
Urea Decomposition Mechanism by Dinuclear Nickel Complexes
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
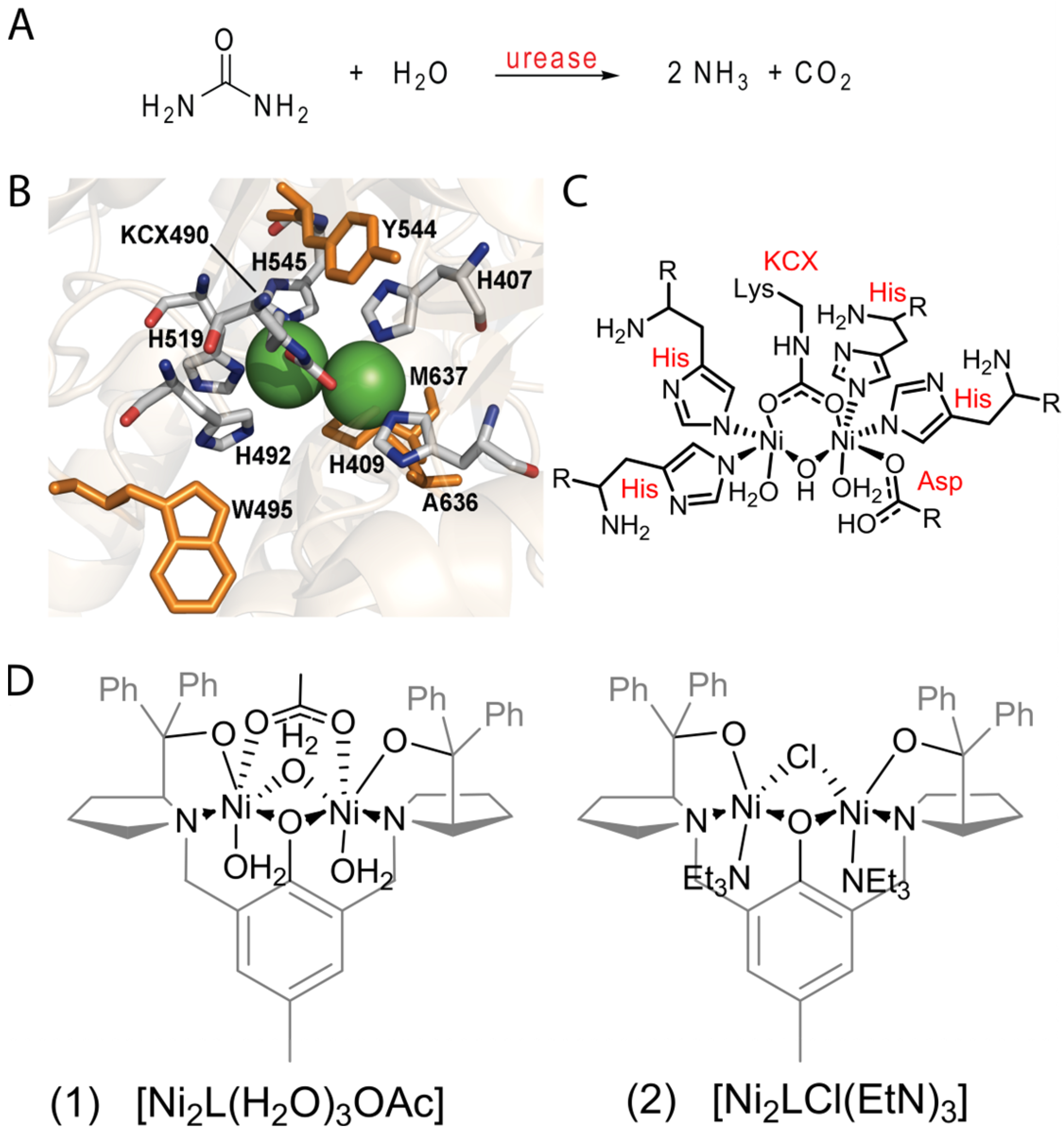
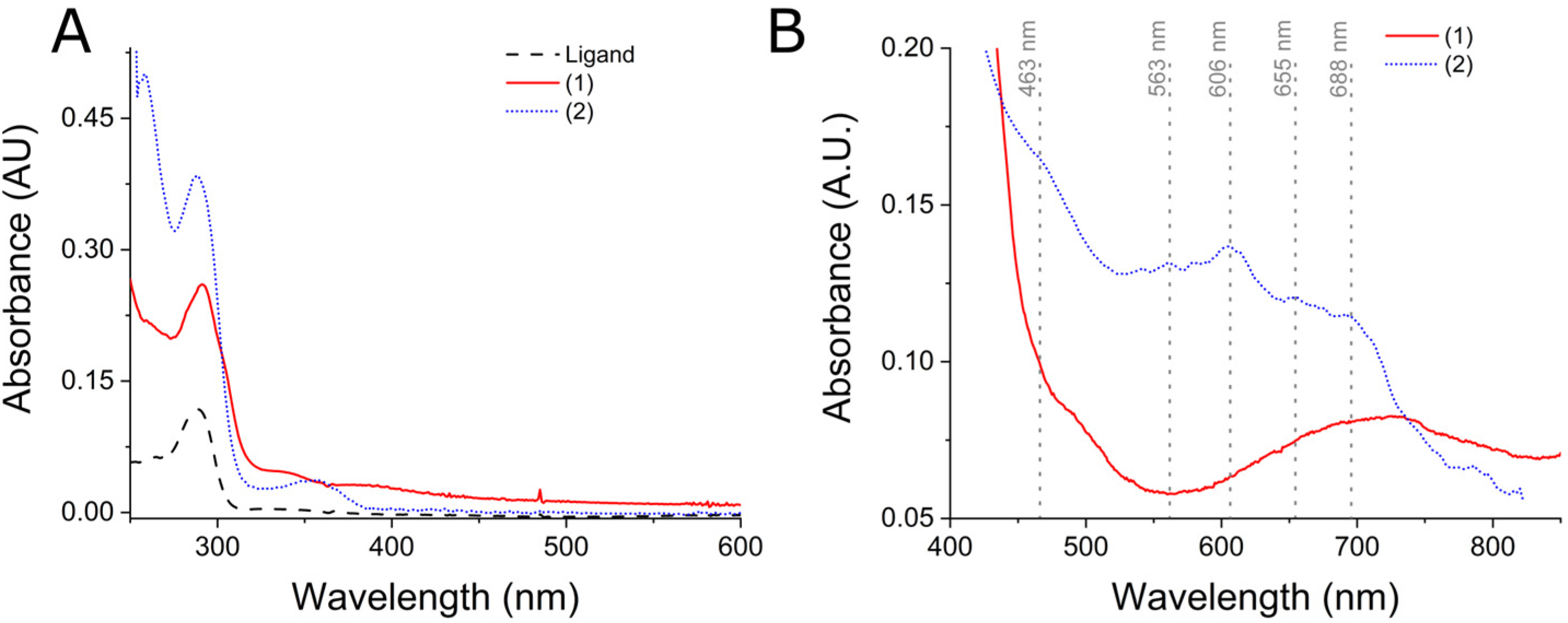
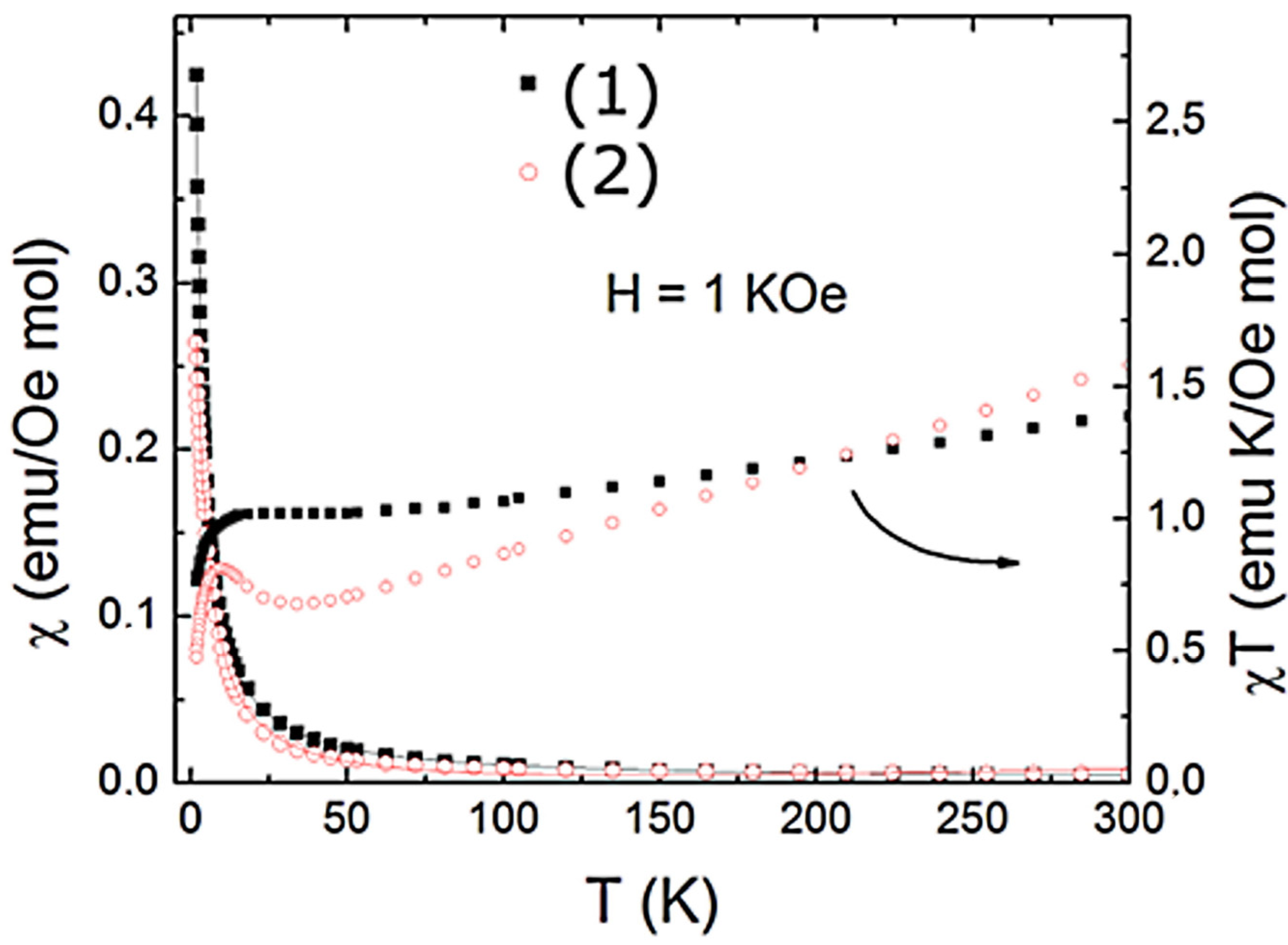
2.1. General Characteristics of (1) and (2) Complexes
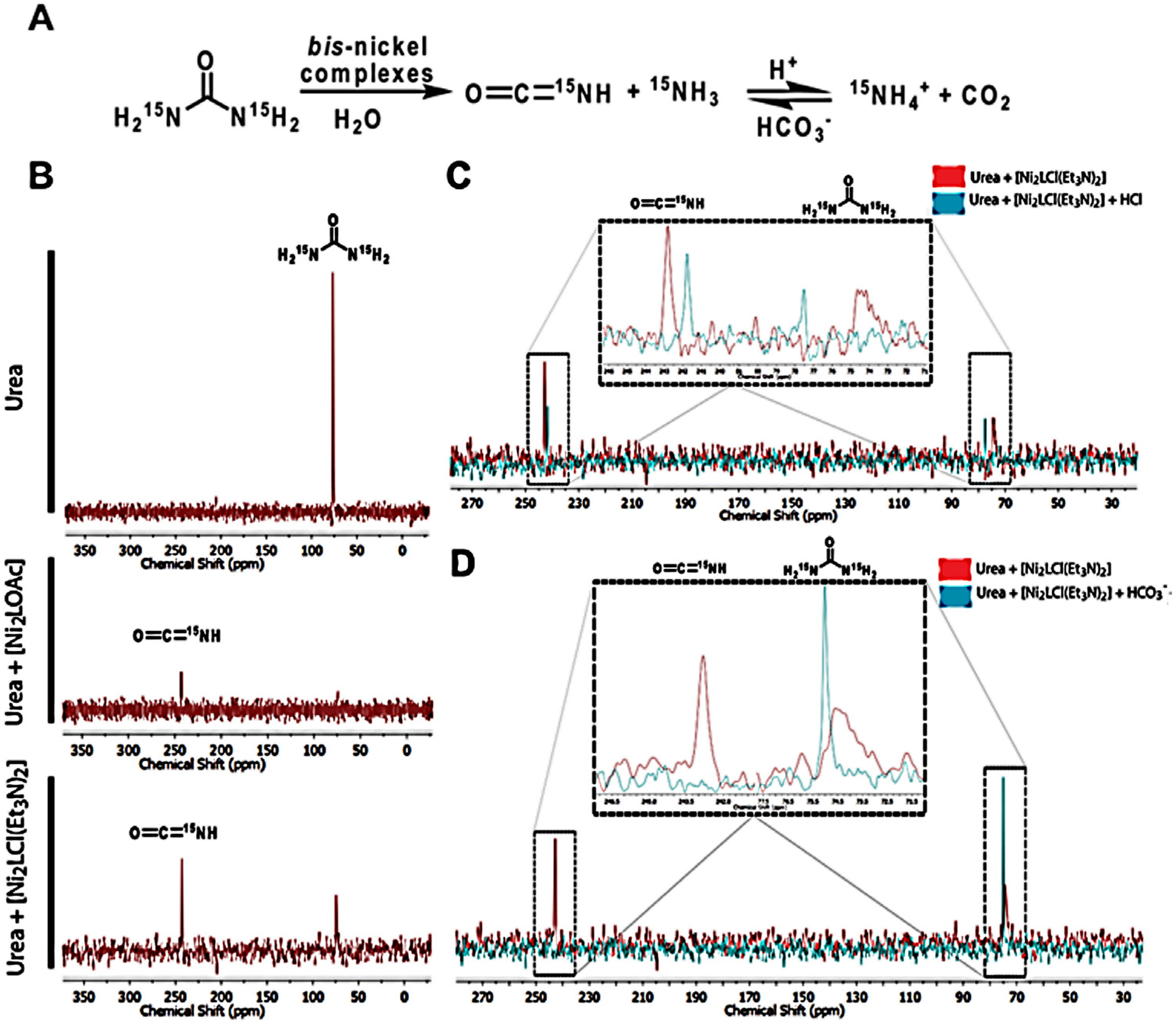
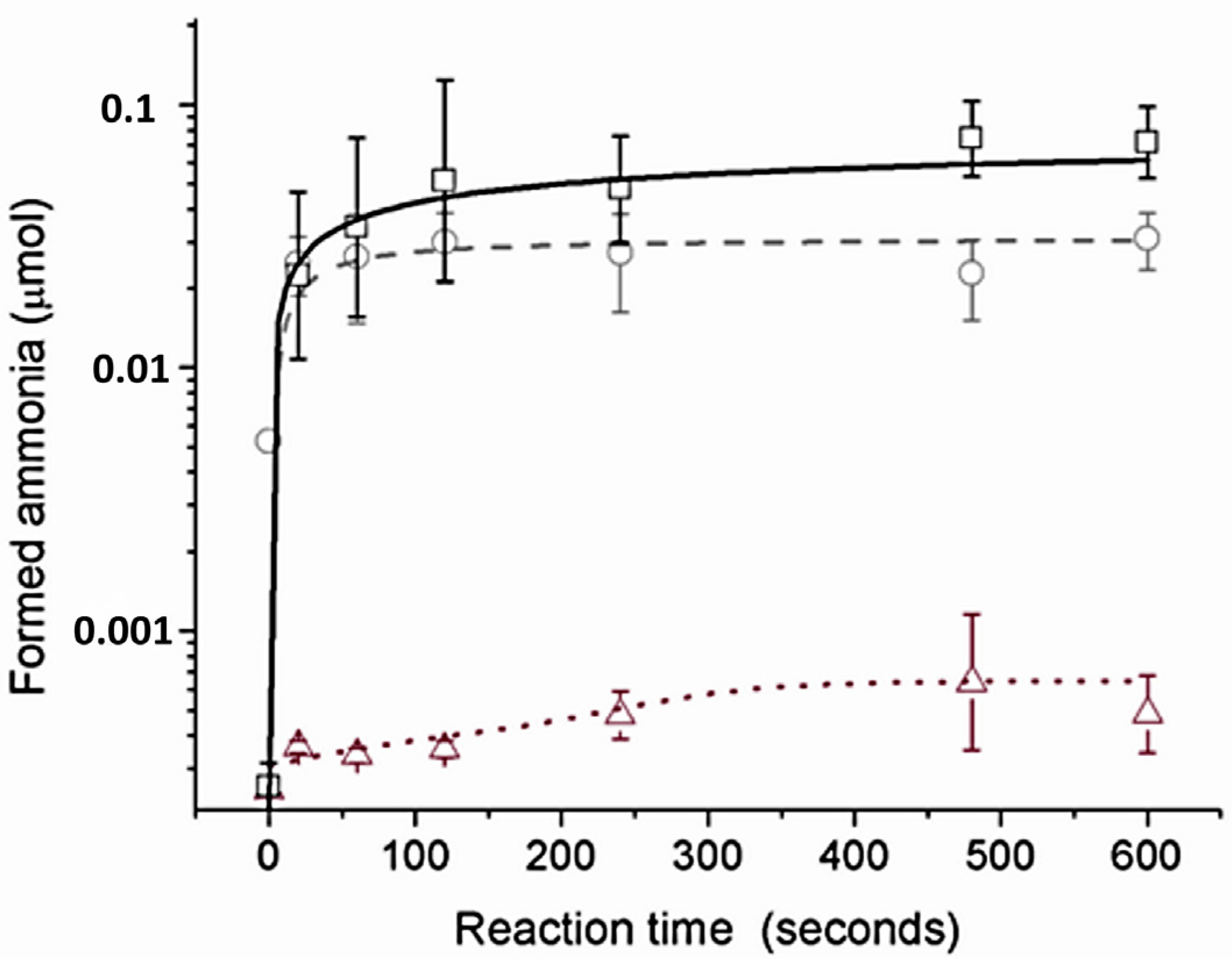
2.2. Catalytic Behavior of the Biomimetic Complexes
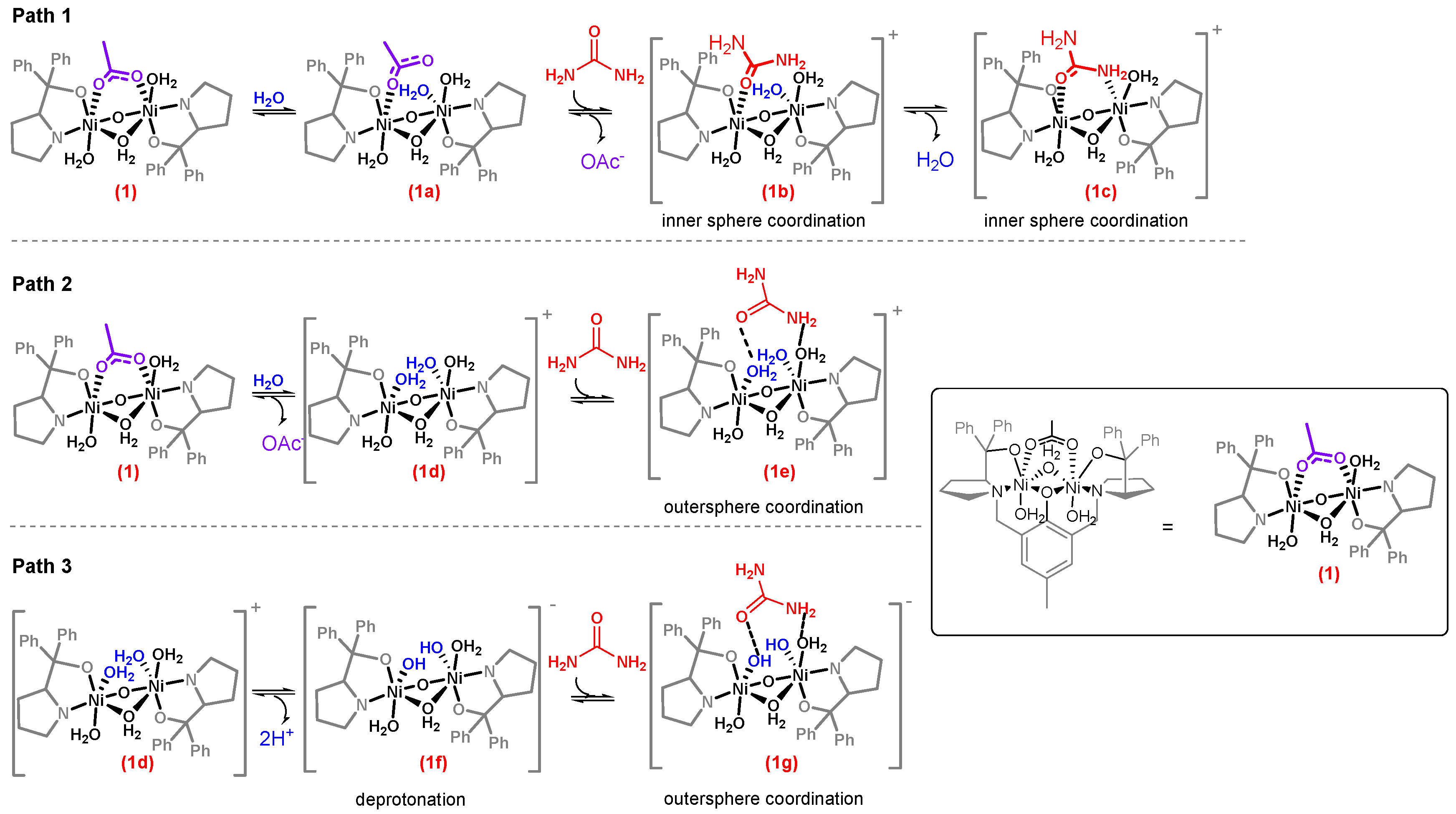
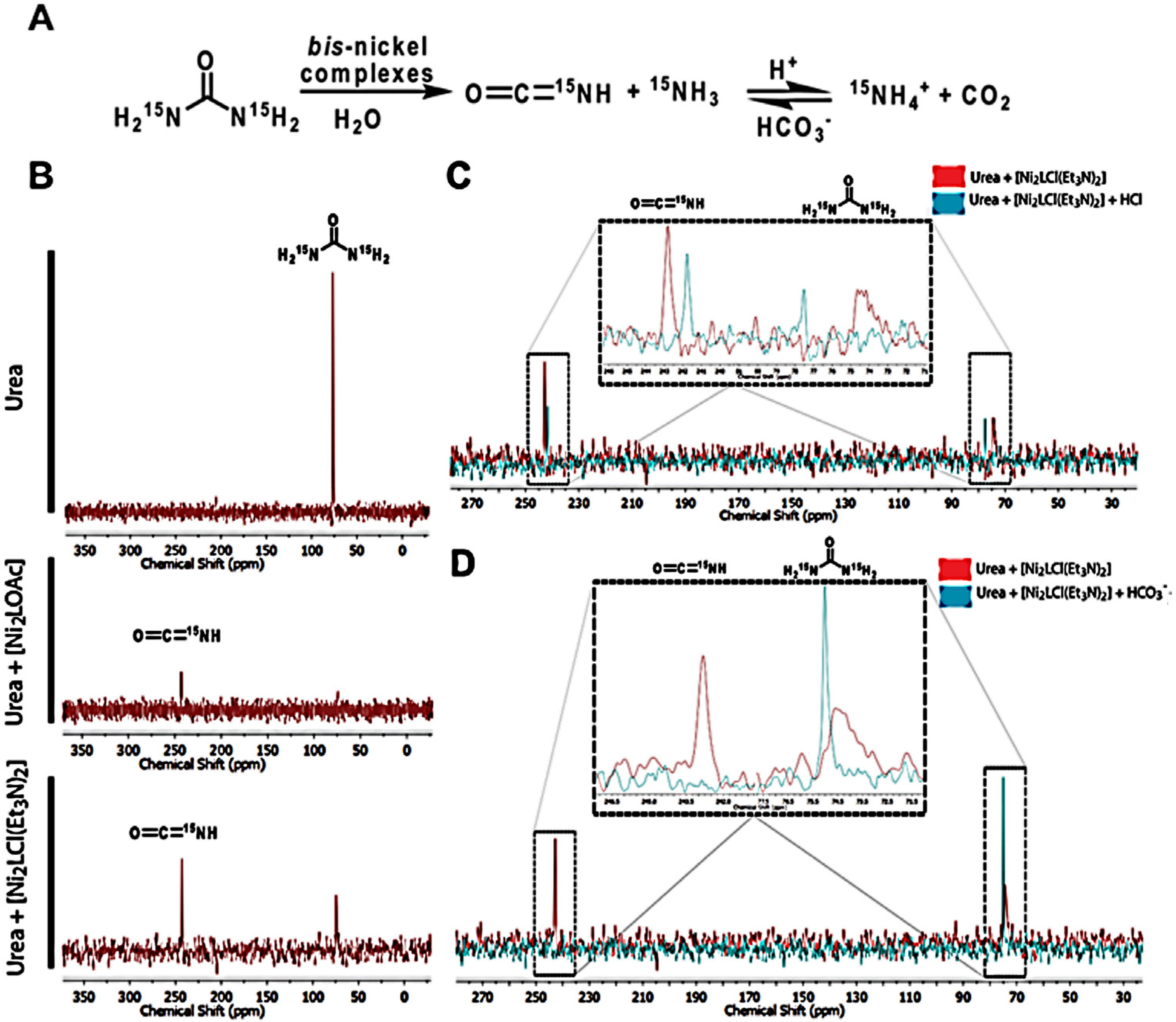
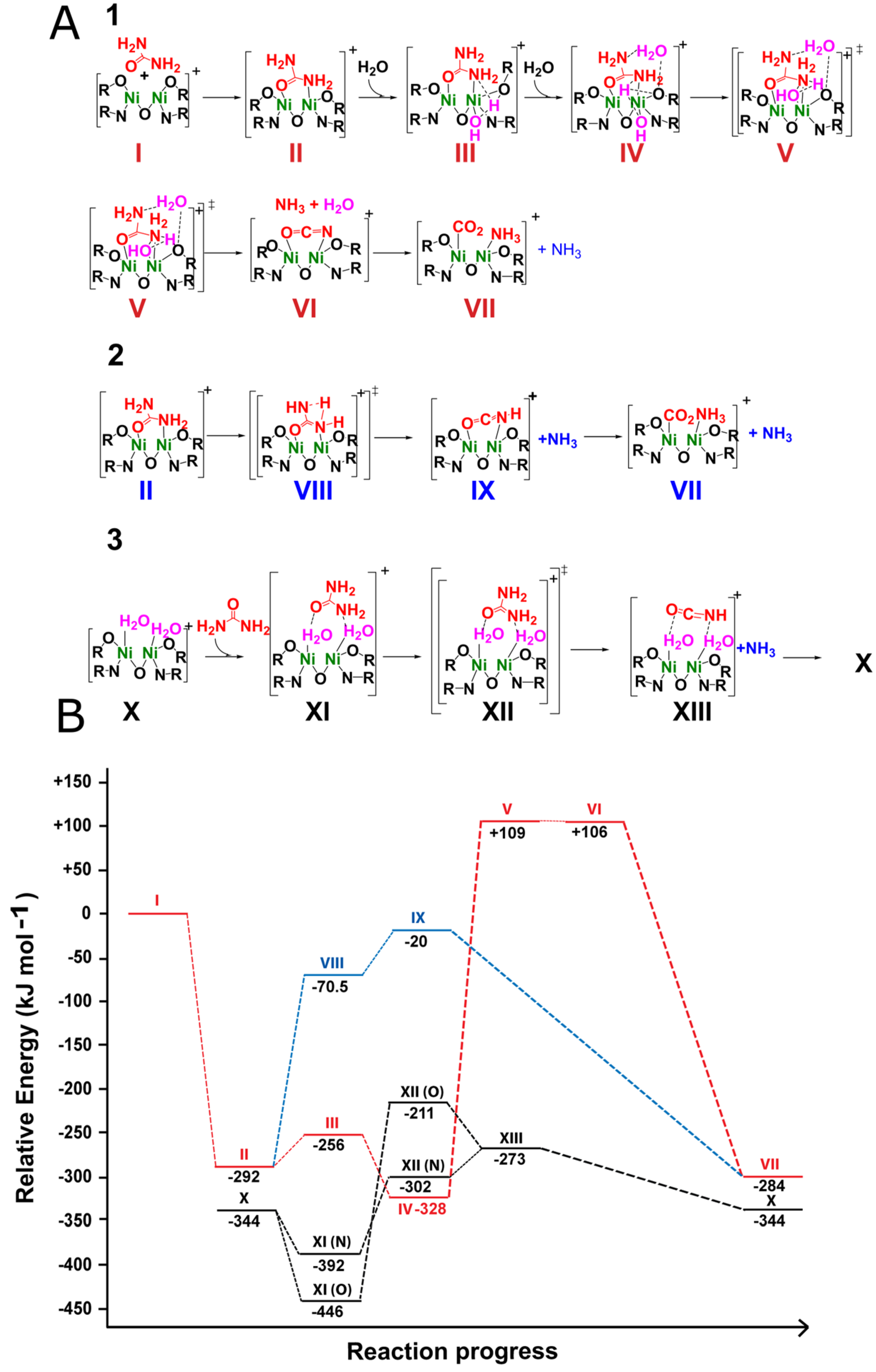
2.3. Theoretical Calculations of the Possible Mechanisms
3. Materials and Methods
3.1. Synthesis of (1)
3.2. Synthesis of (2)
3.3. Synthesis of [Ni2L(OH)2]− (5)
3.4. Urea Hydrolysis with [Ni2L(OH)2]− (5)
3.5. Determination of Urea Hydrolysis Using Bethelot Method
3.6. General Protocol for Substrates Hydrolysis
3.7. 15N-NMR Experiments
3.8. Susceptibility Measurements
3.9. Evaluation of Substrate Hydrolysis by GC-MS
3.10. Computational—Geometry Optimization and Transition States
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hasnain, S.S.; Piggott, B. An EXAFS study of jack bean urease, a nickel metalloenzyme. Biochem. Biophys. Res. Commun. 1983, 112, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Alagna, L.; Hasnain, S.S.; Piggott, B.; Williams, D.J. The nickel ion environment in jack bean urease. Biochem. J. 1984, 220, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, L.; Cianci, M.; Benini, S.; Ciurli, S. The structure of the elusive urease-urea complex unveils a paradigmatic case of metallo-enzyme catalysis. Angew. Chem. Int. Ed. 2019, 58, 7415–7419. [Google Scholar] [CrossRef]
- Mazzei, L.; Musiani, F.; Ciurli, S. The structure-based reaction mechanism of urease, a nickel dependent enzyme: Tale of a long debate. J. Biol. Inorg. Chem. 2020, 25, 829–845. [Google Scholar] [CrossRef]
- Lyu, Y.; Scrimin, P. Mimicking Enzymes: The quest for powerful catalysts from simple molecules to nanozymes. ACS Catal. 2021, 11, 11501–11509. [Google Scholar] [CrossRef]
- Kirby, A.J. Enzyme mechanisms, models, and mimics. Angew. Chem. Int. Ed. 1996, 35, 706–724. [Google Scholar] [CrossRef]
- Rakopoulou, L.; Papatriantafyllopoulou, C.; Terzis, A.; Perlepes, S.P.; Manessi-Zoupa, E.; Papaefstathiou, G.S. Synthesis, X-ray structure, and characterization of a complex containing the Hexakis(urea) cobalt(II) cation and lattice urea molecules. Hydrogen-Bonded Networks Based on Cobalt(II), Nickel(II), and Zinc(II) Complexes of N,N′-Diethylurea. Bioinorg. Chem. Appl. 2007, 11, 051567. [Google Scholar] [CrossRef]
- Meyer, F.; Konrad, M.; Kaifer, E. Novel mu(3)-coordination of urea at a nickel(II) site: Structure, reactivity and ferromagnetic superexchange. EurJIC. 1999, 11, 1851–1854. [Google Scholar]
- Koga, T.; Furutachi, H.; Nakamura, T.; Fukita, N.; Ohba, M.; Takahashi, K.; Okawa, H. Dinuclear nickel (II) complexes of phenol-based “end-off” compartmental ligands and their urea adducts relevant to the urease active site. Inorg. Chem. 1998, 37, 989–996. [Google Scholar] [CrossRef]
- Barrios, A.M.; Lippard, S.J. Decomposition of alkyl-substituted urea molecules at a hydroxide-bridged dinickel center. Inorg. Chem. 2001, 40, 1250–1255. [Google Scholar] [CrossRef]
- Mochizuki, K.; Takahashi, J.; Ishima, Y.; Shindo, T. Synthesis and structures of urea-coordinated dinickel(II) complexes with binucleating ligand 1,3-bis(N-(2-(dimethylamino)ethyl)-N-methylamino)propan-2-ol (HL1) and its analogs. Inorg. Chem. Acta 2013, 400, 151–158. [Google Scholar] [CrossRef]
- Meyer, F.; Konrad, M.; Kaifer, E. Synthesis and structures of urea coordinated dinickel(II) complexes with dinucleating ligand. Eur. J. Inorg. Chem. 1999, 11, 1851–1854. [Google Scholar] [CrossRef]
- Konrad, M.; Meyer, F.; Jacobi, A.; Kircher, P.; Rutsch, P.; Zsol, L. Coordination and conversion of urea at Dinuclear μ-Acetato nickel (II) complexes with symmetric and asymmetric cores. Inorg. Chem. 1999, 38, 4559–4566. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F. A bridging coordination mode of urea and carbamate at a dinuclear nickel(II) centre. Chem. Commun. 1998, 17, 1555–1556. [Google Scholar] [CrossRef]
- Barrios, A.M.; Lippard, S.J. Interaction of urea with a hydroxide-bridged dinuclear nickel center: An alternative model for the mechanism of urease. J. Am. Chem. Soc. 2000, 122, 9172–9177. [Google Scholar] [CrossRef]
- Kundu, B.K.; Biswas, S.; Mondal, A.; Mazumdar, S.; Mobin, S.M.; Mukhopadhyay, S. Unveiling the urease like intrinsic catalytic activities of two dinuclear nickel complexes towards the in situ syntheses of aminocyanopyridines. Dalton Trans. 2021, 50, 4848–4858. [Google Scholar] [CrossRef]
- Uprety, B.; Arderne, C.; Bernal, I. Catalytic Cleavage of the Amide Bond in Urea Using a Cobalt(III) Amino-Based Complex. Eur. J. Inorg. Chem. 2018, 47, 5058–5067. [Google Scholar]
- Estiu, G.; Merz, K.M. The hydrolysis of urea and the proficiency of urease. J. Am. Chem. Soc. 2004, 126, 6932–6944. [Google Scholar] [CrossRef]
- Callahan, B.P.; Yuan, Y.; Wolfenden, R. The burden borne by urease. J. Am. Chem. Soc. 2005, 127, 10828–10829. [Google Scholar] [CrossRef]
- Wojnar, M.K.; Laorenza, D.W.; Schaller, R.D.; Freedman, D.E. Nickel (II) metal complexes as optically addressable qubit candidates. J. Am. Chem. Soc. 2020, 142, 14826–14830. [Google Scholar] [CrossRef]
- Dorn, M.; Mack, K.; Carrella, L.M.; Rentschler, E.; Förster, C.; Heinze, K. Structure and electronic properties of an expanded terpyridine complex of nickel (II)[Ni (ddpd)2](BF4)2. Z. Anorg. Allg. Chem. 2018, 644, 706–712. [Google Scholar] [CrossRef]
- Chandramouli GV, R.; Manoharan, P.T. Electronic spectrum of dichlorobis [o-phenylenebis (dimethylphosphine)] nickel (III) hexafluorophosphate. Inorg Chem 1987, 26, 3291–3293. [Google Scholar] [CrossRef]
- Lampeka, Y.D.; Gavrish, S.P. Spectral characteristics of the copper (III) and nickel (III) coordination compounds with open-chain and macrocyclic dioxotetraamines. Polyhedron 2000, 19, 2533–2538. [Google Scholar] [CrossRef]
- Rhine, E.D.; Mulvaney, R.L.; Pratt, E.J.; Sims, G.K. Improving the Berthelot reaction for determining ammonium in soil extracts and water. Soil Sci. Soc. Am. 1998, 62, 473–480. [Google Scholar] [CrossRef]
- Keuleers, R.; Desseyn, H.O.; Rousseau, B.; Van Alsenoy, C. Vibrational analysis of urea. J. Phys. Chem. A 1999, 103, 4621–4630. [Google Scholar] [CrossRef]
- Cui, J.W.; Liu, Y.G.; Hao, Z.C.; Dong, G.Y. Synthesis, characterization, and crystal structures of 2D cobalt (II) and nickel (II) coordination polymers containing flexible bis (benzimidazole) ligands. Transition Met. Chem. 2015, 40, 565–571. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds: Part B; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Krajewska, B. A combined temperature-pH study of urease kinetics. Assigning pKa values to ionizable groups of the active site involved in the catalytic reaction. J. Mol. Catal. B Enzym. 2016, 124, 70–76. [Google Scholar] [CrossRef]
- Barrios, A.M.; Lippard, S.J. Amide hydrolysis effected by a hydroxo-bridged dinickel (II) complex: Insights into the mechanism of urease. J. Am. Chem. Soc. 1999, 121, 11751–11757. [Google Scholar] [CrossRef]
- Benini, S.; Rypniewski, W.; Wilson, K.; Ciurli, S.; Mangani, S. Structure-based rationalization of urease inhibition by phosphate: Novel insights into the enzyme mechanism. J. Biol. Inorg. Chem. 2001, 6, 778–790. [Google Scholar] [CrossRef]
- Benini, S.; Rypniewski, W.R.; Wilson, K.S.; Mangani, S.; Ciurli, S. Molecular details of urease inhibition by boric acid: Insights into the catalytic mechanism. J. Am. Chem. Soc. 2004, 126, 3714–3715. [Google Scholar] [CrossRef]
- Todd, M.J.; Hausinger, R.P. Fluoride inhibition of Klebsiella aerogenes urease: Mechanistic implications of a pseudo-uncompetitive, slow-binding inhibitor. Biochemistry 2000, 39, 5389–5396. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.A.; Moodie, R.B.; Sansom, P.J. The kinetics of hydrolysis of methyl and phenyl lsocyanates. J. Chem. Soc. Perkin Trans. 1985, 5, 737–742. [Google Scholar] [CrossRef]
- Beddie, C.; Webster, C.E.; Hall, M.B. Urea decomposition facilitated by a urease model complex: A theoretical investigation. Dalton Trans. 2005, 21, 3542–3551. [Google Scholar] [CrossRef]
- Blakeley, R.L.; Treston, A.; Andrews, R.K.; Zerner, B. Nickel (II)-promoted ethanolysis and hydrolysis of N-(2-pyridylmethyl) urea. A model for urease. J. Am. Chem. Soc. 1982, 104, 612–614. [Google Scholar] [CrossRef]
- Trost, B.M.; Bartlett, M.J. ProPhenol-catalyzed asymmetric additions by spontaneously assembled dinuclear main group metal complexes. Acc. Chem. Res. 2015, 48, 688–701. [Google Scholar] [CrossRef]
- Hrycyna, C.A.; Bergo, M.O.; Tamanoi, F. The Enzymes: Protein Prenylation Part B; Elsevier Inc.: London, UK, 2011; Volume 30. [Google Scholar]
- Martin, P.R.; Hausinger, R.P. Site-directed mutagenesis of the active site cysteine in Klebsiella aerogenes urease. J Biol Chem 1992, 267, 20024–20027. [Google Scholar] [CrossRef] [PubMed]
- Dixon, N.E.; Riddles, P.W.; Gazzola, C.; Blakeley, R.L.; Zerner, B. Jack bean urease (EC 3.5. 1.5). V. On the mechanism of action of urease on urea, formamide, acetamide, N-methylurea, and related compounds. Can. J. Biochem. 1980, 58, 1335–1344. [Google Scholar] [CrossRef]
- Uozumi, S.; Furutachi, H.; Ohba, M.; Ōkawa, H.; Fenton, D.E.; Shindo, K.; Murata, M.S.; Kitko, D.J. Dinuclear nickel (II) complexes of an unsymmetric “end-off” compartmental ligand: Conversion of urea into cyanate at a dinuclear nickel core. Inorg. Chem. 1998, 37, 6281–6287. [Google Scholar] [CrossRef]
- Kryatov, S.V.; Rybak-Akimova, E.V.; Meyer, F.; Pritzkow, H. A study of the equilibrium and kinetics of urea binding by a biomimetic dinickel (II) complex. Eur. J. Inorg. Chem. 2003, 2003, 1581–1590. [Google Scholar] [CrossRef]
- Bremer, C.; Ruf, H.; Grell, E. Kinetics and mechanism of complex formation between Mg2+ and methylthymol blue. J. Phys. Chem. A 1998, 102, 146–152. [Google Scholar] [CrossRef]
- Parr, R.G.; Weitao, Y. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1986. [Google Scholar]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Frisch, G.W.T.M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X. Gaussian16 Revision A. 03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Pires, D.A.; Arake, L.M.; Silva, L.P.; Lopez-Castillo, A.; Prates, M.V.; Nascimento, C.J.; Bloch, C., Jr. A previously undescribed hexapeptide His-Arg-Phe-Leu-Arg-His-NH2 from amphibian skin secretion shows CO2 and metal biding affinities. Peptides 2018, 106, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Catão, A.J.L.; López-Castillo, A. On the degradation pathway of glyphosate and glycine. Environ. Sci. Process. Impacts 2018, 20, 1148–1157. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (1) | (2) | Urease | |||||
|---|---|---|---|---|---|---|---|
| Reaction Yield (%) | (±2) | ||||||
| Time | 5 min | 12 h | 5 min | 12 h | 5 min | 12 h | |
| Substrate | |||||||
| Urea | 67 | 64 | 58 | 48 | 92 | 99 | |
| Formamide | 70 | 66 | 62 | 87 | 73 | 21 | |
| Acetamide | 6 | 4 | 0 | 4 | 12 | 82 | |
| N-methylurea | 4 | 6 | 0 | 6 | 6 | 9 | |
| N-phenylurea c | 13 a | 13.7 a | 13.2 a | 14 a | 0 | 0 | |
| 8.6 b | 10.6 b | 9.9 b | 9.6 b | ||||
| Butilcarbamate | 0 | 0 | 0 | 0 | 0 | 0 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, C.O.; Sebastiany, L.K.; Lopez-Castillo, A.; Freitas, R.S.; Andrade, L.H.; Toma, H.E.; Netto, C.G.C.M. Urea Decomposition Mechanism by Dinuclear Nickel Complexes. Molecules 2023, 28, 1659. https://doi.org/10.3390/molecules28041659
Martins CO, Sebastiany LK, Lopez-Castillo A, Freitas RS, Andrade LH, Toma HE, Netto CGCM. Urea Decomposition Mechanism by Dinuclear Nickel Complexes. Molecules. 2023; 28(4):1659. https://doi.org/10.3390/molecules28041659
Chicago/Turabian StyleMartins, Christian O., Leticia K. Sebastiany, Alejandro Lopez-Castillo, Rafael S. Freitas, Leandro H. Andrade, Henrique E. Toma, and Caterina G. C. Marques Netto. 2023. "Urea Decomposition Mechanism by Dinuclear Nickel Complexes" Molecules 28, no. 4: 1659. https://doi.org/10.3390/molecules28041659
APA StyleMartins, C. O., Sebastiany, L. K., Lopez-Castillo, A., Freitas, R. S., Andrade, L. H., Toma, H. E., & Netto, C. G. C. M. (2023). Urea Decomposition Mechanism by Dinuclear Nickel Complexes. Molecules, 28(4), 1659. https://doi.org/10.3390/molecules28041659