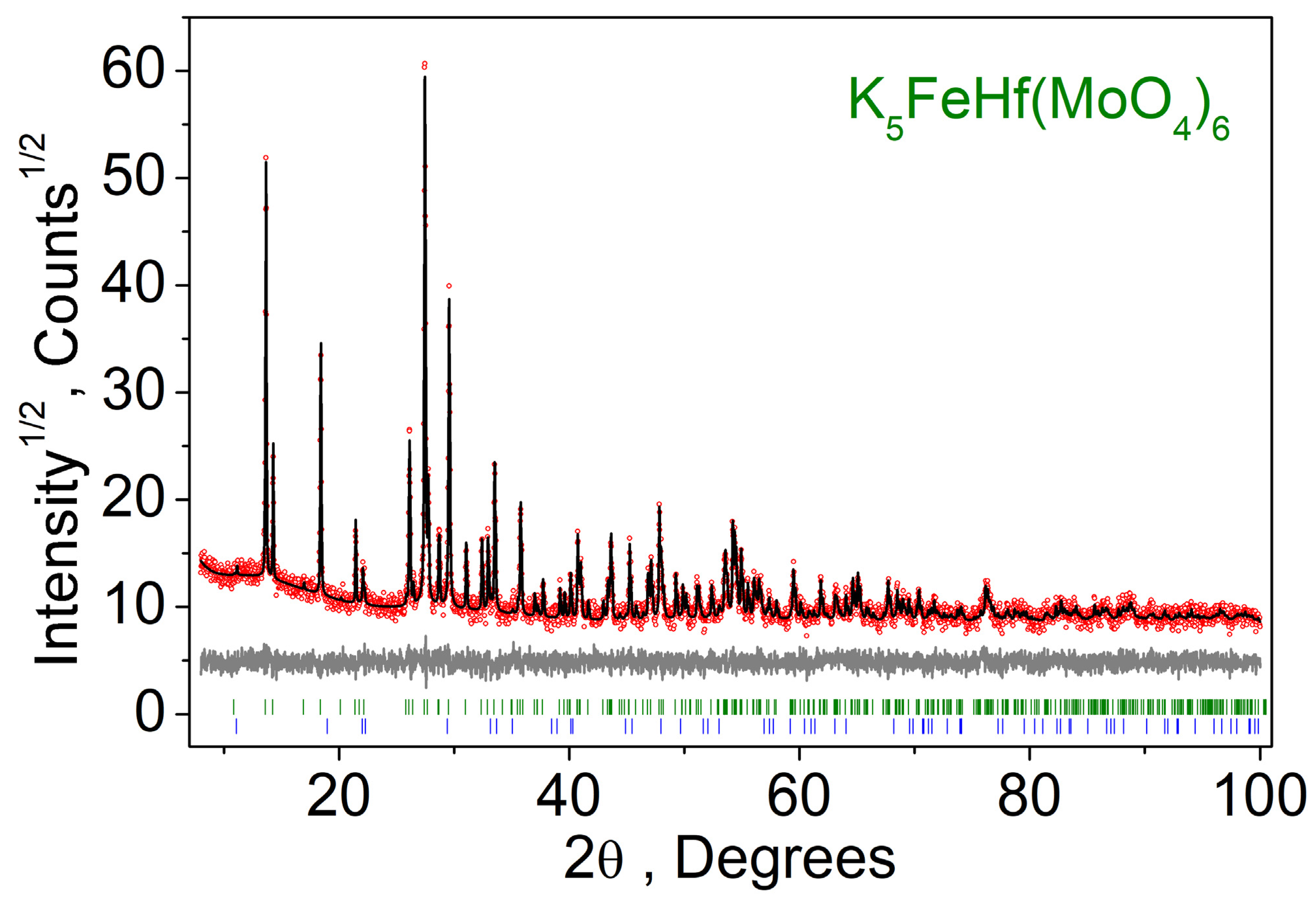
2.2. Crystal Structure of K5FeHf(MoO4)6

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | FeHfK5Mo6O24 |
|---|---|
| Formula wt, g × mol−1 | 1389.48 |
| Crystal system | trigonal |
| Space group | Rc |
| Crystal cell dimensions, Å | a = b = 10.4633(3) |
| c = 37.3113(9) | |
| Cell volume, Å3 | 3537.60(17) |
| Z | 6 |
| dcalc, g/cm3 | 3.913 |
| μ, mm−1 | 9.043 |
| F(000) | 3822 |
| Crystal size, mm | 0.06 × 0.05 × 0.04 |
| θ range for data collection, deg. | 2.50–27.49 |
| Index ranges | −13 ≤ h ≤ 13 −13 ≤ k ≤ 13 −32 ≤ l ≤ 48 |
| Ihkl coll | 8381 |
| Ihkl > 2σI (Rint) | 908 (Rint = 0.0371) |
| GOOF for F2 | 1.099 |
| R (I > 2σI) | R1 = 0.0137, wR2 = 0.0317 |
| R (Ihkl coll) | R1 = 0.0145, wR2 = 0.0319 |
| Largest diff peak, hole e/Å3 | 1.038 and −0.525 |
| Mo Tetrahedron | (Fe,Hf) Octahedra | ||
|---|---|---|---|
| Bond | d, Å | Bond | d, Å |
| Mo(1)-O(1) | 1.800(2) | M(1)–O(1) | 2.051(2) × 6 |
| Mo(1)-O(2) | 1.801(2) | M(2)–O(2) | 2.019(2) × 6 |
| Mo(1)-O(3) | 1.735(2) | K1 polyhedron | |
| Mo(1)-O(4) | 1.716(2) | K(1)–O | 2.795(2)–3.143(2) |
| 〈Mo(1)-O〉 | 1.763(2) | K2 polyhedron | |
| K(2)–O | 2.766(2)–3.015(2) | ||
| 3.278(2)–3.292(2) | |||
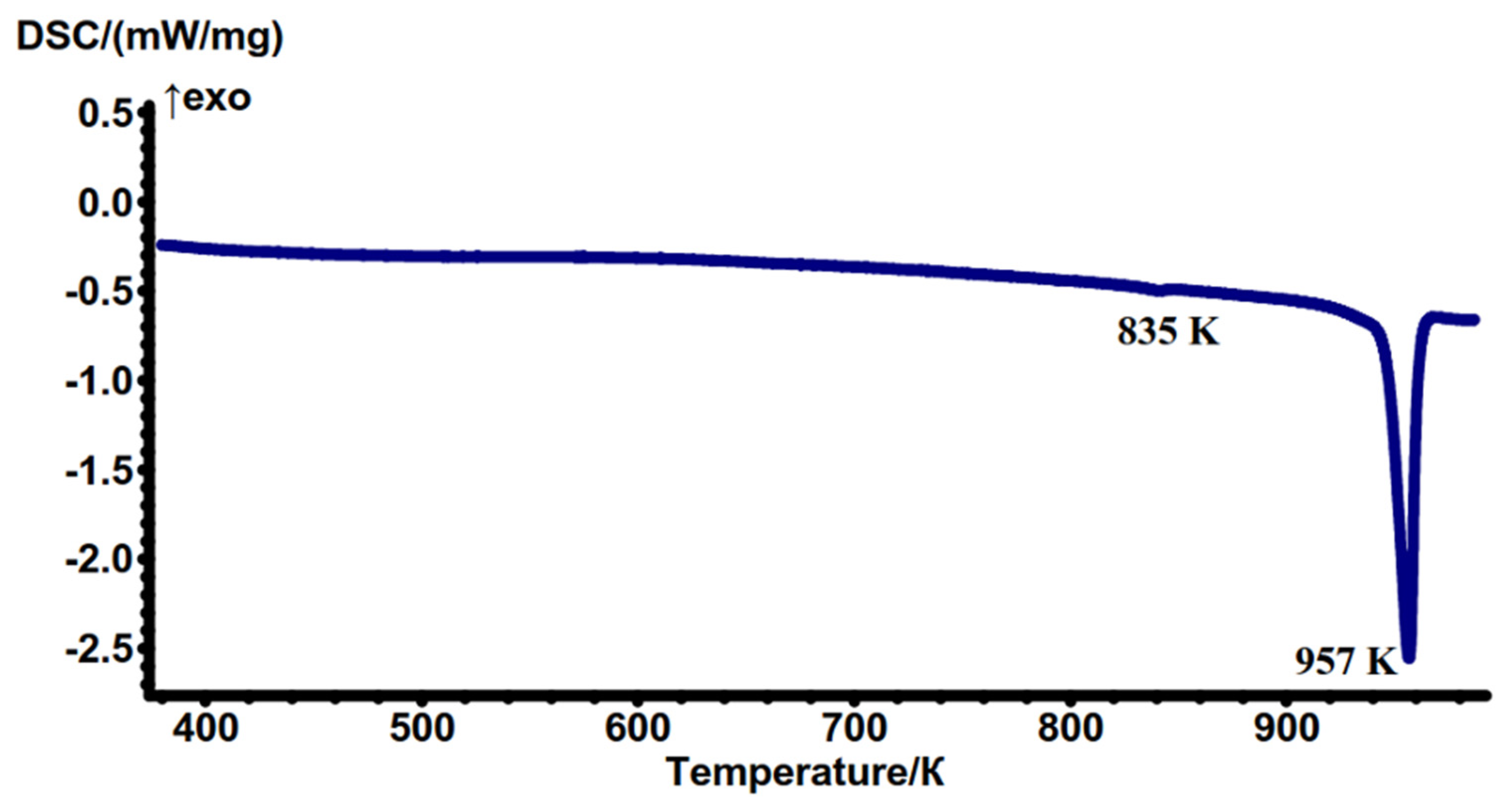
2.3. Thermochemical Properties
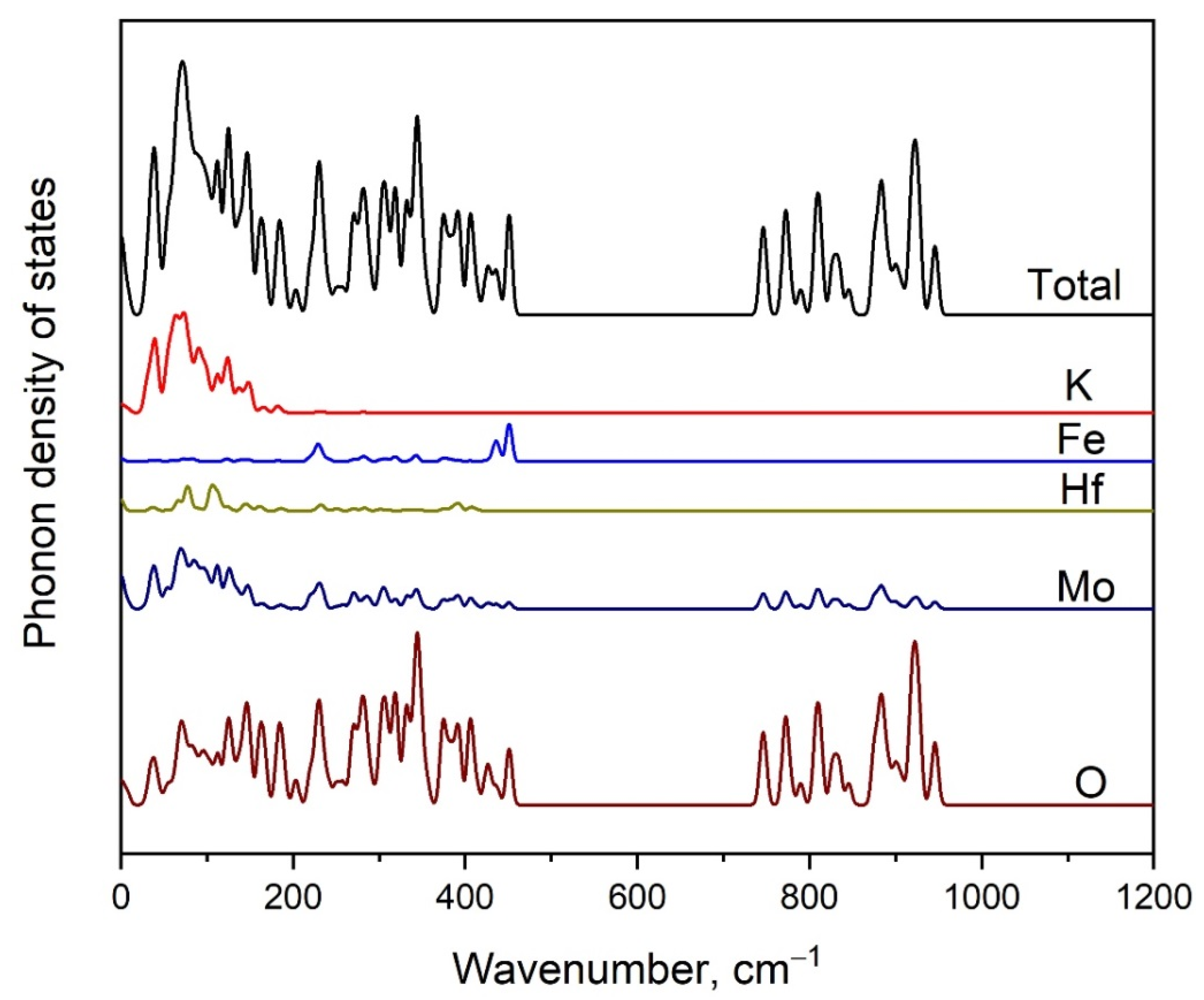
2.4. Vibration Dynamics
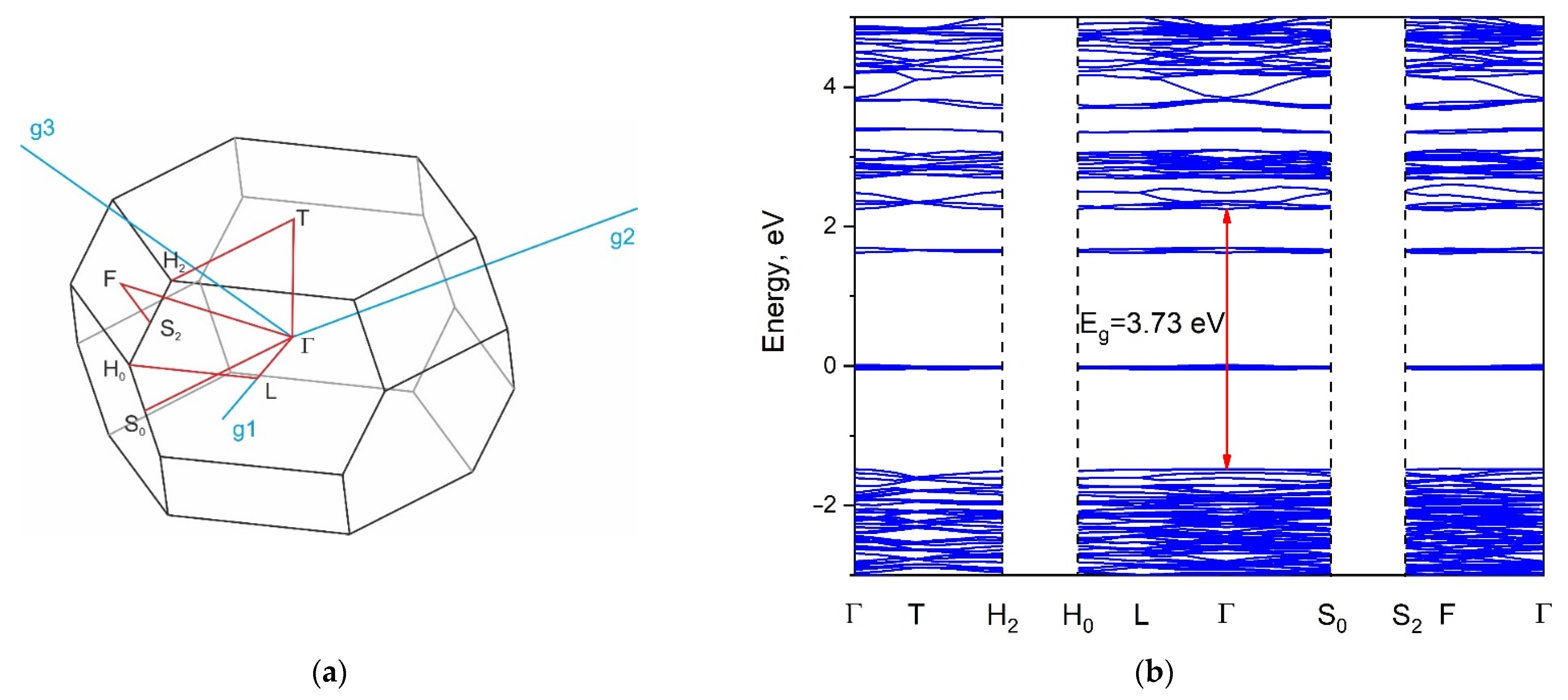
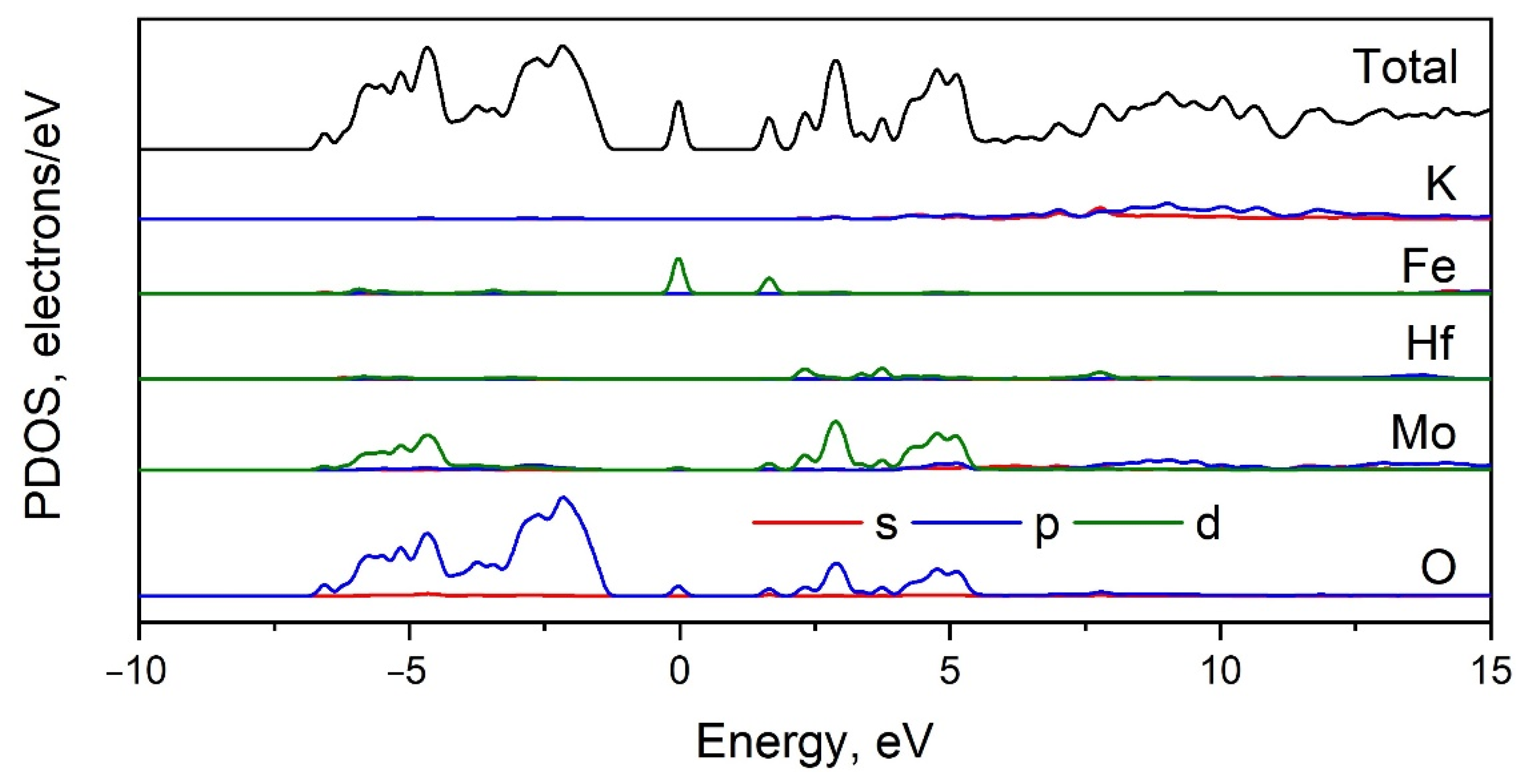
2.5. Calculated Electronic Structure
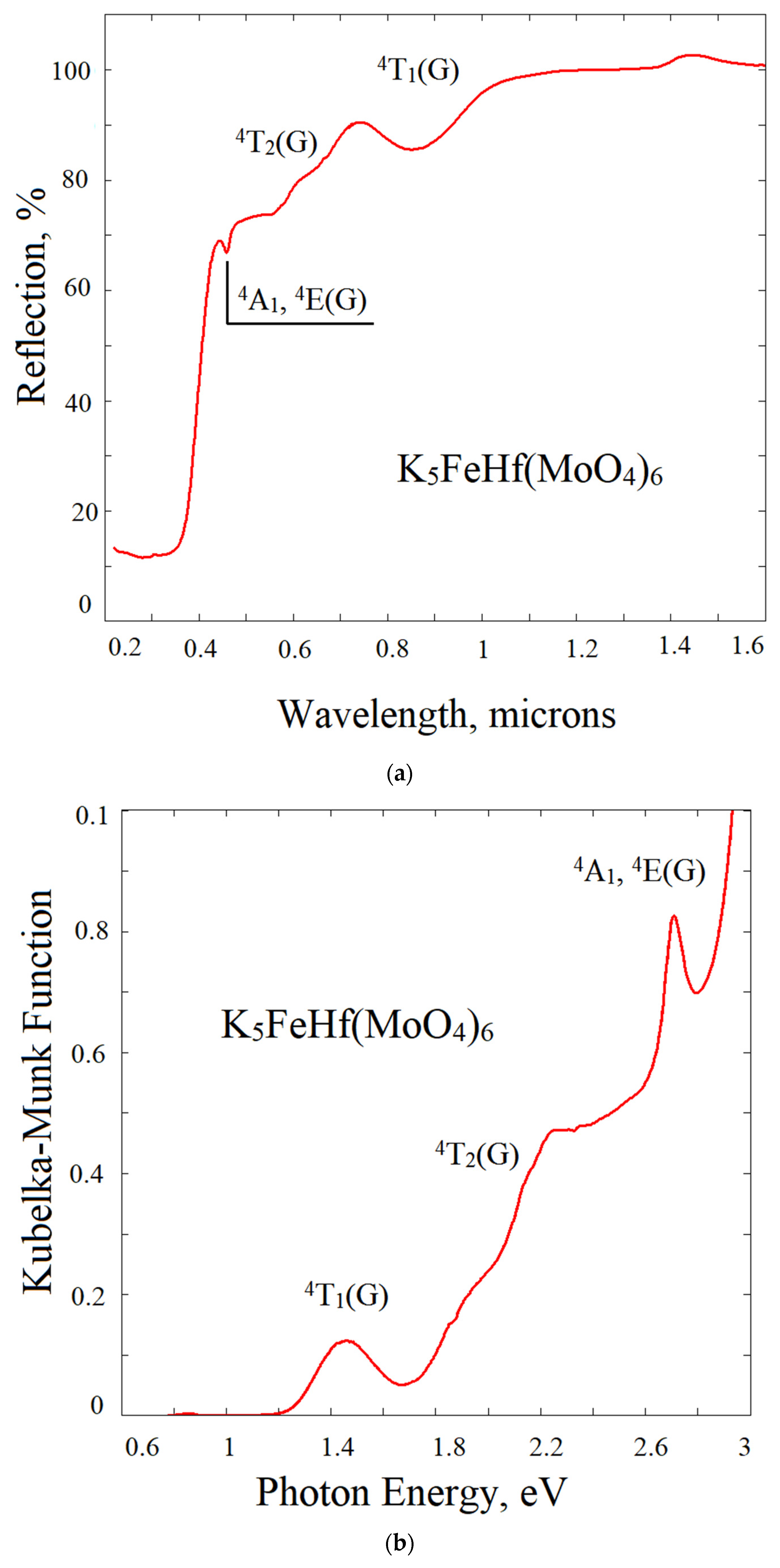
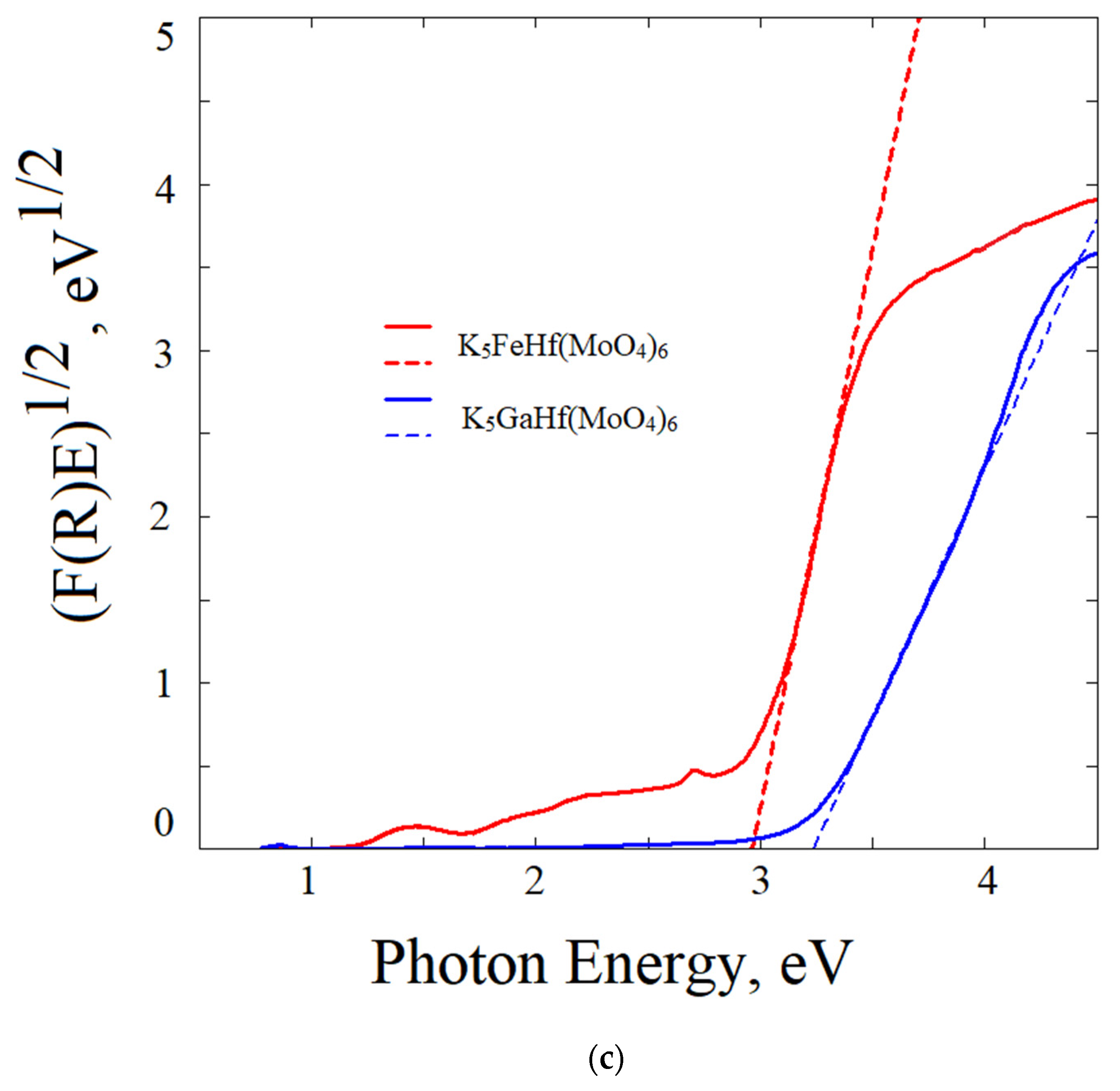
2.6. Reflection Spectrum and Experimental Bandgap
2.7. Electrical Properties
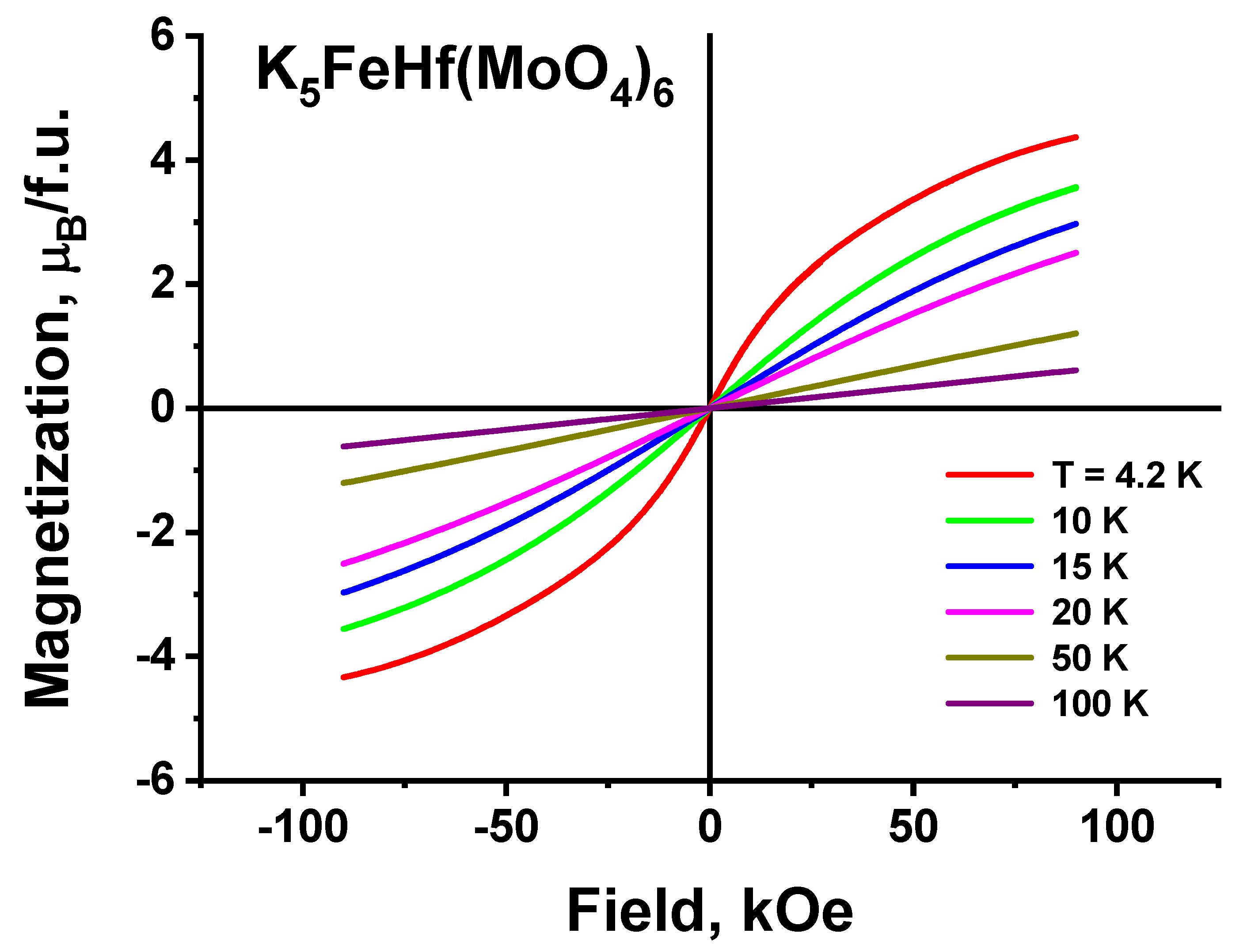
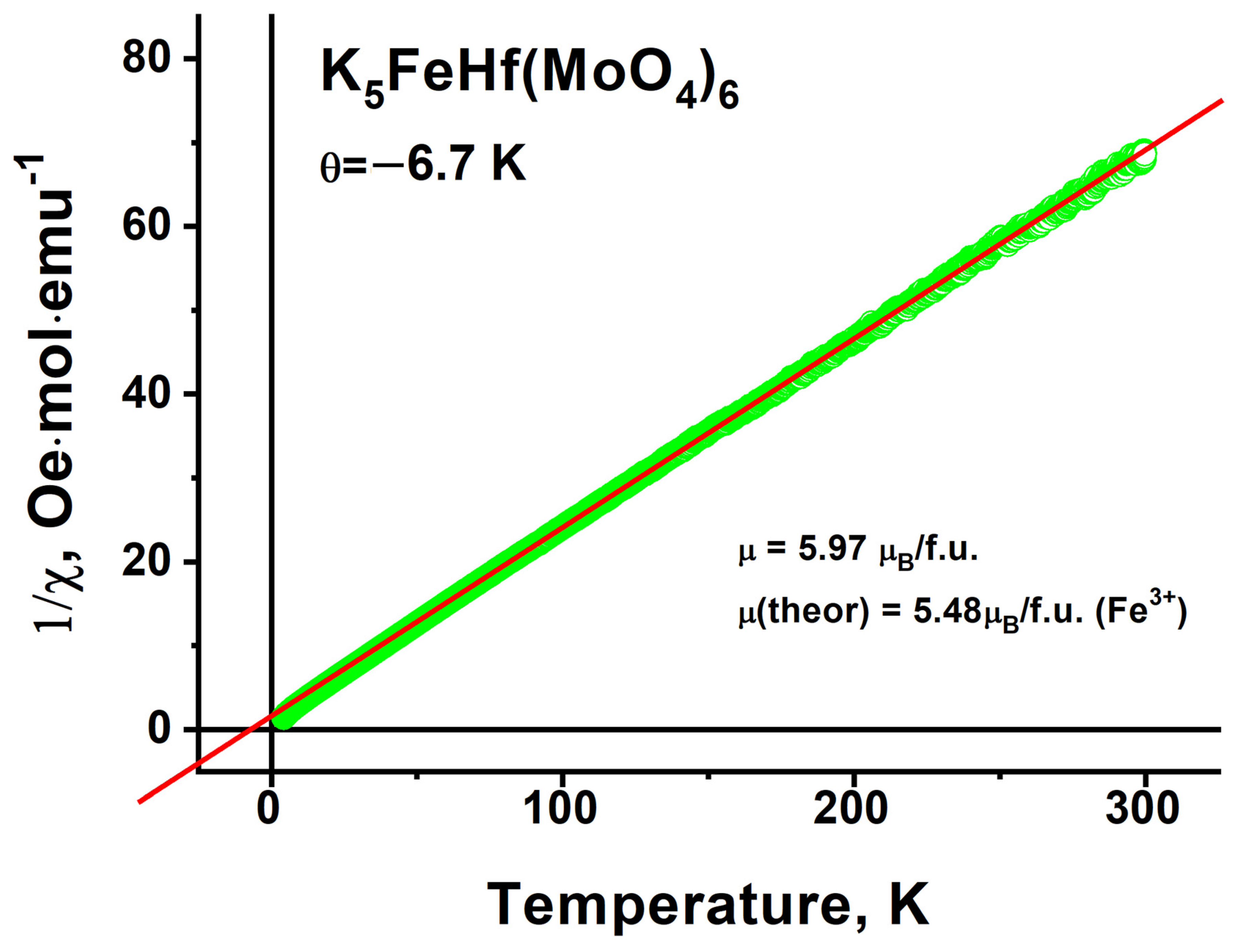
2.8. Magnetic Properties
3. Materials and Methods
4. Computation Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Klevtsova, R.F.; Bazarova, Z.G.; Glinskaya, L.A.; Alekseev, V.I.; Arkhincheeva, S.I.; Bazarov, B.G.; Klevtsov, P.V.; Fedorov, K.N. Synthesis of ternary potassium, magnesium, and zirconium molybdates. The crystal structure of K5(Mg0.5Zr1.5)·(MoO4)6. J. Struct. Chem. 1994, 35, 286–290. [Google Scholar] [CrossRef]
- García-Cortés, A.; Serrano, M.; Zaldo, C.; Cascales, C.; Strömqvist, G.; Pasiskevicius, V. Nonlinear refractive indices of disordered NaT(XO4)2 T=Y, La, Gd, Lu and Bi, X=Mo, W femtosecond laser crystals. Appl. Phys. B Laser Opt. 2008, 91, 507–510. [Google Scholar] [CrossRef]
- Zhou, D.; Clive, A.R.; Pang, L.-X.; Wang, H.; Wu, X.-G.; Guo, J.; Zhang, G.-Q.; Shui, L.; Yao, X. Microwave dielectric properties of Li2(M2+)2Mo3O12 and Li3(M3+)Mo3O12 (M = Zn, Ca, Al, and In) lyonsite-related-type ceramics with ultra-low sintering temperatures. J. Am. Ceram. Soc. 2011, 94, 802–805. [Google Scholar] [CrossRef]
- Atuchin, V.; Grossman, V.; Adichtchev, S.; Surovtsev, N.; Gavrilova, T.; Bazarov, B. Structural and vibrational properties of microcrystalline TlM(MoO4)2 (M = Nd, Pr) molybdates. Opt. Mater. 2012, 34, 812–816. [Google Scholar] [CrossRef]
- Shi, P.; Xia, Z.; Molokeev, M.S.; Atuchin, V.V. Crystal chemistry and luminescence properties of red-emitting CsGd1-xEux(MoO4)2 solid-solution phosphors. Dalton Trans. 2014, 43, 9669–9675. [Google Scholar] [CrossRef]
- Savina, A.; Atuchin, V.; Solodovnikov, S.; Solodovnikova, Z.A.; Krylov, A.; Maximovskiy, E.; Molokeev, M.; Oreshonkov, A.; Pugachev, A.; Khaikina, E. Synthesis, structural and spectroscopic properties of acentric triple molybdate Cs2NaBi(MoO4)3. J. Solid State Chem. 2015, 225, 53–58. [Google Scholar]
- Atuchin, V.; Aleksandrovsky, A.; Chimitova, O.; Diao, C.-P.; Gavrilova, T.; Kesler, V.; Molokeev, M.; Krylov, A.; Bazarov, B.; Bazarova, J.; et al. Electronic structure of β-RbSm(MoO4)2 and chemical bonding in molybdates. Dalton Trans. 2015, 44, 1805–1815. [Google Scholar] [CrossRef]
- Kurilchik, S.; Loiko, P.; Yasukevich, A.; Trifonov, V.; Volokitina, A.; Vilejshikova, E.; Kisel, V.; Mateos, X.; Baranov, A.; Goriev, O.; et al. Orthorombic Yb:Li2Zn2(MoO4)3—A novel potential crystal for broadly tunable lasers. Laser Phys. Lett. 2017, 14, 085804. [Google Scholar] [CrossRef]
- Solodovnikov, S.F.; Atuchin, V.V.; Solodovnikova, Z.A.; Khyzhun, O.Y.; Danylenko, M.I.; Pishchur, D.P.; Plyusnin, P.E.; Pugachev, A.M.; Gavrilova, T.A.; Yelisseyev, A.P.; et al. Synthesis, structural, thermal, and electronic properties of palmierite-related double molybdate α-Cs2Pb(MoO4)2. Inorg. Chem. 2017, 56, 3276–3286. [Google Scholar]
- Atuchin, V.V.; Aleksandrovsky, A.S.; Bazarov, B.G.; Bazarova, J.G.; Chimitova, O.D.; Denisenko, Y.G.; Gavrilova, T.A.; Krylovk, A.S.; Maximovskiy, E.A.; Molokeev, M.S.; et al. Exploration of structural, vibrational and spectroscopic properties of self-activated orthorhombic double molybdate RbEu(MoO4)2 with isolated MoO4 units. J. Alloys Compd. 2019, 785, 692–697. [Google Scholar] [CrossRef]
- Lim, C.-S.; Aleksandrovsky, A.; Molokeev, M.; Oreshonkov, A.; Atuchin, V. Structural and spectroscopic effects of Li+ substitution for Na+ in LixNa1-xCaGd0.5Ho0.05Yb0.45(MoO4)3 scheelite-type upconversion phosphors. Molecules 2021, 26, 7357. [Google Scholar] [CrossRef]
- Begam, K.; Taufiq-Yap, Y.; Michael, M.; Prabaharan, S. A new NASICON-type polyanion, LixNi2(MoO4)3 as 3-V class positive electrode material for rechargeable lithium batteries. Solid State Ion. 2004, 172, 47–52. [Google Scholar] [CrossRef]
- Chimitova, O.D.; Bazarov, B.G.; Fedorov, K.N.; Bazarova, Z.G. Electrical properties of triple molybdates Rb5LnHf(MoO4)6. Russ. J. Appl. Chem. 2008, 81, 1928–1929. [Google Scholar] [CrossRef]
- Sorokin, N.I. Ionic conductivity of double sodium-scandium and cesium-zirconium molybdates. Phys. Solid State 2009, 51, 1128–1130. [Google Scholar] [CrossRef]
- Mikhailova, D.; Sarapulova, A.; Voss, A.; Thomas, A.; Oswald, S.; Gruner, W.; Trots, D.M.; Bramnik, N.N.; Ehrenberg, H. Li3V(MoO4)3: A New Material for Both Li Extraction and Insertion. Chem. Mater. 2010, 22, 3165–3173. [Google Scholar] [CrossRef]
- Souilem, A.; Zid, M.F. Synthèse, étude et validation structurale d’un triple bis-molybdate en couches, Ag0.60Na0.40Fe(MoO4)2lié à yavapaiite. Acta Crystallogr. Sect. E Crystallogr. Commun. 2016, 72, 737–740. [Google Scholar] [CrossRef]
- Kotova, I.Y.; Solodovnikov, S.F.; Solodovnikova, Z.A.; Belov, D.A.; Stefanovich, S.Y.; Savina, A.A.; Khaikina, E.G. New series of triple molybdates AgA3R(MoO4)5 (A = Mg, R = Cr, Fe; A = Mn, R = Al, Cr, Fe, Sc, In) with framework structures and mobile silver ion sublattices. J. Solid State Chem. 2016, 238, 121–128. [Google Scholar] [CrossRef]
- Mhiri, M.; Badri, A.; Ben Amara, M. Synthesis and crystal structure of NaMgFe(MoO4)3. Acta Crystallogr. Sect. E Crystallogr. Commun. 2016, 72, 864–867. [Google Scholar] [CrossRef]
- Sorokin, N.I. Ionic conductivity of KMgCr(MoO4)3 molybdate. Crystallogr. Rep. 2017, 62, 416–418. [Google Scholar] [CrossRef]
- Jendoubi, I.; Ptak, M.; Pikul, A.; Chmielowiec, J.; Ciupa, A.; Mączka, M.; Zid, M.F. Synthesis, crystal structure, phonon, magnetic and electrical properties of new molybdate Na2Mn2(MoO4)3. J. Solid State Chem. 2019, 277, 738–750. [Google Scholar] [CrossRef]
- Grossman, V.; Adichtchev, S.V.; Atuchin, V.V.; Bazarov, B.G.; Bazarova, J.G.; Kuratieva, N.; Oreshonkov, A.S.; Pervukhina, N.V.; Surovtsev, N.V. Exploration of the structural and vibrational properties of the ternary molybdate Tl5BiHf(MoO4)6 with isolated MoO4 units and Tl+ conductivity. Inorg. Chem. 2020, 59, 12681–12689. [Google Scholar] [CrossRef]
- Chimitova, O.; Bazarov, B.; Bazarova, J.; Atuchin, V.; Azmi, R.; Sarapulova, A.; Mikhailova, D.; Balachandran, G.; Fiedler, A.; Geckle, U.; et al. The crystal growth and properties of novel magnetic double molybdate RbFe5(MoO4)7 with mixed Fe3+/Fe2+ states and 1D negative thermal expansion. CrystEngComm 2021, 23, 3297–3307. [Google Scholar] [CrossRef]
- BBazarov, G.; Klevtsova, R.; Bazarova, T.; Glinskaya, L.; Fedorov, K.; Bazarova, Z. Synthesis and crystal structure of triple molybdate K5InHf(MoO4)6. Russ. J. Inorg. Chem. 2005, 50, 1146–1149. [Google Scholar]
- Bazarov, B.G.; Klevtsova, R.F.; Chimitova, O.D.; Glinskaya, L.A.; Fedorov, K.N.; Tushinova, Y.L.; Bazarova, Z.G. Phase formation in the Rb2MoO4-Er2(MoO4)3-Hf(MoO4)2 system and the crystal structure of new triple molybdate Rb3ErHf(MoO4)6. Russ. J. Inorg. Chem. 2006, 51, 800–804. [Google Scholar]
- Romanova, E.Y.; Bazarov, B.G.; Klevtsova, R.F.; Glinskaya, L.A.; Tushinova, Y.L.; Fedorov, K.N.; Bazarova, Z.G. Phase formation in the K2MoO4-Lu2(MoO4)-Hf(MoO4)2 system and the structural study of triple molybdate K5LuHf(MoO4)6. Russ. J. Inorg. Chem. 2007, 52, 815–818. [Google Scholar] [CrossRef]
- Chimitova, O.D.; Bazarov, B.G.; Klevtsova, R.F.; Fedorov, K.N.; Glinskaya, L.A.; Kuznetsov, M.V.; Bazarova, Z.G. Synthesis, crystal structure, and electrical properties of the new ternary molybdate Rb5NdHf(MoO4)6. Russ. Chem. Bull. 2007, 56, 2135–2139. [Google Scholar]
- Bazarov, B.G.; Namsaraeva, T.V.; Klevtsova, R.F.; Anshits, A.; Vereshchagina, T.A.; Kurbatov, R.V.; Glinskaya, L.A.; Fedorov, K.N.; Bazarova, Z.G. Phase equilibrium in the Cs2MoO4-Bi2(MoO4)3-Zn(MoO4)2 system and the crystal structure of new triple molybdate Cs5BiZr(MoO4)6. Russ. J. Inorg. Chem. 2008, 53, 1484–1488. [Google Scholar] [CrossRef]
- Bazarov, B.G.; Chimitova, O.D.; Klevtsova, R.F.; Tushinova, Y.L.; Glinskaya, L.A.; Bazarova, Z.G. Crystal structure of a new ternary molybdate in the Rb2MoO4-Eu2(MoO4)3-Hf(MoO4)2 system. J. Struct. Chem. 2008, 49, 53–57. [Google Scholar] [CrossRef]
- Chimitova, O.; Bazarov, B.; Klevtsova, R.; Anshits, A.; Fedorov, K.; Dubentsov, A.; Vereshchagina, T.; Tushinova, Y.; Glinskaya, L.; Bazarova, Z.; et al. Crystal structure of triple molybdate in the Rb2MoO4–Nd2(MoO4)3–Zr(MoO4)2 system. J. Struct. Chem. 2010, 51, 173–176. [Google Scholar]
- Gongorova, L.; Bazarov, B.; Chimitova, O.; Anshits, A.; Vereschagina, T.; Klevtsova, R.; Glinskaya, L.; Bazarova, Z. Crystal structure of a new ternary molybdate Rb5CeZr(MoO4)6. J. Struct. Chem. 2012, 53, 329–333. [Google Scholar] [CrossRef]
- Grossman, V.G.; Bazarova, J.G.; Molokeev, M.S.; Bazarov, B.G. New triple molybdate K5ScHf(MoO4)6: Synthesis, properties, structure and phase equilibria in the M2MoO4–Sc2(MoO4)3–Hf(MoO4)2 (M = Li, K) systems. J. Solid State Chem. 2020, 283, 121143. [Google Scholar] [CrossRef]
- Grossman, V.G.; Bazarov, B.G.; Bazarova, Z.G. Subsolidus phase diagrams for the Tl2MoO4-Ln2(MoO4)3-Hf(MoO4)2 systems, where Ln = La-Lu. Russ. J. Inorg. Chem. 2008, 53, 1788–1794. [Google Scholar] [CrossRef]
- Logvinova, A.; Bazarov, B.; Tushinova, Y.; Bazarova, J. Phase Relations in the K2MoO4–Ln2(MoO4)3–Zr(MoO4)2 (Ln = La–Lu, Y) systems. Inorg. Mater. 2017, 53, 1286–1292. [Google Scholar] [CrossRef]
- Grossman, V.; Bazarov, B.; Bazarova, T.; Glinskay, L.; Bazarova, J.; Temuujin, J. Phase equilibria in the Tl2MoO4–Ho2(MoO4)3–Zr(MoO4)2 system and the crystal structure of Ho2Zr2(MoO4)7 and TlHoZr0.5(MoO4)3. J. Ceram. Proc. Res. 2017, 18, 875–881. [Google Scholar]
- Grossman, V.; Bazarov, B.; Bazarova, J. K5InHf(MoO4)6: A solid state conductor. IOP Conf. Ser. Earth Environ. Sci. 2019, 320, 012050. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A 1976, 32, 751–766. [Google Scholar] [CrossRef]
- Andersson, A.; Kalska, B.; Eyob, P.; Aernout, D.; Häggström, L.; Thomas, J. Lithium insertion into rhombohedral Li3Fe2(PO4)3. Solid State Ion. 2001, 140, 63–70. [Google Scholar] [CrossRef]
- Kotova, I.Y.; Kozhevnikova, N.M. Phase Relations and Electrical Properties of Phases in Systems Na2MoO4-AMoO4-R2(MoO4)3(A = Mg, Mn, Co, Ni; R = Cr, Fe). Russ. J. Appl. Chem. 2003, 76, 1572–1576. [Google Scholar] [CrossRef]
- Grossman, V.G.; Bazarov, B.G.; Klevtsova, R.F.; Glinskaya, L.A.; Bazarova, Z.G. Phase equilibria in the Tl2MoO4-Fe2(MoO4)3-Hf(MoO4)2 system and the crystal structure of ternary molybdate Tl(FeHf0.5)(MoO4)3. Russ. Chem. Bull. 2012, 61, 1546–1549. [Google Scholar] [CrossRef]
- Atuchin, V.; Vinnik, D.; Gavrilova, T.; Gudkova, S.; Isaenko, L.; Jiang, X.; Pokrovsky, L.; Prosvirin, I.; Mashkovtseva, L.; Lin, Z. Flux crystal growth and the electronic structure of BaFe12O19 hexaferrite. J. Phys. Chem. C 2016, 120, 5114–6123. [Google Scholar] [CrossRef]
- Vinnik, D.; Prosvirin, I.; Zhivulin, V.; Wang, N.; Jiang, X.; Trofimov, E.; Zaitseva, O.; Gudkova, S.; Nemrava, S.; Zherebtsov, D.; et al. Crystal growth, structural characteristics and electronic structure of Ba1-xPbxFe12O19 (x = 0.23–0.80) hexaferrites. J. Alloys Compd. 2020, 844, 156036. [Google Scholar] [CrossRef]
- Ramana, C.V.; Bandi, M.; Nair, A.; Manciu, F.S.; Sreenivasan, S.; Shutthanandan, V. Electronic structure, chemical bonding, and electrocatalytic activity of Ba(Fe0.7Ta0.3)O3−δ compounds. ACS Appl. Energy Mater. 2021, 4, 1313–1322. [Google Scholar] [CrossRef]
- Pinaeva, L.; Prosvirin, I.; Chesalov, Y.; Atuchin, V. High-temperature abatement of N2O over FeOx/CeO2-Al2O3 catalysts: The effects of oxygen mobility. Catalysts 2022, 12, 938. [Google Scholar] [CrossRef]
- Trunov, K.; Efremov, V.; Velikodny, Y. Crystal Chemistry and Properties of Double Molybdates and Tungstates; Nauka: Saint Petersburg, Russia, 1986. [Google Scholar]
- Mokhosoev, M.; Alekseev, F.; Lutsyk, V. State Diagrams of Molybdate and Tungstate Systems; Nauka: Novosibirsk, Russia, 1978. [Google Scholar]
- Klevtsova, R.; Klevtsov, P. Synthesis and crystal structure of the double moibdates KR(MoO4)2 for R3+ = Al, Sc, and Fe, and of the tungstate KSc(WO4)2. Sov. Phys. Crystallogr. (Engl. Transl.) 1970, 15, 953–959. [Google Scholar]
- Kadyrova, Y. Abstract of the Dissertation… Candidate of Chemical Sciences; Irkutsk State Iniv: Irkutsk, Russia, 2010; 24p. (In Russian) [Google Scholar]
- Lazoryak, B.; Efremov, V. The double molybdates Me5Tr(MoO4)4. Kristallografiya 1987, 32, 378–384. [Google Scholar]
- Tissot, R.G.; Rodriguez, M.A.; Sipola, D.L.; Voigt, J.A. X-ray powder diffraction study of synthetic Palmierite, K2Pb(SO4)2. Powder Diffr. 2001, 16, 92–97. [Google Scholar] [CrossRef]
- Otko, A.I.; Nesterenko, N.M.; Povstyanyi, L.V. Phenomenological approach to structural phase transitions in trigonal double molybdates and tungstates. Phys. Status Solidi (A) 1978, 46, 577–587. [Google Scholar] [CrossRef]
- Nesterenko, N.M.; Fomin, V.I. Soft modes and the raman spectrum in KSc(MoO4)2 at ferroelastic phase transitions. Phys. Status Solidi (A) 1979, 51, K101–K104. [Google Scholar] [CrossRef]
- Zapart, W.; Zapart, M.B. Incommensurate phase in KSc(MoO4)2 by EPR of Cr3+. Phys. Status Solidi (A) 1990, 121, K43–K45. [Google Scholar] [CrossRef]
- Zapart, W. Possibility of simultaneously incommensurate and ferroelastic phase in RbIn(MoO4)2 by EPR of Cr3+ ion. Phys. Status Solidi (A) 1990, 118, 447–454. [Google Scholar] [CrossRef]
- Zapart, M.B.; Zapart, W. Investigations of incommensurate phase in KSc(WO4)2. Phase Transit. 1993, 43, 173–178. [Google Scholar] [CrossRef]
- Svistov, L.E.; Smirnov, A.I.; Prozorova, L.A.; Petrenko, O.A.; Shapiro, A.Y.; Dem’Yanets, L.N. On the possible coexistence of spiral and collinear structures in antiferromagnetic KFe(MoO4)2. J. Exp. Theor. Phys. Lett. 2004, 80, 231–235. [Google Scholar] [CrossRef]
- Maczka, M.; Pietraszko, A.; Saraiva, G.D.; Filho, A.G.S.; Paraguassu, W.; Lemos, V.; A Perottoni, C.; Gallas, M.R.; Freire, P.T.C.; E Tomaszewski, P.; et al. High pressure effects on the structural and vibrational properties of antiferromagnetic KFe(MoO4)2. J. Phys. Condens. Matter 2005, 17, 6285–6300. [Google Scholar] [CrossRef]
- Maczka, M.; Pietraszko, A.; Paraguassu, W.; Filho, A.G.S.; Freire, P.T.C.; Filho, J.M.; Hanuza, J. Structural and vibrational properties of K3Fe(MoO4)2(Mo2O7)—A novel layered molybdate. J. Phys. Condens. Matter 2009, 21, 095402. [Google Scholar] [CrossRef]
- Zolotova, E. Abstract of the Dissertation… Candidate of Chemical Sciences; Novosibirsk State University: Novosibirsk, Russia, 1986; 25p. (In Russian) [Google Scholar]
- Tushinova, Y.; Bazarova, J.; Arhincheeva, S. Phase equilibria in R2(MoO4)3–Zr(MoO4)2 systems. In All-Russian Scientific Conference with International Participation; BSC SB RAS Press: Ulan-Ude, Russia, 2002; pp. 90–91. [Google Scholar]
- Bruker AXS. TOPAS V4: General Profile and Structure Analysis Software for Powder Diffraction Data—User’s Manual; Bruker AXS: Karlsruhe, Germany, 2008. [Google Scholar]
- Gongorova, L. Phase Equilibrium, Structure and Properties of New Molybdates in the Systems Rb2MoO4–Ln2(MoO4)3–Zr(MoO4)2 (Ln = La–Lu). Ph.D. Thesis, Irkutsk State University, Irkutsk, Russia, 2012. [Google Scholar]
- Lim, C.S.; Aleksandrovsky, A.; Atuchin, V.; Molokeev, M.; Oreshonkov, A. Microwave-Employed Sol–Gel Synthesis of Scheelite-Type Microcrystalline AgGd(MoO4)2:Yb3+/Ho3+ Upconversion Yellow Phosphors and Their Spectroscopic Properties. Crystals 2020, 10, 1000. [Google Scholar] [CrossRef]
- Tillard, M.; Granier, D.; Daenens, L.; Reibel, C.; Armand, P. Crystal structure, Raman characterization, and magnetic properties of the hydrate RbYb(MoO4)2, H2O. J. Solid State Chem. 2022, 309, 122953. [Google Scholar] [CrossRef]
- Armand, P.; Granier, D.; Reibel, C.; Tillard, M. Growth, crystal structure, and properties of the Li3Ba2Ln3(MoO4)8 (Ln = Er, Tm) molybdates. Solid State Sci. 2022, 123, 106796. [Google Scholar] [CrossRef]
- Reshak, A.H.; Alahmed, Z.A.; Bila, J.; Atuchin, V.V.; Bazarov, B.G.; Chimitova, O.D.; Molokeev, M.S.; Prosvirin, I.P.; Yelisseyev, A.P. Exploration of the Electronic Structure of Monoclinic α-Eu2(MoO4)3: DFT-Based Study and X-ray Photoelectron Spectroscopy. J. Phys. Chem. C 2016, 120, 10559–10568. [Google Scholar] [CrossRef]
- Malakhovskii, A.V.; Sukhachev, A.L.; Vasil’Ev, A.D.; Leont’Ev, A.A.; Kartashev, A.V.; Temerov, V.L.; Gudim, I.A. Nature of optical properties of GdFe3(BO3)4 and GdFe2.1Ga0.9(BO3)4 crystals and other 3d5 antiferromagnets. Eur. Phys. J. B 2012, 85, 80. [Google Scholar] [CrossRef]
- Voronkova, V.; Kharitonova, E.; Orlova, E.; Gorshkov, N.; Goffman, V. Synthesis and Unusual Properties of Tetragonal Pb-Contained Oxymolybdates Based on La2MoO6. Eur. J. Inorg. Chem. 2017, 47, 5582–5587. [Google Scholar] [CrossRef]
- Zouaoui, M.; Jendoubi, I.; Zid, M.F.; Bourguiba, N.F. Synthesis, crystal structure and physico-chemical investigations of a new lyonsite molybdate Na0.24Ti1.44(MoO4)3. J. Solid State Chem. 2021, 300, 122221. [Google Scholar] [CrossRef]
- Fedorov, D.; Buzlukov, A.; Baklanova, Y.; Suetin, D.; Tyutyunnik, A.; Korona, D.; Maksimova, L.; Ogloblichev, V.; Denisova, T.; Medvedeva, N. Sodium diffusion in scheelite-type Na2Zr(MoO4)3 and Na4Zr(MoO4)4. Ceram. Int. 2022, 48, 32338–32347. [Google Scholar] [CrossRef]
- Ben Nasr, W.; Mahmoud, A.; Boschini, F.; Ben Rhaiem, A. Optical and AC conductivity studies on Li2-xRbx MoO4 (x = 0, 0.5, 1) compounds. J. Alloys Compd. 2019, 788, 522–532. [Google Scholar] [CrossRef]
- Nasri, R.; Larbi, T.; Amlouk, M.; Zid, M.F. Highly efficient K0.4Na3.6Co(MoO4)3 new alluaudite type structure for photocatalytic degradation of methylene blue and green diamine B dyes. J. Mater. Sci. Mater. Electron. 2019, 30, 9642–9651. [Google Scholar] [CrossRef]
- Grossman, V.G.; Bazarova, J.G.; Molokeev, M.S.; Bazarov, B.G. Thallium ionic conductivity of new thallium indium hafnium molybdate ceramics. Ionics 2020, 26, 6157–6165. [Google Scholar] [CrossRef]
- Grossman, V.G.; Molokeev, M.S.; Bazarov, B.G.; Bazarova, J.G. Potassium and thallium conductors with a trigonal structure in the M2MoO4–Cr2(MoO4)3–Hf(MoO4)2 (M = K, Tl) systems: Synthesis, structure, and ionic conductivity. J. Alloys Compd. 2021, 873, 159828. [Google Scholar] [CrossRef]
- Grossman, V.G.; Molokeev, M.S.; Bazarova, J.G.; Bazarov, B.G.; Sorokin, N.I. Structural, thermal and electrical studies of thallium-scandium-hafnium(zirconium) molybdates. J. Solid State Chem. 2021, 307, 122832. [Google Scholar] [CrossRef]
- Namsaraeva, T.; Bazarov, B.; Mikhailova, D.; Kuratieva, N.; Sarapulova, A.; Senyshyn, A.; Ehrenberg, H. Orthomolybdates in the Cs-FeII,III-Mo-O system: Cs4Fe(MoO4)3, Cs2Fe2(MoO4)3 and CsFe5(MoO4)7. Eur. J. Inorg. Chem. 2011, 7, 2832–2841. [Google Scholar] [CrossRef]
- Chen, H.-Y. The crystal structure and twinning behavior of ferric molybdate, Fe2(MoO4)3. Mater. Res. Bull. 1979, 14, 1583–1590. [Google Scholar] [CrossRef]
- Auray, M.; Quarton, M.; Tarte, P. Crystal data for two molybdates M(MoO4)2 with M = Zr, Hf. Power Diffr. 1987, 2, 36–39. [Google Scholar] [CrossRef]
- Bruker AXS Inc. APEX2 (Version 1.08), SAINT (Version 7.03), and SADABS (Version 2.11). Bruker Advanced X-ray Solutions; Bruker AXS Inc.: Madison, WI, USA, 2004. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. Kristallogr. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]


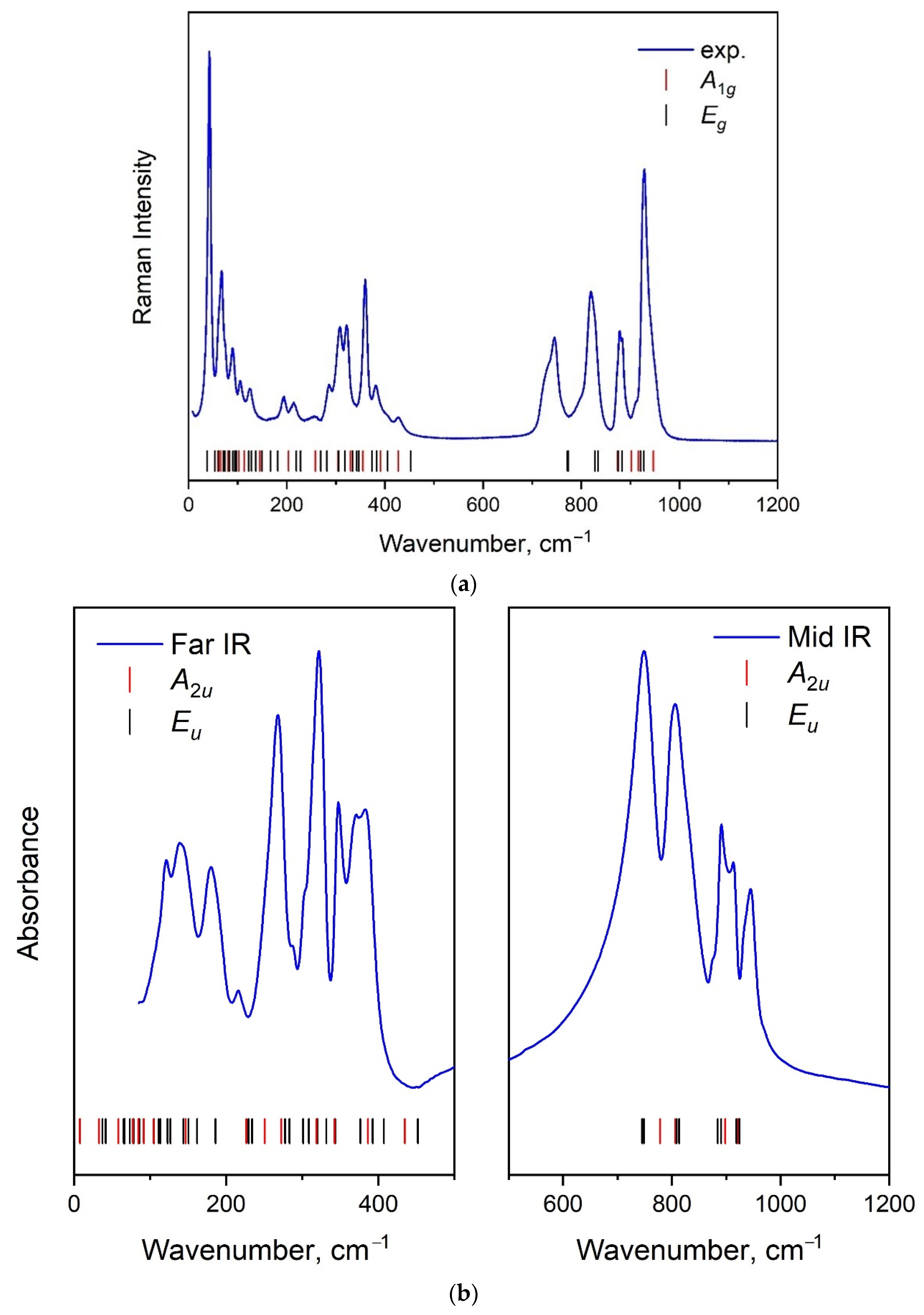



| Molecular Group Symmetry. | Site Symmetry. | Space Group Symmetry | ||
|---|---|---|---|---|
| Td | C1 | D3d | ||
| ν1, A1 | → | A | → | A1g + A1u + A2g + A2u + 2Eg + 2Eu |
| ν2, E | → | 2A | → | 2A1g + 2A1u + 2A2g + 2A2u + 4Eg + 4Eu |
| ν3, ν4, F2 | → | 3A | → | 3A1g + 3A1u + 3A2g + 3A2u + 6Eg + 6Eu |
| Molybdate | Conductivity, S cm−1 at 773 K | The Activation Energy of Electrotransfer, eV | Reference |
|---|---|---|---|
| K5CrHf(MoO4)6 | 5.22 × 10−4 | 0.73 (640–830 K) | [73] |
| K5FeHf(MoO4)6 | 4 × 10−4 | 0.8 (740–893 K) | this study |
| K5ScHf(MoO4)6 | 2.6 × 10−4 | 0.84 (750–920 K) | [31] |
| K5InHf(MoO4)6 | 1.59 × 10−4 | 0.85 (750–920 K) | [35] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grossman, V.; Atuchin, V.; Bazarov, B.G.; Aleksandrovsky, A.; Eremin, E.; Krylov, A.; Kuratieva, N.; Bazarova, J.G.; Maximov, N.; Molokeev, M.; et al. Structural, Spectroscopic, Electric and Magnetic Properties of New Trigonal K5FeHf(MoO4)6 Orthomolybdate. Molecules 2023, 28, 1629. https://doi.org/10.3390/molecules28041629
Grossman V, Atuchin V, Bazarov BG, Aleksandrovsky A, Eremin E, Krylov A, Kuratieva N, Bazarova JG, Maximov N, Molokeev M, et al. Structural, Spectroscopic, Electric and Magnetic Properties of New Trigonal K5FeHf(MoO4)6 Orthomolybdate. Molecules. 2023; 28(4):1629. https://doi.org/10.3390/molecules28041629
Chicago/Turabian StyleGrossman, Victoria, Victor Atuchin, Bair G. Bazarov, Aleksandr Aleksandrovsky, Evgeniy Eremin, Alexander Krylov, Natalia Kuratieva, Jibzema G. Bazarova, Nikolai Maximov, Maxim Molokeev, and et al. 2023. "Structural, Spectroscopic, Electric and Magnetic Properties of New Trigonal K5FeHf(MoO4)6 Orthomolybdate" Molecules 28, no. 4: 1629. https://doi.org/10.3390/molecules28041629
APA StyleGrossman, V., Atuchin, V., Bazarov, B. G., Aleksandrovsky, A., Eremin, E., Krylov, A., Kuratieva, N., Bazarova, J. G., Maximov, N., Molokeev, M., Oreshonkov, A., Pervukhina, N., & Shestakov, N. (2023). Structural, Spectroscopic, Electric and Magnetic Properties of New Trigonal K5FeHf(MoO4)6 Orthomolybdate. Molecules, 28(4), 1629. https://doi.org/10.3390/molecules28041629