
Structure, Vibrational Spectra, and Cryogenic MatrixPhotochemistry of 6-Bromopyridine-2-carbaldehyde: From the Single Molecule of the Compound to the Neat Crystalline Material


Abstract
:
1. Introduction
2. Experimental Methods
3. Computational Details
4. Results and Discussion
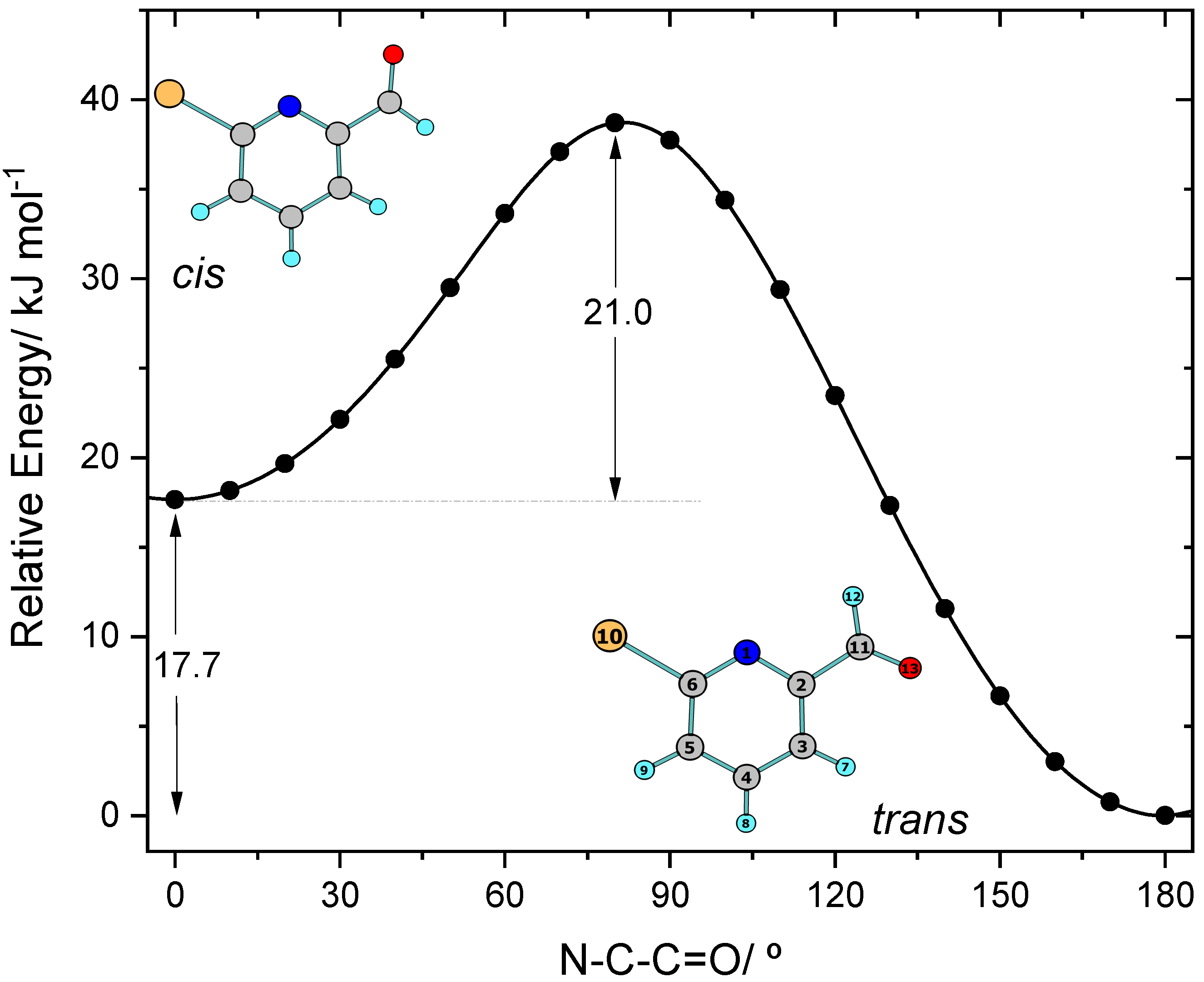
4.1. DFT Calculations: Conformers, Relative Energies and Barriers for Conformational Isomerization
4.2. Infrared Spectra of Matrix-Isolated BPCA
4.3. UV-Induced Rotamerization in Matrix-Isolated BPCA
4.4. Room Temperature IR and Raman Spectra of Crystalline BPCA and Intermolecular Interactions in the Crystal
5. Conclusions

Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, X.; Gibb, C.L.D.; Kuebel, M.E.; Gibb, B.C. Two New ligands for Carbonic Anhydrase Mimicry. Tetrahedron 2001, 57, 1175–1182. [Google Scholar] [CrossRef]
- Orita, A.; Nakano, T.; Yokoyama, T.; Babu, G.; Otera, J. Double Elimination Protocol for Access to Pyridine-Containing Arylene-Ethynylenes. Chem. Lett. 2004, 33, 1298–1299. [Google Scholar] [CrossRef]
- Yamada, Y.; Tanaka, K. Metal-Induced Structural Switching of a Folded Quinone-Sandwiched Porphyrin. J. Inorg. Organomet. Polym. 2013, 23, 180–185. [Google Scholar] [CrossRef]
- Doddipatla, S.; Ferreira, G.I.; Gülseven Sıdır, Y.; Nunes, C.M.; Fausto, R. Breaking and Formation of Intramolecular Hydrogen Bonds in Dihydroxybenzaldehydes through UV-Induced Conformational Changes in a Low-Temperature Matrix. J. Phys. Chem. A 2022, 126, 8645–8657. [Google Scholar] [CrossRef]
- Brito, A.L.B.; Roque, J.P.L.; Sıdır, İ.; Fausto, R. Low-Temperature Infrared Spectra and Ultraviolet-Induced Rotamerization of 5-Chlorosalicylaldehyde. J. Phys. Chem. A 2022, 126, 5148–5159. [Google Scholar] [CrossRef]
- Ogruc Ildiz, G.; Konarska, J.; Fausto, R. UV-induced Conformational Isomerization and Photochemistry of 3-Chloro-4-methoxybenzaldehyde in Cryogenic Inert Matrices. J. Chem. Phys. 2019, 151, 104303. [Google Scholar] [CrossRef]
- Ogruc Ildiz, G.; Konarska, J.; Fausto, R. Structural Transformations of 3-Fluoro and 3-Fluoro-4-methoxy Benzaldehydes under Cryogenic Conditions: A Computational and Low Temperature Infrared Spectroscopy Investigation. Chem. Phys. 2018, 508, 7–19. [Google Scholar] [CrossRef]
- Cluyts, K.; Sharma, A.; Kuş, N.; Schoon, K.; Fausto, R. Matrix Isolation Infrared Spectroscopic Study of 4-Pyridinecarboxaldehyde and of its UV-induced Photochemistry. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 171, 207–212. [Google Scholar] [CrossRef]
- Lopes, S.; Gómez-Zavaglia, A.; Fausto, R. Low Temperature IR Spectroscopy and Photochemistry of Matrix-isolated a-Pyridil. J. Photochem. Photobiol. A Chem. 2008, 200, 169–180. [Google Scholar] [CrossRef]
- Hees, U.; Vogelbacher, U.-J.; Michels, G.; Regitz, M. Antiaromatic Compounds—24. Steric Effects on Valence Isomerizations in the Dewar Pyridine/Azaprismane/Pyridine System. Tetrahedron 1989, 45, 3115–3130. [Google Scholar] [CrossRef]
- Zhong, D.; Diau, E.W.-G.; Bernhardt, T.M.; De Feyter, S.; Roberts, J.D.; Zewail, A.H. Direct Observation of the Femtosecond Nonradiative Dynamics of Azulene in a Molecular Beam: The Anomalous Behavior in the Isolated Molecule. Chem. Phys. Lett. 1998, 298, 9785–9788. [Google Scholar]
- Han, K.-L.; He, G.-Z. Photochemistry of Aryl Halides: Photodissociation Dynamics. J. Photochem. Photobiol. C Photochem. Rev. 2007, 8, 55–66. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Persson, P.; Lunell, S. Theoretical Study of the Fast Photodissociation Channels of the Monohalobenzenes. J. Phys. Chem. A 2004, 108, 2339–2345. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Qin, D.-B.; Jing, L.-H.; Gua, S.-J.; Mao, Z.-H. 6-Bromopyridine-2-carbaldehyde. Acta Cryst. E Struct. Rep. 2006, 62, o1715–o1716. [Google Scholar] [CrossRef]
- John Wiley & Sons, Inc. SpectraBase. Available online: https://spectrabase.com/ (accessed on 2 January 2023).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Hratchian, H.P.; et al. Gaussian 09 (Revision A.02); Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li-F. J. Comput. Chem. 1883, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.; Binkley, J.S. Self-Consistent Molecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Schachtschneider, J.H.; Mortimer, F.S. Vibrational Analysis of Polyatomic Molecules; VI. FORTRAN IV Programs for Solving the Vibrational Secular Equation and for the Least-Squares Refinement of Force Constants. Project No. 31450. Structural Interpretation of Spectra; Shell Development Co.: Emeryville, CA, USA, 1969. [Google Scholar]
- Pulay, P.; Fogarasi, G.; Pang, F.; Boggs, J.E. Systematic Ab Initio Gradient Calculation of Molecular Geometries, Force Constants, and Dipole Moment Derivatives. J. Am. Chem. Soc. 1979, 101, 2550–2560. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Jaquemin, D.; Perpète, E.A.; Scuseria, G.E.; Ciofini, I.; Adamo, C. TD-DFT Performance for the Visible Absorption Spectra of Organic Dyes: Conventional versus Long-Range Hybrids. J. Chem. Theory Comput. 2008, 4, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Miertus, C.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilizaion of Ab initio Molecular Potentials for the Prevision of Solvent Effects. J. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Spackman, M.A.; Byrom, P.G. A Novel Definition of a Molecule in a Crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Turner, M.J.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer 17.5; University of Western Australia: Crawley, Australia, 2013. [Google Scholar]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Shahi, A.; Arunan, E. Why are Hydrogen Bonds Directional? J. Chem. Sci. 2016, 128, 1571–1577. [Google Scholar] [CrossRef]
- Fausto, R.; de Carvalho, L.A.E.B.; Teixeira-Dias, J.J.C.; Ramos, M.N. The s-cis and s-trans Conformers of Formic, Thioformic and Dithioformic Acids: An Ab Initio Study. J. Chem. Soc. Faraday Trans. 2 1989, 85, 1945–1962. [Google Scholar] [CrossRef]
- Chandra, A.K.; Nguyen, M.T.; Zeegers-Huyskens, T. Density Functional Calculations on Simple Carbonyl Bases: Protonation and Hydrogen Bond Formation with Water. Chem. Phys. 2000, 255, 149–163. [Google Scholar] [CrossRef]
- Dinu, D.F.; Podewitz, M.; Grothe, H.; Loerting, T.; Liedl, K.R. Decomposing Anharmonicity and Mode-coupling from Matrix Effects in the IR Spectra of Matrix-isolated Carbon Dioxide and Methane. Phys. Chem. Chem. Phys. 2020, 22, 17932–17947. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Yamada, K.M.T. Infrared Spectra of Carbon Monoxide in Kr and Xe Matrices: Shifts of the Vibrational Line Positions. Struct. Chem. 2003, 14, 211–215. [Google Scholar] [CrossRef]
- Kuş, N.; Reva, I.; Fausto, R. Photoisomerization and Photochemistry of Matrix-Isolated 3-Furaldehyde. J. Phys. Chem. A 2010, 114, 12427–12436. [Google Scholar] [CrossRef]
- Shemesh, D.; Lan, Z.; Gerber, R.B. Dynamics of Triplet-State Photochemistry of Pentanal: Mechanisms of Norrish I, Norrish II, and H Abstraction Reactions. J. Phys. Chem. A 2013, 117, 11711–11724. [Google Scholar] [CrossRef]
- Zalat, O.A.; Elsayed, M.A. A Study on Microwave Removal of Pyridine from Wastewater. J. Environ. Chem. Eng. 2013, 1, 137–143. [Google Scholar] [CrossRef]
- Sauer, J.D. The Synthesis of Heteromacrocycles Containing the 2,6-Pyridine Moiety; Louisiana State University and Agricultural & Mechanical College, Historical Dissertations and Theses: Baton Rouge, LA, USA, 1976; pp. 138–139. Available online: https://digitalcommons.lsu.edu/gradschool_disstheses/2938 (accessed on 7 December 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | cis | trans | Crystal (XRD) 14, b (trans) | Parameter | cis | trans | Crystal (XRD) 14 (trans) |
|---|---|---|---|---|---|---|---|
| C–Br | 1.925 | 1.924 | 1.901 (5) | C2–N–C6 | 117.7 | 117.7 | 117.1 (4) |
| C=O | 1.203 | 1.208 | 1.207 (6) | N–C6–C5 | 124.8 | 124.4 | 124.6 (4) |
| C–H12 | 1.111 | 1.106 | 1.05 (6) | N–C2–C3 | 122.7 | 123.1 | 122.7 (4) |
| C2–N | 1.345 | 1.345 | 1.341 (6) | N–C2–C11 | 118.1 | 115.3 | 114.8 (4) |
| C6–N | 1.307 | 1.312 | 1.296 (6) | C3–C2–C11 | 119.2 | 121.6 | 122.5 (4) |
| C2–C11 | 1.498 | 1.494 | 1.481 (7) | C–C=O | 125.3 | 123.7 | 123.6 (5) |
| C2–C3 | 1.393 | 1.393 | 1.376 (7) | C–C–H12 | 113.5 | 113.7 | 110 (3) |
| C3–H7 | 1.084 | 1.082 | 1.04 (4) | O=C–H | 121.2 | 122.5 | 127 (3) |
| C3–C4 | 1.396 | 1.392 | 1.364 (7) | C2–C3–H7 | 120.3 | 119.5 | 122 (3) |
| C4–C5 | 1.386 | 1.391 | 1.368 (7) | N–C–Br | 117.1 | 117.0 | 116.4 (4) |
| C5–C6 | 1.403 | 1.399 | 1.390 (8) | C5–C6–Br | 118.1 | 118.6 | 119.0 (4) |
| N–C–C=O | 0.0 | 180.0 | 178.4 |
| Ar Matrix | Kr Matrix | Xe Matrix | Calculated | Assignment (PED) c | ||
|---|---|---|---|---|---|---|
| IIR | ||||||
| 3130 | 3108, 3100 | 3096, 3088 | 3070.6 | 0.4 | A′ | νC4–H (91) |
| 3130 | 3108, 3100 | 3096, 3088 | 3063.1 | 1.0 | A′ | νC2/3–H s (68) + νC2/3–H a (28) |
| 3075, 3069 | 3061.3055 | 3052, 3048 | 3038.4 | 4.5 | A′ | νC2/3–H a (72) + νC2/3–H s (24) |
| 2856, 2853, 2852, 2845, 2836, 2824 b | 2847, 2843, 2839, 2829, 2821 b | 2847, 2842, 2838, 2833, 2828, 2818 b | 2818.3 | 63.9 | A′ | νC–Hal (100) |
| 1736, 1733, 1731, 1728, 1727, 1725, 1723, 1720, 1718, 1713, 1708, 1704 b | 1736, 1732, 1729, 1723, 1718, 1716, 1714, 1708, 1706 b | 1730, 1728, 1725, 1724, 1722, 1717, 1710, 1709, 1703, 1701 b | 1751.4 | 269.6 | A′ | νC=O (91) |
| 1592, 1590 | 1591, 1588 | 1589, 1586 | 1578.5 | 8.7 | A′ | νring1 (56) + δCHr 3 (15) + νring2 (13) |
| 1587, 1584, 1580, 1568, 1565, 1562, 1560, 1556 b | 1584, 1581, 1570, 1564, 1561, 1558, 1557 b | 1580, 1577, 1566, 1560, 1557, 1552 b | 1569.3 | 95.8 | A′ | νring2 (61) + δCHr 2 (11) |
| 1437, 1435, 1432, 1430, 1427, 1423, 1420, 1418 | 1438, 1434, 1430, 1426, 1423, 1420, 1418 | 1436, 1434, 1432, 1429, 1427, 1423, 1420, 1416 | 1431.0 | 52.5 | A′ | δCHr 1 (54) + νring4 (30) |
| 1415, 1411, 1409 | 1413, 1411, 1408 | 1411, 1410, 1405 | 1420.5 | 21.5 | A′ | νring5 (37) + δCHr 3 (36) + δCHal (17) |
| 1349, 1348, 1345, 1343 | 1351, 1347, 1346, 1342 | 1348, 1347, 1345, 1340 | 1346.3 | 6.9 | A′ | δCHal (82) |
| 1284, 1281, 1279 | 1285, 1283, 1280 | 1290, 1286, 1283, 1282 | 1297.3 | 29.4 | A′ | νring6 (66) |
| 1223, 1220, 1219, 1213, 1211 b | 1223, 1221, 1216, 1215, 1212, 1210 b | 1223, 1221, 1216, 1213, 1211, 1206 b | 1220.1 | 46.1 | A′ | δCHr 1 (33) + νC–C (24) + νring6 (14) + νring4 (13) |
| 1156, 1153 | 1155, 1153 | 1154, 1152 | 1158.8 | 17.5 | A′ | δCHr 2 (61) + νring6 (15) |
| 1128, 1125 | 1126, 1124 | 1123, 1120 | 1117.8 | 108.1 | A′ | νring4 (33) + νring3 (16) + δring1 (14) + δCHr 2 (12) |
| 1076, 1073 | 1075, 1074 | 1074, 1072 | 1077.5 | 4.7 | A′ | δCHr 3 (43) + νring5 (40) |
| 1008, 1005 | 1005, 1004 | 1007, 1002 | 1011.0 | 1.7 | A″ | γCHal (71) + γCHr 2 (20) |
| n.obs. | n.obs. | 993 | 997.7 | 0.03 | A″ | γCHr 2 (87) + γCHal (16) |
| 990, 989, 987 | 991, 988 | 989, 988, 986 | 986.8 | 7.4 | A′ | δring1 (53) + νring3 (43) |
| 916, 913 | 913, 910 | 910 | 922.4 | 0.2 | A″ | γCHr 3 (100) |
| 863, 861, 859, 855, 853, 849, 846 | 861, 858, 852, 844 | 862, 859, 857, 850, 841, 839 | 849.1 | 41.7 | A′ | νC–C (26) + δring3 (18) + νring4 (13) + νring3 (10) |
| 802, 800, 795, 793, 792 b | 799, 796, 793, 790 b | 798, 795, 792, 789 b | 797.1 | 40.9 | A″ | γCHr 1 (84) |
| 732, 725, 721 | 731, 723, 719 | 730, 723, 720, 717 | 723.7 | 4.4 | A″ | τring2 (94) |
| 714, 712 | 715, 712 | 713, 711 | 705.6 | 64.8 | A′ | δring3 (27) + δC=O (27) + νC–Br (13) |
| 633 | 632 | 631, 629 | 634.0 | 18.1 | A′ | δring2 (64) + δC=O (19) |
| n.obs. | n.obs. | n.obs. | 541.5 | 0.002 | A″ | γCBr (37) + τring1 (32) + γCHO (29) |
| 470 | 474 | 475 | 468.3 | 3.8 | A′ | δring3 (37) + νC–C (25) + wCHO (13) |
| n.obs. | n.obs. | n.obs. | 431.0 | 0.6 | A″ | τring3 (91) |
| n.i. | n.i. | n.i. | 296.7 | 8.6 | A′ | νC–Br (54) + δCBr (15) |
| n.i. | n.i. | n.i. | 271.2 | 6.6 | A′ | δCBr (33) + δC=O (22) + wCHO (16) + νC–Br (14) |
| n.i. | n.i. | n.i. | 225.0 | 15.9 | A″ | τC–CHO (38) + γCHO (31) + γCBr (14) + τring1 (12) |
| n.i. | n.i. | n.i. | 163.2 | 0.8 | A″ | τring1 (65) + γCHO (25) |
| n.i. | n.i. | n.i. | 151.9 | 2.9 | A′ | wCHO (43) + δCBr (38) |
| n.i. | n.i. | n.i. | 94.9 | 3.6 | A″ | τC–CHO (52) + γCHO (17) + τring3 (13) |
| Ar Matrix | Kr Matrix | Xe Matrix | Calculated | Assignment (PED) c | ||
|---|---|---|---|---|---|---|
| IIR | ||||||
| n.obs | n.obs | n.obs | 3071.0 | 0.3 | A′ | νC4–H (94) |
| n.obs. | n.obs. | n.obs. | 3047.0 | 5.0 | A′ | νC2/3–H s (95) |
| n.obs. | n.obs. | n.obs. | 3034.0 | 2.8 | A′ | νC2/3–H a (98) |
| 2744 | 2730 | 2732 | 2752.9 | 117.6 | A′ | νC–Hal (100) |
| 1759, 1727 b | 1745, 1734 b | 1734, 1728 b | 1769.6 | 242.3 | A′ | νC=O (92) |
| n.obs. | n.obs. | n.obs. | 1577.8 | 16.9 | A′ | νring1 (65) + δCHr 3 (18) |
| 1573 | 1566 | 1560, 1558 | 1572.5 | 82.9 | A′ | νring2 (71) + δCHr 2 (13) |
| 1434 | 1430 | 1430 | 1429.7 | 5.1 | A′ | νring5 (30) + δCHr 3 (19) + δCHal (16) |
| 1434 | 1430 | 1427 | 1427.9 | 79.2 | A′ | δCHr 1 (47) + νring4 (24) |
| 1389 | 1389, 1387 | 1386 | 1390.4 | 41.8 | A′ | δCHal (84) |
| 1288 | n.obs. | 1285 | 1287.8 | 9.1 | A′ | νring6 (77) |
| n.obs. | 1195 | 1193 | 1192.0 | 31.3 | A′ | νC–C (24) + δCHr 1 (18) + νring4 (14) + δring1 (14) + νring3 (13) |
| 1160 | 1158, 1157 | 1157, 1156 | 1161.9 | 33.4 | A′ | δCHr 2 (59) + νring6 (15) |
| 1131, 1129 | 1129, 1127 | 1127 | 1122.3 | 123.5 | A′ | νring4 (40) + δCHr 1 (12) + δCHr 2 (12) |
| n.obs. | n.obs. | 1089 | 1085.1 | 8.1 | A′ | νring5 (40) + δCHr 3 (38) + νring3 (11) |
| n.obs. | n.obs. | n.obs. | 1002.7 | 1.5 | A″ | γCHal (81) |
| n.obs. | n.obs. | n.obs. | 991.4 | 0.003 | A″ | γCHr 2 (100) |
| 986 | 985 | 986 | 984.0 | 9.5 | A′ | δring1 (57) + νring3 (39) |
| n.obs. | n.obs. | n.obs. | 909.1 | 0.1 | A″ | γCHr 3 (100) |
| 860, 852, 848 | 867 | 860, 857, 854 | 871.9 | 23.8 | A′ | νC–C (28) + δC=O (15) + δring3 (12) |
| 804, 798 | 788, 785 | 790, 783 | 790.4 | 41.8 | A″ | γCHr 1 (82) |
| n.obs. | 719 | 727 | 723.1 | 6.0 | A″ | τring2 (90) |
| 692 | 691 | 690 | 688.7 | 74.9 | A′ | δring3 (45) + δC=O (30) |
| n.obs. | 647 | 630 | 647.7 | 7.2 | A′ | δring2 (63) |
| n.obs. | n.obs. | n.obs. | 545.2 | 0.2 | A″ | γCBr (37) + γCHO (31) + τring1 (30) |
| n.obs. | n.obs. | n.obs. | 435.5 | 0.001 | A″ | τring3 (84) |
| 445 | 432 | 434 | 435.5 | 1.9 | A′ | νC–C (29) + δring3 (20) + δring2 (14) + wCHO (10) |
| n.i. | n.i. | n.i. | 309.8 | 10.9 | A′ | νC–Br (35) + δCBr (25) + wCHO (13) + δC=O (13) |
| n.i. | n.i. | n.i. | 292.7 | 2.6 | A′ | νC–Br (36) + δCBr (31) |
| n.i. | n.i. | n.i. | 218.5 | 8.0 | A″ | τring1 (40) + τC–CHO (28) + γCHO + τring3 (12) |
| n.i. | n.i. | n.i. | 153.5 | 2.6 | A″ | γCBr (44) + τring1 (19) + γCHO (13) + τring3 (12) |
| n.i. | n.i. | n.i. | 136.5 | 3.0 | A′ | wCHO (43) + δCBr (34) + δC=Ol (12) |
| n.i. | n.i. | n.i. | 90.2 | 3.5 | A″ | τC–CHO (64) + τring1 (20) + γCHO (13) |
| Ar Matrix | Kr Matrix | Xe Matrix | Calculated | Assignment (PED) b | ||
|---|---|---|---|---|---|---|
| IIR | ||||||
| n.obs. | n.obs. | n.obs. | 3069.3 | 0.8 | A′ | νCHr 1 (48) + νCHr 2 (33) |
| 3098 | 3088 | 3082 | 3053.9 | 10.3 | A′ | νCHr 1 (42) + νCHr 4 (36) + νCHr 2 (22) |
| 3080 | n.obs. | n.obs. | 3034.2 | 7.0 | A′ | νCHr 3 (47) + νCHr 2 (33) + νCHr 4 (20) |
| 3080 | n.obs. | n.obs. | 3020.9 | 11.2 | A′ | νCHr 3 (44) + νCHr 4 (35) + νCHr 2 (11) |
| 1580 | n.obs. | 1577 | 1580.9 | 33.9 | A′ | νring1 (60) + νring2 (14) |
| 1580 | n.obs. | 1577 | 1576.7 | 86.2 | A′ | νring2 (57) + δCHr 4 (13) + νring1 (10) |
| 1452, 1440 | 1462 | 1451 | 1455.4 | 61.8 | A′ | δCHr 2 (50) + νring5 (27) + δCHr 3 (11) |
| 1427, 1423, 1416 | 1417 | 1417 | 1420.0 | 69.3 | A′ | δCHr 1 (58) + νring4 (25) |
| n.obs. | n.obs. | n.obs. | 1290.6 | 0.9 | A′ | δCHr 4 (35) + δCHr 1 (27) + νring1 (14) + νring5 (11) |
| n.obs. | 1270 | 1260 | 1266.5 | 5.4 | A′ | νring6 (80) + δCHr 3 (10) |
| n.obs. | n.obs. | 1133 | 1152.8 | 3.2 | A′ | δCHr 3 (70) + νring6 (16) |
| 1113, 1097 | 1113 | 1113, 1109 | 1106.5 | 46.4 | A′ | νring4 (42) + δCHr 4 (23) + δCHr 1 (11) |
| 1082 | 1082 | 1080 | 1077.3 | 60.6 | A′ | νring5 (29) + δring 1 (21) + δCHr 2 (16) + δCHr 4 (15) |
| 1046 | 1051 | 1044 | 1042.0 | 17.8 | A′ | νring3 (37) + νring4 (16) + νring5 (15) + ring 1 (15) |
| n.obs. | n.obs. | n.obs. | 988.5 | 0.001 | A″ | γCHr 3 (100) |
| 986 | 985 | 986 | 985.0 | 7.4 | A′ | δring 1 (54) + νring3 (42) |
| n.obs. | n.obs. | n.obs. | 961.9 | 0.3 | A″ | γCHr 4 (95) |
| n.obs. | n.obs. | n.obs. | 881.2 | 0.01 | A″ | γCHr 2 (88) |
| 768 | 770 | 760 | 760.0 | 57.2 | A″ | γCHr 1 (85) |
| n.obs. | n.obs. | n.obs. | 729.6 | 1.0 | A″ | τring2 (100) |
| 704 | 703 | 703 | 701.3 | 35.9 | A′ | δring 3 (67) + νC–Br (16) |
| n.obs. | n.obs. | n.obs. | 614.0 | 2.4 | A′ | δring 2 (77) + δring 3 (12) |
| n.obs. | n.obs. | n.obs. | 470.9 | 4.0 | A″ | γCBr (43) + τring1 (40) + τring3 (13) |
| n.i. | n.i. | n.i. | 408.3 | 4.7 | A″ | τring3 (89) + τring1 (19) |
| n.i. | n.i. | n.i. | 304.2 | 6.3 | A′ | νC–Br (74) + δring 3 (13) |
| n.i. | n.i. | n.i. | 255.2 | 1.9 | A′ | δCBr (91) |
| n.i. | n.i. | n.i. | 154.2 | 1.3 | A″ | τring1 (50) + γCBr (41) |
| Excited State | Energy (eV) | λcalc.a (nm) | f a | Main Contributions (%) b |
|---|---|---|---|---|
| T1 (A′) | 3.17 | 391 | HOMO→LUMO (55%) + HOMO-3→LUMO (30%) + HOMO→LUMO+1 (17%) | |
| T2 (A″) | 3.24 | 383 | HOMO-1→LUMO (64%) | |
| S1 (A″) | 3.81 | 326 | 0.000 | HOMO-1→LUMO (65%)) |
| T3 (A′) | 4.20 | 295 | HOMO→LUMO+1 (47%) + HOMO-3→LUMO (23%) | |
| T4 (A″) | 4.40 | 282 | HOMO-4→LUMO (43%) + HOMO-2→LUMO (37%) | |
| S2 (A′) | 4.50 | 275 | 0.130 | HOMO→LUMO (65%) |
| T5 (A′) | 4.58 | 271 | HOMO-3→LUMO (47%) + HOMO-5→LUMO (16%) | |
| S3 (A″) | 4.61 | 269 | 0.001 | HOMO-4→LUMO (49%) + HOMO-2→LUMO (43%) |
| T6 (A′) | 5.29 | 234 | HOMO-3→LUMO+1 (53%) + HOMO-3→LUMO (29%) | |
| T7 (A″) | 5.38 | 231 | HOMO→LUMO+3 (29%) + HOMO-1→LUMO+1 (28%) | |
| T8 (A″) | 5.43 | 228 | HOMO→LUMO+3 (36%) + HOMO-4→LUMO+1 (23%) | |
| T9 (A′) | 5.47 | 227 | HOMO-7→LUMO (44%) + HOMO-5→LUMO (27%) | |
| S4 (A′) | 5.51 | 223 | 0.067 | HOMO→LUMO+1 (52%) + HOMO-3→LUMO (39%) + HOMO→LUMO (21%) |
| S5 (A″) | 5.71 | 217 | 0.004 | HOMO-1→LUMO+1 (45%) |
| T10 (A′) | 5.78 | 215 | HOMO-2→LUMO+3 (45%) + HOMO-4→LUMO+2 (18%) | |
| S6 (A″) | 5.96 | 208 | 0.000 | HOMO→LUMO+3 (48%) |
| Experimental | Calculated | Assignment d | |||
|---|---|---|---|---|---|
| IR b | Raman | (trans) | IIR | IR | |
| 3081 | 3083 | 3070.6 | 0.4 | 488.7 | νC4–H |
| 3074 | 3075 | 3063.1 | 1.0 | 352.5 | νC2/3–H s |
| 3039 | 3042 | 3038.4 | 4.5 | 395.3 | νC2/3–H a |
| 2871, 2846 c | 2872 | 2818.3 | 63.9 | 598.1 | νC–Hal |
| 1726, 1712, 1701 c | 1722, 1711, 1700 | 1751.4 | 269.6 | 1622.0 | νC=O |
| 1572 | 1574, 1563 | 1578.5 | 8.7 | 1698.9 | νring1 |
| 1553, 1542 | 1543 | 1569.3 | 95.8 | 361.9 | νring2 |
| 1434 | 1436 | 1431.0 | 52.5 | 357.9 | δCHr 1; νring4 |
| 1413 | 1414 | 1420.5 | 21.5 | 274.0 | νring5; δCHr 3 |
| 1351 | 1352 | 1346.3 | 6.9 | 34.1 | δCHal |
| 1289 | 1291 | 1297.3 | 29.4 | 107.1 | νring6 |
| 1214 | 1214 | 1220.1 | 46.1 | 1050.4 | δCHr 1; νC–C |
| 1163 | 1163 | 1158.8 | 17.5 | 67.1 | δCHr 2 |
| 1116 | 1120 | 1117.8 | 108.1 | 202.8 | νring4 |
| 1078 | 1078 | 1077.5 | 4.7 | 142.5 | δCHr 3; νring5 |
| 1013 | 1011 | 1011.0 | 1.7 | 130.6 | γCHal |
| 998 | 999 | 997.7 | 0.03 | 12.4 | γCHr 2 |
| 985 | 986 | 986.8 | 7.4 | 1184.2 | δring1; νring3 |
| 914 | 914 | 922.4 | 0.2 | 9.2 | γCHr 3 |
| 854 | 858 | 849.1 | 41.7 | 264.4 | νC–C; δring3 |
| 794 | 795 | 797.1 | 40.9 | 10.1 | γCHr 1 |
| 716 | n.obs. | 723.7 | 4.4 | 0.1 | τring2 |
| 706 | 708 | 705.6 | 64.8 | 320.5 | δring3; δC=O |
| 632 | 634 | 634.0 | 18.1 | 148.2 | δring2 |
| n.obs. | n.obs. | 541.5 | 0.002 | 9.6 | γCBr; τring1 |
| 471 | 473 | 468.3 | 3.8 | 316.2 | δring3 |
| 417 | 422 | 431.0 | 0.6 | 11.3 | τring3 |
| n.i. | 301 | 296.7 | 8.6 | 1322.2 | νC–Br |
| n.i. | 276 | 271.2 | 6.6 | 1561.5 | δCBr |
| n.i. | 229 | 225.0 | 15.9 | 309.7 | γCHO; τC–CHO |
| n.i. | 181 | 163.2 | 0.8 | 1075.8 | τring1 |
| n.i. | 167 | 151.9 | 2.9 | 855.3 | wCHO; δCBr (38) |
| n.i. | 79 | 94.9 | 3.6 | 2697.8 | τC–CHO |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brito, A.L.B.; Lopes, S.; Ogruc Ildiz, G.; Fausto, R. Structure, Vibrational Spectra, and Cryogenic MatrixPhotochemistry of 6-Bromopyridine-2-carbaldehyde: From the Single Molecule of the Compound to the Neat Crystalline Material. Molecules 2023, 28, 1673. https://doi.org/10.3390/molecules28041673
Brito ALB, Lopes S, Ogruc Ildiz G, Fausto R. Structure, Vibrational Spectra, and Cryogenic MatrixPhotochemistry of 6-Bromopyridine-2-carbaldehyde: From the Single Molecule of the Compound to the Neat Crystalline Material. Molecules. 2023; 28(4):1673. https://doi.org/10.3390/molecules28041673
Chicago/Turabian StyleBrito, Anna Luiza B., Susy Lopes, Gulce Ogruc Ildiz, and Rui Fausto. 2023. "Structure, Vibrational Spectra, and Cryogenic MatrixPhotochemistry of 6-Bromopyridine-2-carbaldehyde: From the Single Molecule of the Compound to the Neat Crystalline Material" Molecules 28, no. 4: 1673. https://doi.org/10.3390/molecules28041673