Abstract
This paper thoroughly explores the formation of Schiff bases derived from salicylaldehydes and a conformationally restricted amino alcohol (1-amino-2-indanol), as well as the generation of 1,3-oxazolidines, a key heterocyclic core present in numerous bioactive compounds. We provide enough evidences, both experimental-including crystallographic analyses and DFT-based calculations on imine/enamine tautomerism in the solid state and solution. In the course of imine formation, a pentacyclic oxazolidine–oxazine structure could be isolated with complete stereocontrol, whose configuration has been determined by merging theory and experiment. Mechanistic studies reveal that, although oxazolidines can be obtained under kinetic conditions, the prevalence of imines obeys to thermodynamic control as they are the most stable structures. The stereochemical outcome of imine cyclization under acylating conditions leads to formation of 2,4-trans-oxazolidines.
1. Introduction
For more than a decade, we have extensively investigated the chemistry of Schiff bases derived from aminopolyols and arylaldehydes, and disclosing a series of stereoelectronic effects that correlate with structure. It is particularly relevant to the interconversion between acyclic and heterocyclic frameworks with varied stereocontrol. Thus, the condensation of tris(hydroxymethyl) methylamine (TRIS, 1) with substituted benzaldehydes gives rise to 1,3-oxazolidines (2), which afford the corresponding per-O-acetylated imines (3) upon conventional acetylation conditions (Scheme 1) [1,2]. However, the reaction of TRIS with salicylaldehydes produces Schiff bases (4), which lead to a mixture of peracetylated imines (5) and N-acyl-1,3-oxazolidines (6) after acetylation (Scheme 1).
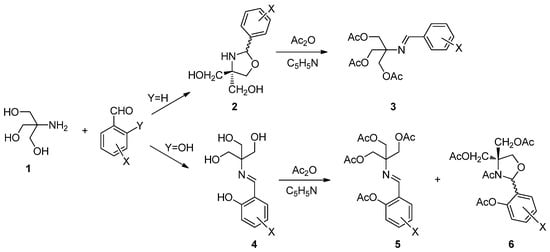
Scheme 1.
Ring-closure and ring-opening reactions under acetylating conditions.
The above results are evidence that product selection is substrate-dependent and we were prone to investigate the stereochemical outcome under acylating conditions. In general, the formation of oxazolidines and related heterocycles proceed with high diastereoselectivity (often close to 100%). A key point lies in the configuration at the new chiral center formed (C-2), whose assignment in previous literature has been misleading. In fact, due to the reversibility of this transformation, the stereoselectivity can be controlled thermodynamically, thus results are strongly dependent on the experimental conditions. It has now been established that 2,4-cis-oxazolidine is the major diastereomer obtained through equilibrium conditions (Figure 1). Since N-alkyloxazolidines are the only stable products (NH-oxazolidines exist mainly as open imines), one can conclude that the most stable isomer with substituents at positions 2, 3 and 4 is the one showing 2,3-trans and 3,4-trans relationships.

Figure 1.
2,4-Oxazolidine diastereomers.
Surprisingly, the first X-ray analysis of a chiral oxazolidine, described as early as 1971, was controversial because the authors reported that the condensation of ephedrine and 4-bromobenzaldehyde in refluxing benzene gave rise to a 2,4-trans-oxazolidine as the sole product [3]. A subsequent synthetic and crystallographic revision proved this assignment to be wrong [4], and evidenced the opposite cis configuration. A plausible explanation for this discrepancy would be that the first authors crystallized the minor diastereoisomer present in the reaction mixture, enriched however in the other isomer. The relative 2,4-cis configuration has now been widely accepted and is usually observed under various experimental conditions, as exemplified by the condensation of aryl and alkyl aldehydes with ephedrine (7), N-methyl or N-benzyl-phenylglycinol (8) and N-methylvalinol (9) (Figure 2).
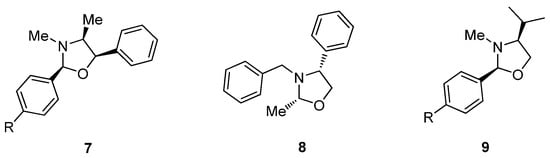
Figure 2.
Previous examples of 2,4-cis-configured oxazolidines.
That said, some exceptions to the 2,4-cis configuration have been reported as well. Thus, the formation of the 2,4-trans isomer (10) has been observed in the reaction of methyl hemiacetal derived from ethyl glyoxylate with N-benzyl-phenylglycinol in CH2Cl2 at reflux and in the presence of molecular sieves [5]. In contrast, the cis configuration has been reported by other research groups in the condensation of glyoxylate with N-Boc-ephedrine under different conditions [6,7]. A trans configuration was also proposed for compound 11, obtained by double condensation of ephedrine with glyoxal in THF-water [8] (Figure 3). These exceptions could be justified on the basis of both structural and experimental factors. In a comprehensive study, Agami and Rizk showed that condensation with aromatic aldehydes bearing electron-withdrawing groups led to trans-oxazolidines as kinetic products, and the rate of isomerization to the more stable cis isomer is largely influenced by the solvent system [9].
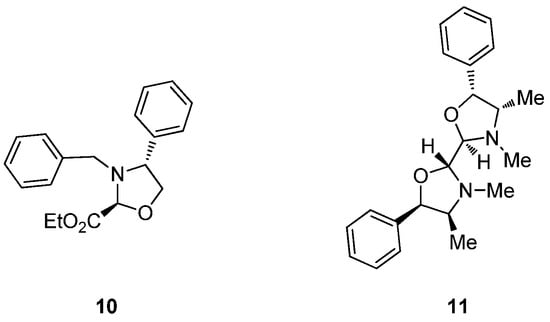
Figure 3.
Reported examples of 2,4-trans-configured oxazolidines.
On the other hand, little data are available on the relative stereochemistry between the substituents at C-2 and C-5 after ring closure. To our knowledge, we described the only antecedent on the synthesis of chiral oxazolidines (14, 15) from 1-amino-1-deoxyhexitols with aromatic aldehydes [10]. Condensation with d-glucamine (1-amino-1-deoxy-d-glucitol, 12) leads initially to the corresponding imines 13, which under acylating conditions produce chiral N-acyl oxazolidines 14 (Scheme 2). In this case, the stereochemistry with which the formation of oxazolidine takes place is 2,5-trans. The same is true for oxazolidines derived from N-methyl-d-glucamine [11].
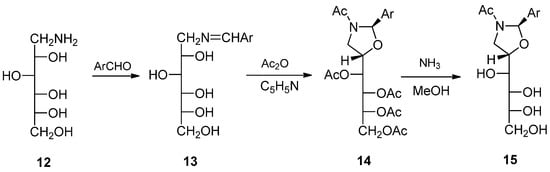
Scheme 2.
Chiral trans-configured oxazolidines obtained from d-glucamine and aryl aldehydes.
In all the above cases, the aminopolyols employed were conformationally flexible substances. We reasoned a conformationally restricted partner might provide a different bias in the steric outcome and, to this end, 1-amino-2-indanol (16) was chosen. In principle, Scheme 3 provides a general strategy to the target compounds in line with the aforementioned results, i.e., Schiff bases 17 should be obtained by reaction of 16 with substituted salicylaldehydes. Subsequent acetylation would possibly lead to a mixture of the acetylated imine 18 and/or the corresponding oxazolidine 19. As we shall see, however, through this detailed analysis, the transformation and products are endowed with tautomeric and configurational complexity, whose elucidation in both solution and solid state required a combination of spectroscopic and computational methods, which ultimately unveiled a marked stereoselection supplied by the rigid framework.
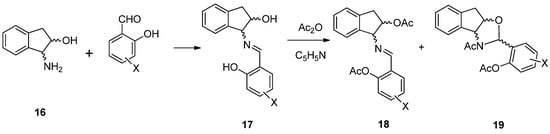
Scheme 3.
General synthesis of imines and oxazolidines derived from 1-amino-2-indanol.
2. Results and Discussion
2.1. Imine Synthesis
Because compound 16 has two stereogenic centers, this chiral aminopolyol can exist in different stereoisomeric structures. Accordingly, the synthesis of the Schiff bases was carried out with (1R,2S)-1-amino-2-indanol (20), (1S,2R)-1-amino-2-indanol (21) and (1R,2R)-1-amino-2-indanol (22) (Figure 4). The choice is based on the relative disposition of the amino and hydroxyl groups, which lie in a cis arrangement in the first two and therefore they are spatially close; conversely, the trans disposition in 22 keeps them away. These configurational variations allow us to evaluate the influence of such spatial arrangements on the stereochemistry of ring-closing reactions.
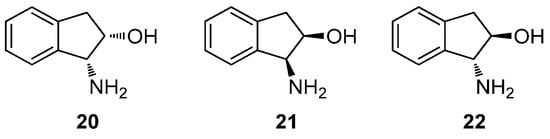
Figure 4.
1-Amino 2-indanols used in the synthesis of Schiff bases.
All reactions were conducted by mixing equimolar amounts of the aminoalcohol and the corresponding substituted salicylaldehyde in absolute ethanol. The mixture was stirred at room temperature until the appearance of a solid, which can easily be isolated by filtration and usually purified by recrystallization, such as in the case of adducts 23–34. To isolate salicylimines 29 and 34, the reaction had to be carried out in ethyl ether because the use of ethanol as solvent resulted in the formation of an unexpected side product, whose structure will be discussed later. Moreover, the trans-configured imine 37 could be synthesized from the corresponding substituted salicylaldehyde, whereas enantiomerically pure cis imines 35 and 36 are commercially available. Yields ranged from moderate to good, although they were not optimized (see Experimental and Supplementary Information, Table S1) (Figure 5).
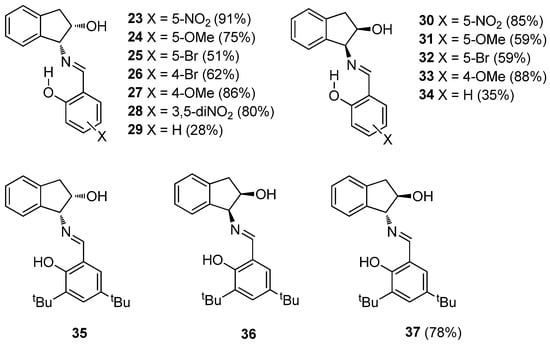
Figure 5.
Synthesized Schiff bases.
2.2. Solid-State Structures
The structures assigned to the above-mentioned Schiff bases are supported by their elemental microanalyses and spectroscopic data. The main problem associated with an accurate structural elucidation of Schiff bases stems from tautomerism because these substances can exhibit imine or enamine structure, both in solid state and in solution, which are not necessarily identical.
Solid-state structures can be inferred from FT-IR and Raman spectroscopies and a conclusive statement of these vibrational spectra is that the actual solid-state structures for compounds 23, 26–30, 33 and 34 correspond to enamine tautomers 38–45, respectively (see Supplementary Material) (Figure 6).
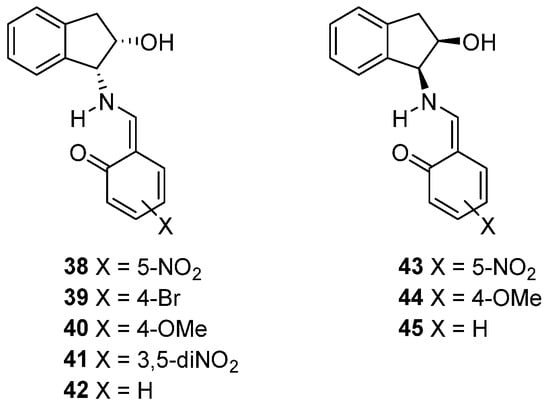
Figure 6.
Synthesized Schiff bases with an enamine structure.
Gratifyingly, crystals suitable for X-ray diffraction could be obtained by slow evaporation for compounds 24 and 40–42, thereby determining with confidence their crystal structures. Crystallographic analyses evidenced an imine structure for 24, while enamine tautomers were found for 40–42. The exact position of the amine/enamine hydrogen atom was determined from the corresponding Fourier maps of electron density differences for the four refinements without including the hydrogen in the model. The simplest Schiff base, 42, derives from unsubstituted salicylaldehyde (X = H), whose electron density difference map shows that the proton is located on the nitrogen atom (Figure 7).
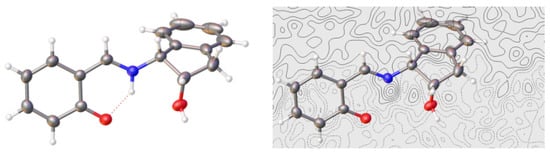
Figure 7.
X-Ray diffraction pattern and electron density difference map for compound 42 (Color code: carbon, black; oxygen, red; nitrogen, blue; hydrogen, white).
For compound 24, the structural elucidation by single-crystal diffraction showed the presence of one water molecule in the crystal lattice (Figure 8).
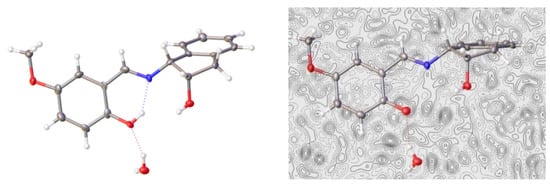
Figure 8.
X-Ray diffraction pattern and electron density difference map of 24 (Color code: carbon, black; oxygen, red; nitrogen, blue; hydrogen, white).
The imine structure obtained for 24 and enamine for 40 (Figure 9) matched with the differences observed in their IR spectra, as noted above (Figures S17 and S24). It is noteworthy that the only difference between compounds 24 and 40 is the position occupied by the methoxy group at the aromatic ring, a key structural element favoring the formation of either imine or enamino tautomers (vide infra).
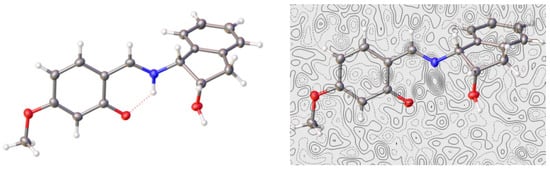
Figure 9.
X-Ray diffraction pattern and electron density difference map of 40 (Color code: carbon, black; oxygen, red; nitrogen, blue; hydrogen, white).
In both cases, the methoxy group is essentially coplanar with the ring to which it is attached, as shown by dihedral angles: −0.21° in 24 and 0.08° in 40. In this arrangement, the delocalization provided by a lone pair from the oxygen atom is maximal. However, even though the Fourier map of electron density differences for 40 shows the proton located mainly on the nitrogen atom, a residual electron density is observed near the oxygen atom, indicating that the proton in question is delocalized between the two positions—that is, it is partially bound to both atoms: oxygen and nitrogen (Figure 9). This suggests the coexistence of imine and enamine tautomers in rapid equilibrium within the crystal lattice. For dinitro derivative 41, X-ray diffraction analysis shows the presence of two molecules in the unit cell and both molecules have an enamine structure (Figure 10).
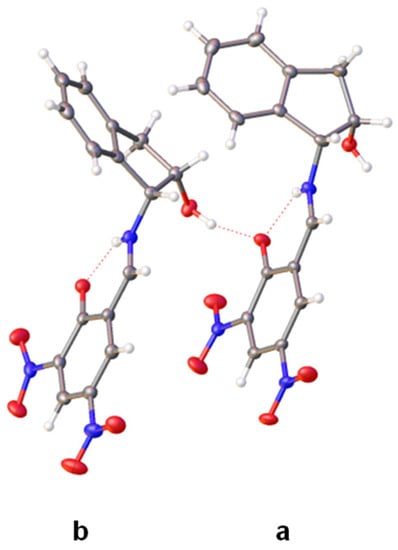
Figure 10.
X-Ray diffraction structure of 41 having two enamine tautomers (molecules a and b) in the unit cell (Color code: carbon, black; oxygen, red; nitrogen, blue; hydrogen, white).
The Fourier difference map places the proton in each molecule primarily on the nitrogen atom, thus evidencing an enamine structure (Figure 11). Like compound 40, however, a residual electronic density is located near the oxygen atom as well. These two molecules differ only by the conformation adopted by the pentagonal ring of indanol moieties. Thus, in one of them, the ring shows a geometry close to an E2 conformation and the angle formed by the two benzene residues is ~67° (Figure 12a). The other molecule has a 2E conformation and the angle between the benzene rings is considerably greater (~106°) (Figure 12b). The nitro groups at C-5 are practically coplanar with the 1,3-cyclohexadiene ring, showing small dihedral angles of 1.62° in the first molecule (41a) and 2.85° in 41b. In contrast, due to steric effects, the nitro groups at C-3 deviate from the planarity forming angles of 9.66° and −12.60°, respectively.
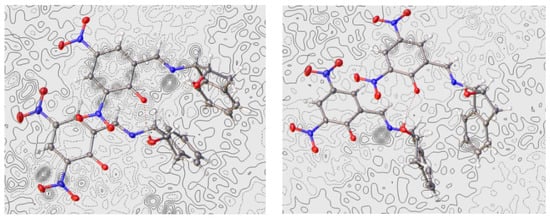
Figure 11.
Electronic density difference maps of the two molecules of 41 (see main text and Figure 12) (Color code: carbon, black; oxygen, red; nitrogen, blue; hydrogen, white).
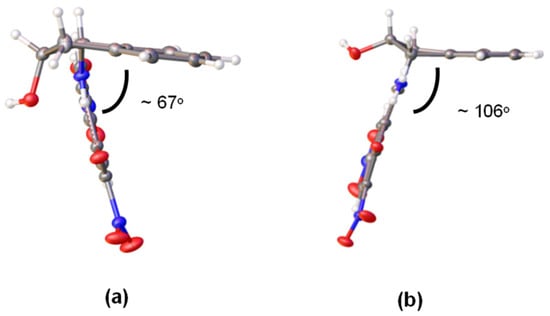
Figure 12.
(a) Conformations and (b) relative dispositions of the aromatic rings of 41 in crystalline state (Color code: carbon, black; oxygen, red; nitrogen, blue; hydrogen, white).
All structures elucidated by single-crystal diffraction exhibit a strong intramolecular hydrogen bond leading to a six-membered ring whose geometric characteristics are collected in Table S2. Table 1 compares some bonding distances obtained by X-ray analyses of 24 and 40–41, with the average values of similar structures described as phenoliminic tautomers [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31] and ketoamines [32,33,34] in the Cambridge Structural Database (Figure 13). The analysis of bond distances in compounds 40 and 42 indicates that the values of dC2-C3 and dC4-O5 are in full agreement with an enamine structure. The value of dN1-C2 for compound 40 is consistent with an enamine structure, while the corresponding value in 42 lies between the extreme values expected for imine and enamine tautomers.

Table 1.
Comparative bond lengths [a] of 24 and 40–41 with average distances [b] of phenoliminic and keto-amino structures.
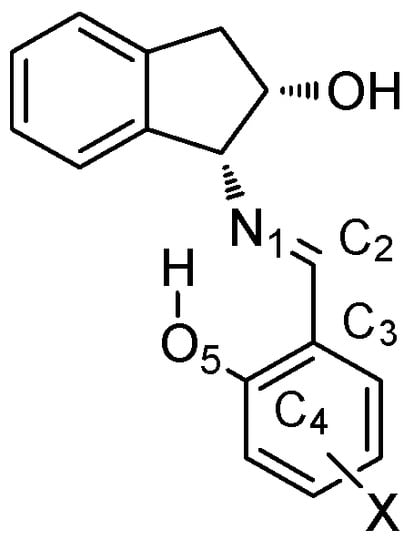
Figure 13.
Atom numbering employed for the tautomeric system.
The dC2-C3 and dC4-O5 distances measured for 24 are between the limiting values of imine and enamine structures, although dN1-C2 clearly agrees with a ketoamine form. It is evident that this ambivalence encountered in the bond distances of 24 shows the existence of a very fast interconversion equilibrium between both structures, even at 100 K, and the conditions employed for recording X-ray diffraction spectra. The coexistence of two molecules in the crystal lattice of 41 is singular. The distances dN1-C2 and dC2-C3 clearly correspond to an iminic structure; however, the short distance dC4-O5 corresponds to a C=O bond. These results do not agree with a neutral enaminic structure such as 41 (Scheme 4), but do agree with a zwitterionic structure such as 41a–41c.
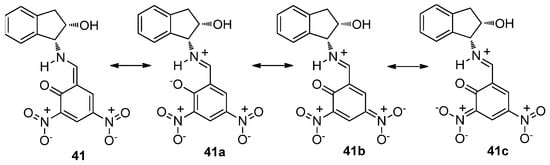
Scheme 4.
Resonance structures (neutral and zwitterionic) for compound 41.
The resonance form 41b should greatly contribute to a global description of this compound, because the nitro group at C-3 is not coplanar with the aromatic ring. Therefore, the delocalization of the negative charge on this group in structure 41c must be less important than in 41b. In the latter, the bonds in which the oxygen and nitrogen atoms are involved show olefinic character, N1=C2 and C4=O5; accordingly, the distances measured by X-ray diffraction are significantly short (1285 Å and 1289 Å for N=C, 1259 Å and 1263 Å for C=O). It should be mentioned that solid-state 13C and 15N NMR spectra have also been used to assign imine/enamine structures, in view of a few diagnostic chemical shifts of such nuclei [35,36,37,38,39,40].
2.3. Structures in Solution
Imine and enamine structures can be assessed in solution through NMR analyses. Thus, for an imine structure, we would observe a singlet signal for the phenolic OH, whereas in an enamine form, the NH proton would be coupled with the protons of neighboring CHs bonds. Nonetheless, in view of a rapid dynamic equilibrium in solution, the recorded signals will average over time, resulting in resonances attributable to both structures. Therefore, the value measured for coupling constants could vary between 0 Hz and a maximum of ~14 Hz. However, the chemical shift of the aromatic carbon linked to the phenolic group is a more reliable indicator of the extent of such an equilibrium. In general, these resonances lie between the limiting chemical shifts of a phenolic carbon for imines and the carbonyl group of enamines (~155 ppm and ~180 ppm, respectively) [36,41,42,43,44,45,].
Table 2 shows the characteristic proton and carbon resonances for compounds 23–34 and 37, for which either imine or enamine structures can be attributed to the prevalent tautomers. Since all spectra are similar, only two-dimensional COSY and HMQC correlations allow us to assign some diagnostic peaks with confidence (2D-NMR spectra for 27 are displayed in Figures S5 and S6).
Table 2.
Selected NMR chemical shifts for 23–34 and 37 [a].
Despite half of the compounds having enamine structures in the solid state, 1H NMR spectra in DMSO-d6 at room temperature evidenced the prevalence of imino tautomers because the HC=N proton remains decoupled. Compound 41 represents a notable exception because, even if the NH proton appears as a broad singlet, the ethylene signal is a doublet with a high coupling constant (3J = 14 Hz) (Figure S7). However, if we look at 13C NMR data, compounds 38 and 43 are compatible with an enamine structure because they exhibit carbon resonances at ~178 ppm, which correspond unequivocally to enamine carbonyls, even though their ethylene protons lack coupling constants.
Nevertheless, a clear-cut structural assignment is not an easy task. As mentioned, the tautomeric equilibrium dictates that the observed chemical shifts (δexp) do actually correspond to average contributions of the imine (δi) and enamine (δe) forms (Equation (1)).
where ni and ne are the populations of molecules with imine and enamine structures, respectively (ni + ne = 1). The same applies to the coupling constant analysis, which can be represented by Equation (2) for the tautomeric equilibrium [1].
However, the presence of acid or basic substances capable of promoting a fast proton exchange can result in partial or totally decoupled spectra. This could account for the behavior of compound 38, which appears to have an imine structure according to its proton spectrum, while carbon resonances point to an enamino tautomer.
2.4. Theoretical Calculations on Imine–Enamine Stability
In order to gain further insight into the ease with which the imine–enamine transformation takes place, DFT calculations [46,47,48,49,50,51,52,53] were performed at the M06-2X/6-311G (d,p) level of theory [54,55,56], as implemented in the Gaussian package [57], and modeling solvation effects with the SMD method [58]. Compounds 24/31, 29/34, 38/43, 40/44 and 41 were used as models because 38, 40 and 41 have an enamine structure and, conversely, 24 and 29 display well-defined imine structures (Scheme 5). Table 3 and Table 4 collect the relative energies for imine and enamine structures, together with the corresponding transition structures (TS‡) characterized by imaginary frequencies, calculated in the gas phase and in DMSO (ε = 46.8).
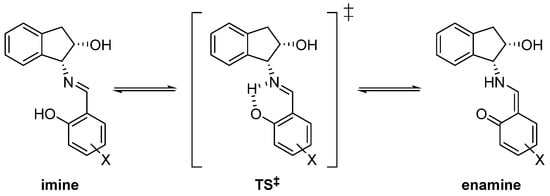
Scheme 5.
Tautomeric imine–enamine equilibrium trough a cyclic transition state (‡).
Table 3.
Relative energies calculated in the gas phase (kcal·mol−1) [a].
Table 4.
Relative energies calculated in DMSO (kcal·mol−1) [a].
Results found in the gas phase indicate that the most stable form is the imine structure. This contrasts with the existence of stable enamino tautomers in the solid state. Because no intermolecular interactions are present in the gas phase, the calculated relative stability expresses the intrinsic stability of each species. Therefore, the inversion of stability encountered in the solid phase reasonably has its origin in packing interactions that stabilize the crystal arrangement. However, results in DMSO (Table 4) agree with those observed experimentally, where compounds 38 and 41 have an enamine structure. That is, the interaction with solvent molecules can also cause the relative imine–enamine stabilities to be reversed.
In addition, calculations suggest that electron-donating groups favor an imine structure, whereas enamines form in the presence of electron-withdrawing substituents. In compound 38, the nitro group at C-5 increases the acidity of the hydroxyl and concomitantly decreases the basicity of the carbonyl oxygen, thus favoring an enamine structure (δC2 = 178.1 ppm). Figure 14 shows how the intense delocalization of the nitro group makes it coplanar with the hexadienic ring.
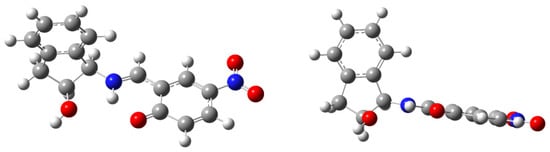
Figure 14.
Computed enamine structure 38, showing coplanarity of aryl and nitro groups.
It could be expected that the presence of two nitro groups would reinforce the aforementioned effect and, as a result, compound 41 should have an enamine structure characterized by δC2 similar to that of 38 or even higher. Surprisingly, this is not the case (δC2 = 170.1 ppm), the reason being that a steric hindrance inhibits delocalization. Thus, the electronic effect of the second nitro group is significantly attenuated by the adjacent carbonyl group, which forces the former to adopt a non-coplanar disposition with the ring (Figure 15).
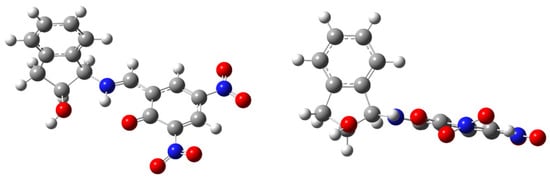
Figure 15.
Computed structure of compound 41 with different arrangements of nitro groups.
The interconversion barriers between the two tautomeric forms are always lower than 6.3 kcal·mol−1 (<4.7 kcal·mol−1 in DMSO), low enough for these transformations to proceed fast at room temperature. This ease of interconversion explains the landscapes in the Fourier maps of electron density differences, in which electron densities at the hydrogen atom are shared between the oxygen and nitrogen atoms. In other words, there must be a rapid equilibrium within the crystal lattice between imine and enamine tautomers.
In the gas phase, the energies of activation in the endothermic direction of the interconversion—that is, when going from enamine to imine—are generally negative (−0.4 ≤ ΔG‡ = ΔGTS‡ − ΔGenamine < 0.0 kcal·mol−1). The saddle points show an almost identical energy to the enaminic tautomers, and therefore the structure of the transition state is essentially coincidental with the ground structure. This curious—but not unusual—behavior has been described for imines derived from salicylaldehydes [59], acetophenones [60], aminoacroleins [61], vinamidines [62], gossypol [63] and a naturally-occurring aldehyde. This variational effect is associated with low activation barriers and high imaginary vibration frequencies (~1000 cm−1), as discussed previously in detail [59,60].
2.5. Electronic Effect of the Substituents on Imine–Enamine Tautomeric Equilibria
The tautomerization constant of the imine–enamine balance in solution [59], defined as KT = [enamine]/[imine] = ne/ni, is determined by:
KT = ne/ni = (δexp − δi)/(δe − δexp)
Constants can be calculated by choosing appropriate δi/δe values that represent “pure” imine or enamine structures.
Scheme 6 shows the equilibrium landscape related to the phenolimine–ketoamine tautomerism in solution. To study the influence exerted by the substituents of the salicylaldehyde core on the tautomeric equilibrium, we have applied the formalism developed for salicylimines [59] to the present case.
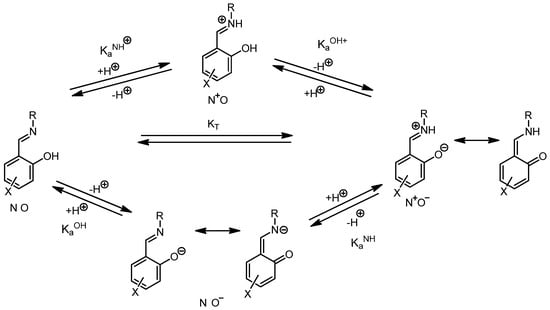
Scheme 6.
General view of phenolimine–ketoamine tautomeric equilibria.
The analysis of substituent effects on these equilibria leads to a modified Hammett Equation (4):
where σxOH = σparax if the substituent is placed at C-5 and σxOH = σmetax if it is at C-4; similarly, σxNH = σparax when the substituent is located at C-4 and σxNH = σmetax when it is at C-5. For polysubstitution, the additive Equation (5) can be used:
log KT = ρ(σxOH − σxNH) + c = ρσef + c
log KT = ρΣσef + c
The quantitative analysis was performed on imines 23–29 using the spectroscopic data collected in Table 2 and Table S3. By inserting the values δe = 178.06 ppm from 23 and δi = 154.62 ppm from 24 into Equation (3), we obtained Equation (6), whose graphical representation is shown in Figure 13a. Despite the limited number of compounds used, a very good fit could be obtained (r = 0.9690) thanks to the large range of σef values (Δσef ~0.65).
KT = ne/ni = (δexp − 154.62)/(178.06 − δexp)
To obtain a more general linear relationship for tautomeric equilibria, the lowest (152.00 ppm) [64] and the highest (180.18 ppm) [59] chemical shifts found in imines/enamines derived from salicylaldehydes were taken into account, leading to Equation (7). A plot of the latter results in Figure 16b exhibiting a good fit (r = 0.9175), yet it is slightly less accurate than that of Figure 16a.
KT = ne/ni = (δexp − 152.00)/(180.18 − δexp)
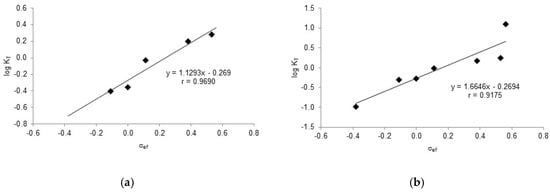
Figure 16.
Linear regression analyses for tautomeric constants of substituted imines/enamines versus 13C NMR chemical shifts. (a) δe = 178.06 ppm, δi = 154.62 ppm. (b) δe = 180.12 ppm, δi = 152.00 ppm.
Similarly, a good correlation could be obtained by plotting the chemical shift of the corresponding phenolic carbon (C2) versus δef, as summarized by Equation (8) and displayed in Figure S1.
δC2 = 20.384 σef + 162.62 (r = 0.9506)
However, the linear relationships involving the imine carbon against σOH and σNH, Equations (9) and (10) respectively, resulted in poorer correlations (Figure S2a,b). In contrast, a plot of log KT against C-2 chemical shifts produces a pretty good correlation (Equation (11), Figure S3); likewise, an accurate fit arises from correlating the chemical shifts of imine carbon and imine hydrogen (r = 0.9482, Figure S4), which evidence similar responses of such atoms to the electronic effects of substituents.
δCN = 1.1595.σOH + 165.04 (r = 0.8216)
δCN = 1.5391.σNH + 165.05 (r = 0.8324)
log KT = 0.084.δC2 + 165.05 (r = 0.9931)
2.6. Reaction of 1-Amino-2-Indanol with Salicylaldehyde
As introduced earlier (Section 2.1), the reaction of (1R,2S)-1-amino-2-indanol (20) with salicylaldehyde (46) in ethanol afforded not only the corresponding imine (29), but also an unknown product detected by thin-layer chromatography. Because the reaction mixture was reluctant to crystallize, it was brought to dryness after one week, giving rise to an oil that crystallized from ethyl ether. The latter was a mixture of white and yellow crystals whose chromatographic analysis (ethyl acetate:petroleum ether 1:5 v/v) showed two spots with Rf = 0.25 and 0.70, which clearly indicated the formation of two structurally different substances. Fortunately, their separation could be achieved after successive recrystallizations. The yellow solid had the lowest Rf value and corresponds to imine 29, while white crystals belonged to the fastest moving spot, further elucidated as 47 and generated by condensation of two salicylaldehyde molecules and one indanol moiety (Scheme 7).
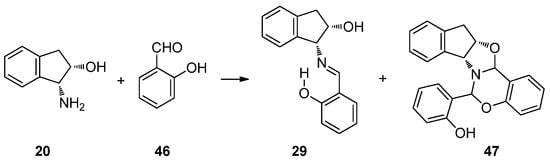
Scheme 7.
Reaction of (1R,2S)-1-amino-2-indanol with salicylaldehyde.
Analogously, starting from (1S,2R)-1-amino-2-indanol, the enantiomeric derivatives 34 and 48 could be isolated (Figure 17). Both analytical and spectroscopic data support the structures assigned to such compounds. In the FT-IR spectra, the main difference is that imine 29 presents an absorption at 1634 cm−1, attributable to the C=N double bond, while for 47, there are no absorptions in the range of 2000–1618 cm−1, thus evidencing the absence of either C=N or C=O bonds (Figures S26 and S31).
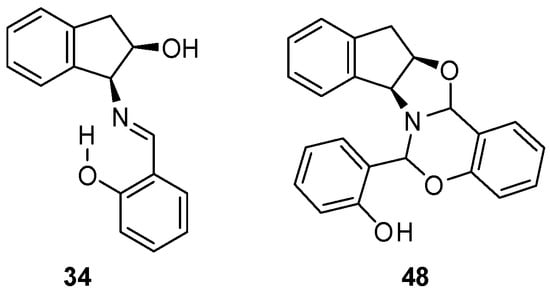
Figure 17.
Structures of 34 and 48, the enantiomers of 29 and 47, respectively.
Figures S10 and S11 collect 1H NMR and 2D-COSY spectra of 47. In the former, the most deshielded signal at 9.91 ppm should be assigned to the only phenolic proton, which can be exchanged with D2O. In the range of 8.0–6.5 ppm, a total of twelve protons are consistent with three aromatic rings. The remaining four protons of the non-aromatic residue of indanol are upfield shifted. An interesting signal appears as a singlet at 5.75 ppm, which, together with the absence of any peak attributable to the imine group (HC=N), points to a saturated carbon. In addition, that chemical shift is similar to that of other oxazolidines described previously [1,2,65,66,67,68,69], which suggests the existence of this heterocycle in 47. However, it is much more relevant a second singlet at 4.92 ppm, which could be consistent with an oxazine ring. Further structural information stems from the 13C NMR spectrum of 47, which has signals corresponding to three aromatic rings between 157–116 ppm. The 2D-experiment provided by the HMQC spectrum correlates the proton signals at 5.75 and 4.92 ppm with those of two carbons at 86.72 and 81.12 ppm, which are typical of saturated carbons and lie in the range observed for the C-2 atom in oxazolidine and oxazine rings (Figures S12 and S13).
To further confirm that the structure of 47 involves a fragment of indanol and two from salicylaldehyde, its high-resolution mass spectrum (chemical ionization mode) was recorded. A peak at m/z 358.1457 was detected, which agrees with the empirical formula C23H20NO3 (calculated m/z 358.1443) corresponding to the [M+H]+ ion of 47.
All the above data allowed us to propose a condensed polycyclic structure for 47, in which an oxazolidine plus a six-membered oxazine ring would have been formed, each containing a new chiral carbon. The fusion of rings generates a rigid structure and, so long as the nitrogen atom is pyramidal, its inversion is impeded and behaves as another stereogenic center. However, spectroscopic data alone are insufficient to determine the stereochemistry of the three new chiral centers, affording potentially up to eight (23) diastereoisomers. At last, X-ray diffractometry unambiguously confirmed the structure proposed for 47, thereby making it possible to establish the absolute configurations (Figure 18).
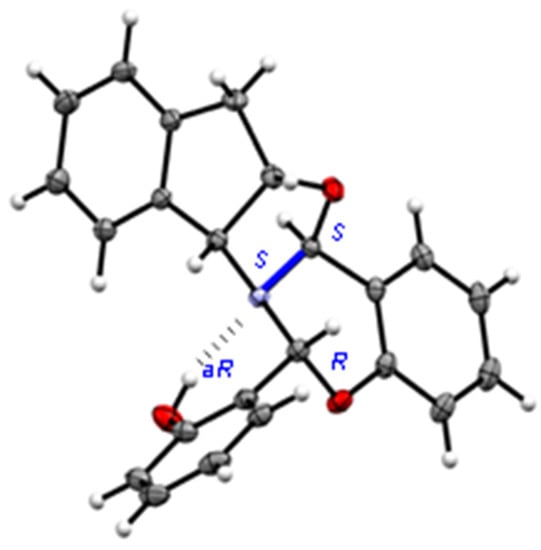
Figure 18.
X-ray diffraction of 47 showing the configuration of chiral centers.
It is worth noting that the fused rings show the following conformational arrangements:
- (a)
- The cyclopentane ring exhibits an E2 conformation [70], i.e., the two benzene carbons and the two saturated carbons forming the cyclopentane ring are virtually coplanar because the dihedral angle ΦC15,C19,C18,C17 is close to zero (0.29°) (Table 5, Figure 19).
Table 5. Empirical and calculated [a] dihedral angles (°) for compound 47.
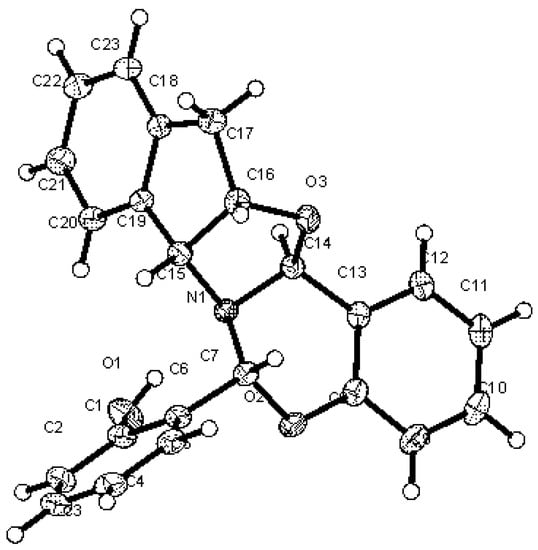 Figure 19. Crystallographic numbering for compound 47.
Figure 19. Crystallographic numbering for compound 47. - (b)
- The oxazolidine fragment shows a 3T2 conformation.
- (c)
- The oxazine ring adopts an unusual conformation for a six-membered heterocycle because five atoms lie approximately in the same plane, which is reflected by the low values of the dihedral angles ΦO2,C8,C13,C14 (2.56°) and ΦO2,C8,C13,C14 (9.12°), whereas the carbon atom between the oxygen and nitrogen atoms lies above that plane.
The nitrogen atom shows a strong pyramidalization, which can be defined as the difference between the sum of the three bond angles sharing a common nitrogen atom (Σθ°) and a planar (360°) system. The calculated pyramidalization (360° − Σθ°) is ~35°, somewhat larger than that of an ideal tetrahedral system such as methane or ammonium ion: 31.5° (Table 6) [71]. As a consequence of pyramidalization and the lack of inversion, the nitrogen atom behaves in practice as a chiral center with absolute (S)-configuration.
Table 6.
Empirical and calculated [a] bonding angles (°) for 47.
The X-ray diffraction pattern of 47 shows a hydrogen bridge between the phenolic hydroxyl and the nitrogen atom. The geometrical data of this hydrogen bridge are gathered in Table S4. This intramolecular hydrogen bond anchors the conformation of the orto-hydroxyphenyl group, strongly restricting its rotational mobility and creating a new source of chirality: an atropisomeric axis of chirality aR [72,73].
2.7. On the Stability of Diastereomeric Combinations
The stereochemical complexity shown by 47 (or 48) arises from the coexistence of four new stereogenic elements, leaving aside the pre-existing chirality of the indanol moiety, namely two new chiral carbons, a chiral pyramidal nitrogen, and a chiral axis due to the intramolecular hydrogen bond that largely restricts the conformational mobility. In other words, there can potentially be up to sixteen diastereoisomers (24), whose discrimination is indeed challenging. Using theoretical calculations, we evaluated the relative stability of all the sixteen diastereoisomers (Figure 20), both in the gas phase and in ethanol, in an attempt to elucidate why one and only one diastereomer was actually isolated. Computational results are summarized in Table 7.
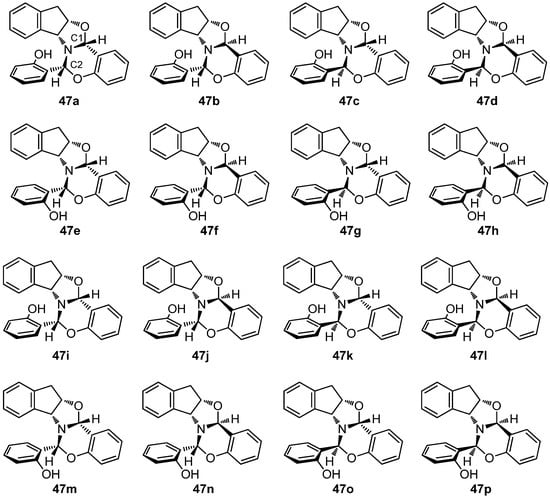
Figure 20.
Diastereomeric combinations of compound 47 having multiple stereogenic elements.
Table 7.
Calculated relative energies (kcal·mol−1) for all diastereomers of 47 [a].
Moreover, from the viewpoint of structural diversity, all diastereoisomers of 47 can also form intramolecular hydrogen bonds with the nitrogen or the oxygen atom, whose geometries are collected in Tables S5 and S6. Of all possible stereoisomeric combinations, the most stable, both in the gas phase and in ethanol, is 47b, which is in fact the experimentally observed diastereomer. The remaining diastereomers are less stable, up to ~15 kcal·mol−1. The calculated structure of 47b (Figure 21) is coincidental with that determined by single-crystal X-ray diffraction analysis (Figure 18).
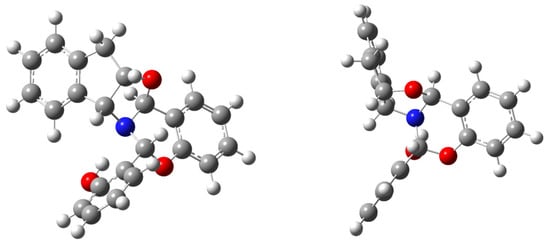
Figure 21.
Calculated structure of 47b (frontal and lateral views).
2.8. Hydrogen Bonding Energy
The chemical shift displacement of the hydroxyl proton can be estimated by Schaefer’s correlation [74] according to Equation (12), giving rise to a value of 6.02 kcal.mol−1 for the strength of the intramolecular bonding in 47b.
EHB = −δexp + 3.89 ± 0.2 = −9.91 + 3.89 ± 0.2 = −6.02 ± 0.2 kcal·mol−1
In contrast, the empirical relationship of Musin and Mariam [75], using the value of dD···A (2.736 Å) as determined by X-ray diffraction, results in Equation (13):
EHB (kcal·mol−1) = −5.554 × 105 e−4.12dD…A = −7.06 kcal·mol−1
The latter (7.06 kcal·mol−1) is slightly higher than the energies calculated in the gas phase and ethanol (6.37 and 6.14 kcal·mol−1, respectively). Furthermore, the stabilizing effect supplied by H-bonding manifests itself by computing the relative energies for 47b with intramolecular hydrogen bond against 49, which is lacking that interaction (Table 8, Figure 22). The latter is generated by 180°-rotation around the phenolic bond. Structure 47b becomes more stable by ~9 kcal·mol−1 in the gas phase and by ~4 kcal·mol−1 when solvation is included. Although the intramolecular H-bond is not especially strong, it suffices to stabilize 47b to a significant extent and reduce the conformational flexibility.
Table 8.
Relative energies calculated for 47b and 49 [a].
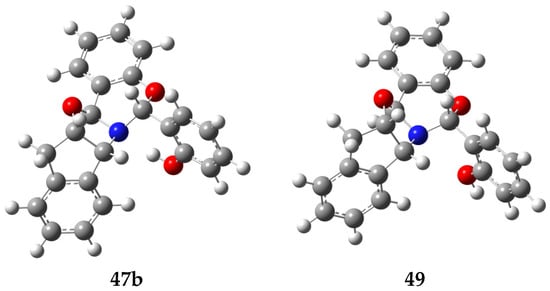
Figure 22.
Computer-generated structures 47b (H-bonded) and 49.
2.9. Reaction Mechanism: Experiment and Rationale
In order to rationalize the formation and isolation of compound 47b, a series of additional experiments and calculations were undertaken. (a) Because this substance contains two units of salicylaldehyde and one indanol fragment, the reaction was repeated in ethanol using a 1:2 indanol-aldehyde ratio and monitored by TLC. Formation of 29 could be observed almost immediately, while compound 47b appeared after five days. (b) When the reaction was conducted in diethyl ether using the aforementioned 1:2 molar ratio, formation of 47b was not detected at all after two weeks, and only imine 29 could be isolated by crystallization. (c) The same reaction was monitored by 1H NMR in MeOH-d4 over time, which evidenced the rapid formation of imine (29). After 24 h, two proton signals at ~5.7 and 4.9 ppm attributable to the chiral carbons of 47b were observed, in a very low proportion nevertheless. After 40 days, the proportion of the latter increased significantly up to a 4:1 ratio between 29 and 47b. Given the low solubility of 47b in methanol, it slowly crystallized on standing. Figure 23 and Figure 24 show the 1H NMR and 13C NMR monitoring of the reaction mixture after 5 min and after 40 days at room temperature. The initial mixture only contained imine and salicylaldehyde mixtures, whereas signals corresponding to 47b appeared progressively after 24 h.
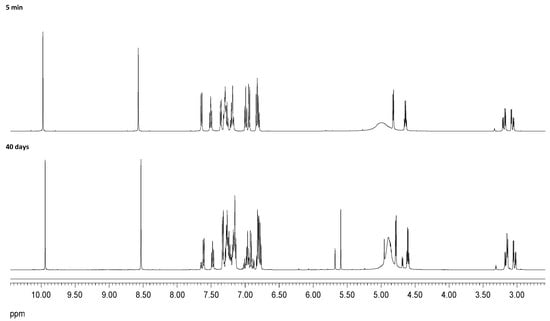
Figure 23.
Temporal recording (1H NMR) for the reaction of 20 and salicylaldehyde in CD3OD.
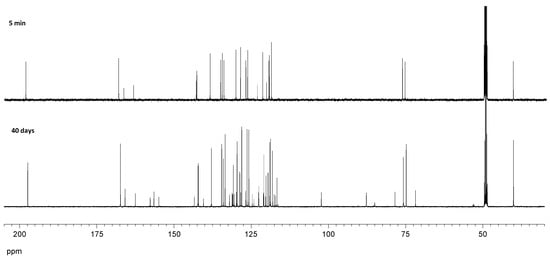
Figure 24.
Temporal recording (13C NMR) for the reaction of 20 and salicylaldehyde in CD3OD.
The singlet signal appearing at ~5.7 ppm correlates (HMQC spectrum) with the carbon signal resonating at 102.3 ppm. Chemical shifts similar to those encountered in oxazolidines suggested the intermediacy of this heterocycle. However, the signal is also accompanied by new peaks in the aromatic zone, which are consistent with the incorporation of salicylaldehyde. In fact, the addition of this substance to the imine in methanol resulted in the formation of 47b after one week. Thus, it is plausible to assume that imine 29 is formed first and subsequent reaction with salicylaldehyde produces 47b. As previously noted, this transformation is highly diastereoselective without observing other isomers. Attempts to synthesize analogs of 47b by varying the substituents at the aromatic ring were unsuccessful.
To shed light upon the ease with which the intermediate is formed with respect to the starting reagents, the relative stability of such candidates, i.e., imine 29 or diastereomeric oxazolidines (50 and/or 51), was evaluated as well (Scheme 8).
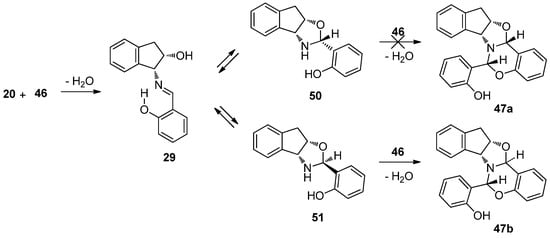
Scheme 8.
Competitive equilibria between imine and oxazolidines toward 47b.
Four different structures should be taken into account depending on whether the chiral center formed is R (50) or S (51), and depending on whether the hydrogen bond involving the aromatic OH is oriented towards the nitrogen (a) or the oxygen atoms of the oxazine ring (b) (Figure 25).
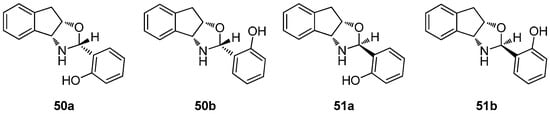
Figure 25.
Structures studied theoretically (DFT method).
Table 9 shows the calculation of relative energies, which favor formation of imine in both the gas phase and ethanol (exothermic pathway). In addition, the imine structure is less stable than the corresponding oxazolidines; however, experiments verified the formation of imine 29 and adduct 47b. The enhanced stability of 50a and 51a could be justified by the intramolecular H-bond with the nitrogen atom (geometrical parameters are tabulated in Table S7).
Table 9.
Relative energies calculated in kcal·mol−1 [a].
Finally, the stability of structure 47b has been calculated with respect to the corresponding reaction intermediates, which are imine 29 and oxazolidines 50 and 51. Although in the gas phase 47b is not the most stable structure, it becomes more stable than the intermediate in ethanol. Formation of 47b is presumably driven by a combination of both its thermodynamic stability and its insolubility in the reaction mixture, thereby leading to spontaneous separation by crystallization. Overall, the equilibria involving other competing species are shifted towards adduct 47b.
2.10. Acetylation of Indanol Imines
The acetylation of compounds 35–37 was carried out with the aim of studying their transformation into N-acetyl oxazolidines under acylating conditions. Compound 35 was chosen as the model to optimize the reaction conditions, which involved the treatment with acetic anhydride in pyridine at room temperature for 24 h, followed by precipitation in ice water. The crude product was found to be a mixture of the acetylated imine 52 and N-acetyl oxazolidine 53. By fractional crystallization from ethanol, compound 52 was isolated as a yellow crystalline solid, followed by 53 as colorless crystals. Likewise, compounds 54 and 55 could be obtained from 36 (Scheme 9). However, when the reaction was carried out with trans-imine 37, only acetyl imine 56 was formed. Probably the origin of this behavior is due to the trans arrangement of the C=N and OH groups, which prevent the closure of a five-membered ring. Interestingly, in all the isolated products 52–56, the phenolic hydroxyl remained unacetylated. At first glance, we conjectured that they underwent deacetylation during the ice water treatment used for isolation; however, as we shall see later, the reason is a strong steric hindrance experienced by the phenolic hydroxyl, flanked by the imine function and a bulky tert-butyl group.
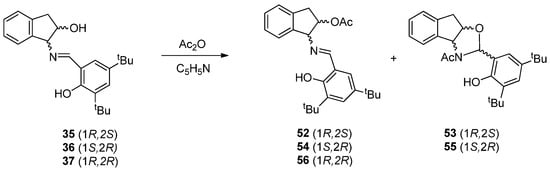
Scheme 9.
Acetylation of indanol imines.
As is customary, structures assigned to 52–56 are supported by analytical and spectroscopic data. The main difference among these heterocyclic derivatives, as inferred from FT-IR spectra (Table 10), is that imines 52, 54 and 56 show two absorption bands at ~1738 cm−1 and ~1627 cm−1, corresponding to the stretching vibrations of the C=O (acetate) and C=N (imine) bonds, respectively; for oxazolidines 53 and 55, that spectral zone only shows the stretching vibration of the amide bond at ~1622 cm−1 (Figure S14 highlights this difference in compounds 52 and 53).
Table 10.
Yields and IR absorptions (cm−1) of 52–56.
Moreover, NMR data recorded in the solution allowed us to differentiate between both structures (Tables S8 and S9). Assignments were aided by COSY and HMQC two-dimensional correlations. 1H NMR spectra of 52, 54 and 56 display a singlet signal at 8.5 ppm corresponding to the iminic proton, while oxazolidines 53 and 55 show the proton of the new chiral center at ~6.6 ppm. The phenolic hydroxyl signal in imines appears at a a much lower field (~13.6 ppm) than in oxazolidines (~9.2 ppm), owing to the existence of a strong intramolecular hydrogen bond in the first compounds. Acetate groups of imines appear at ~2.1 ppm and acetamido groups in oxazolidines at ~2.4 ppm (Tables S8 and S9). Likewise, 13C NMR spectra show the iminic carbon at ~167 ppm, while the C-2 resonance in the oxazolidine derivatives appears at ~87 ppm (see Figures S15 and S16 for 1H and 13C NMR spectra of compounds 52 and 53).
Finally, unequivocal solid-state structures of compounds 52 and 53 could be determined by X-ray diffraction (Figure 26). As noted above, compound 52 arises from monoacetylation of imine 35, while the phenolic hydroxyl remains unprotected as a result of a crowding environment. The position of the hydrogen atom on oxygen evidences an imine tautomer having an intramolecular H-bonded structure.
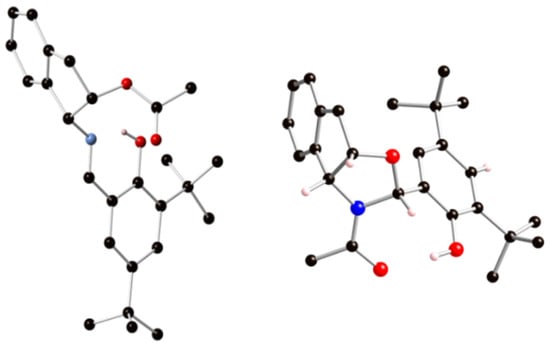
Figure 26.
Crystal structures (ball-and-stick models) of 52 (left) and 53 (right).
When comparing the experimental bond lengths (X-ray data) of 52 collected in Table 11 with those of Table 1, both geometries agree with a phenoliminic structure. The values of 1.288 Å and 1.365 Å correspond to typical C=N and C-O bond lengths of imines.
Table 11.
Crystal and calculated [a] bond distances (Å) of 52.
The crystallographic analysis was also instrumental in unambiguously verifying the N-acetyl oxazolidine skeleton of 53 and the absolute (R)-configuration at the newly created chiral carbon atom. Remarkably, no chirality can be assigned to the nitrogen atom, essentially when it is flat and has a low degree of pyramidalization (<2°) due to the intense delocalization of the lone pair on the carbonyl group.
P = 360 − (120.1 + 128.3 + 109.9) = 1.7°
Like imines, the phenolic OH group of oxazolidines remains unprotected upon acetylation. In lieu of an intramolecular hydrogen bridge involving the endocyclic oxygen leading to a six-membered ring, the OH group is capable of closing an eight-membered ring with the amide carbonyl group (Table S10). In addition, this H-bonding anchors a Z-configuration around the amide bond.
2.11. Imine Formation versus Oxazolidine Rings
The concomitant formation of imines and oxazolidines through acetylation suggests that their synthesis could be dictated by the interplay of thermodynamic and kinetic conditions. To this end, we performed a series of acetylation experiments on compound 35 at different temperatures and conditions. Results are summarized below and illustrated by NMR spectra (Figure 27).
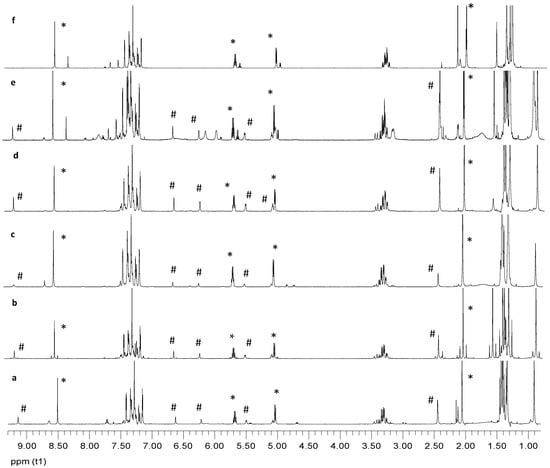
Figure 27.
1H NMR spectra recorded in CDCl3 of acetylation experiments on compound 35 (compounds 52, *, and 53, #).
- (a)
- When imine 35 is acetylated in pyridine at room temperature and worked-up as usual (poured into ice-water after 24 h), the crude product consisted in O-acetylated imine (52) and N-acetylated oxazolidine (53) in a 5:2 ratio.
- (b)
- Acetylation of 35 under the above conditions and then brought to dryness to avoid the aqueous work-up resulted in the same 5:2 ratio of 52 and 53. Clearly, the phenolic hydroxyl substituents do not undergo acetylation, thus evidencing a strong steric hindrance imposed by adjacent groups. It is known that this acetylation reaction is very sensitive to steric crowding when the tetrahedral intermediate leading to acetate is generated [76,77].
- (c)
- Acetylation of imine 35 in pyridine at −20 °C and worked up after 24 h by pouring the reaction into ice water resulted in a crude mixture of 52:53 in 7:1 ratio.
- (d)
- Acetylation of imine 35 in pyridine at 70 °C for 2 h followed by aqueous work-up decreased the 52:53 ratio to 2:1.
- (e)
- When imine 35 is acetylated in pyridine at higher temperatures (110–120 °C) for 2 h followed by aqueous work-up, the 52:53 ratio decreased further (1:1). Moreover, additional signals appeared in the proton NMR spectrum, most likely due to thermal decomposition.
- (f)
- Finally, to determine whether or not imine–oxazolidine interconversion takes place, compound 52 was subjected to acetylation (in pyridine at 110–120 °C) for 2 h, followed by product isolation after aqueous work-up. NMR monitoring gave no indication of oxazolidine (53) formation, albeit peaks accounting for a new side product (4:1 ratio) could be observed. The latter might be ascribed to per-O-diacetylated imine. Thus, at a high temperature, the formation of oxazolidine with respect to imine is favored, which was a behavior pointing to kinetic control during the formation of compounds 52 and 53.
A theoretical analysis to determine the relative stability of imine and oxazolidine structures was also carried out. Table 12 shows such results for imine 52, (R)-configured (53) and (S)-configured (57) oxazolidines, computed in the gas phase and pyridine as solvent (Figure 28). The imine structure is invariably the most stable species, followed by oxazolidine 53, whose configuration is coincidental with the experimental result. It is surprising that the intramolecular hydrogen bond is established with the acetamido group (53) and not with the endocyclic oxygen atom through a six-membered cycle (53a). However, calculations support the former, whose arrangement is more stable than that of 53a. The geometric parameters of hydrogen bonds for structures 52, 53 and 53a are collected in Table S11.
Table 12.
Relative energies for 52, 53, 53a and 57 (kcal·mol−1) [a].
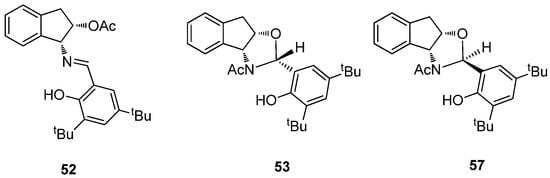
Figure 28.
Stereochemistry of 52, 53 and 57.
Notably, the computed structures for 52 and 53, shown in Figure 29, are practically coincidental with those determined by single-crystal X-ray diffraction. The fact that imine 52 is experimentally formed at a greater extent, and that the proportion of oxazolidine 53 increases as temperature increases, would apparently be consistent with a kinetically controlled transformation. However, calculations show that 52 is actually more stable than 53 and the overall transformation proceeds in fact under thermodynamic control.
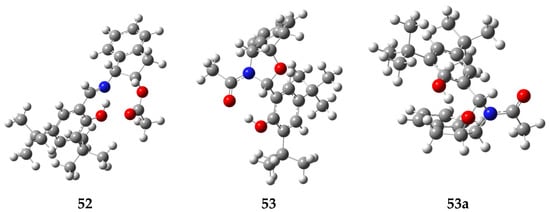
Figure 29.
DFT-calculated structures of 52, 53 and 53a.
3. Materials and Methods
3.1. General Methods
All solvents and chemicals were purchased from commercial suppliers and used without further purification. Solvents were evaporated below 40 °C at pressures between 15 and 30 mm Hg. Melting points were measured on an Electrothermal 9100 apparatus in capillary tubes and are uncorrected. Optical rotations were determined on a Perkin-Elmer 141 polarimeter at different wavelengths (Na and Hg lamps), as specified. Elemental analyses for C, H, N and S were performed using a Leco CHNS-932 analyzer. FTIR spectra were recorded on a Thermo IR-300 spectrophotometer (4000–600 cm−1 range) using KBr pellets. All reactions were monitored by thin-layer chromatography (TLC) on Aldrich Polygram Sil G/UV254 plates (7 × 3 cm). 1H NMR (500, 400 MHz) and 13C NMR (125, 100 MHz) spectra were obtained using Bruker Avance spectrometers, which were recorded at room temperature in CDCl3 and DMSO-d6. Chemical shifts are reported in parts per million (δ) downfield from tetramethylsilane (Me4Si, TMS) as internal reference. Coupling constants (J values) are given in Hz and standard abbreviations were used to indicate spin multiplicities, namely s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublet, m = multiplet. Carbon chemical shifts in the decoupled 13C{1H} NMR spectra are reported relative to CHCl3 (δC 77.00 ppm, central line of triplet). The nature of the carbons (C, CH, CH2 and CH3) was determined by recording the DEPT spectra. The latter, along with 2D NMR correlations, enabled the assignment of signals. Computational data were obtained using the Gaussian09 program package [57]. All structures were optimized through density functional theory (DFT) [46,47,48,49,50,51,52,53] using the M06-2X method [54] and the 6-311G(d,p) basis set [55,56]. The stationary points were confirmed by frequency analysis at the above-mentioned level of theory at 298.15 K. Solvent effects were calculated using the solvation model density (SMD) [58]. Intrinsic reaction coordinate (IRC) analysis validated that all the transition structures belonged to the reaction path.
Compounds 35 and 36 were purchased from Aldrich and used as received.
3.2. Computation
Computational data were obtained using the Gaussian09 program package [57]. All structures were optimized through density functional theory (DFT) [46,47,48,49,50,51,52,53] using the M06-2X method [54] and the 6-311G(d,p) basis set [55,56]. In all cases, frequency calculations were also carried out to confirm the existence of true stationary points on the potential energy surface. All thermal corrections were calculated at the standard values of 1 atm at 298.15 K. Solvent effects were calculated using the solvation model density (SMD) [58].
3.3. X-ray Data Collection and Structural Refinement
Suitable crystals were selected and mounted on a MITIGEN holder in perfluoroether oil, using a Rigaku AFC12 FRE-HF diffractometer. The crystal was kept at T = 100(2) K during data collection. Using Olex2 [78], the structure was solved with the ShelXT structure solution program [79], using the Direct Methods solution method. The model was refined with a version of ShelXL [80] using Least Squares minimization (see Tables S12–S18 for a survey of crystallographic data).
3.4. Synthetic Procedures: Method A
A solution of 1-amino-2-indanol (1.0 mmol) in the minimum amount of ethanol was added to a solution of the corresponding salicylaldehyde (1.0 mmol) in ethanol. The mixture was kept under stirring at room temperature and, after a few minutes, the formation of a precipitate could be observed. This solid phase was filtered, washed with cold ethanol, dried, and recrystallized from ethanol.
3.5. Synthetic Procedures: Method B
A solution of 1-amino-2-indanol (1.0 mmol) in diethyl ether was added to a solution of the corresponding salicylaldehyde (2.0 mmol) in diethyl ether. The mixture was stirred at room temperature for 24 h and then evaporated under reduced pressure to afford the corresponding imine.
3.6. Synthetic Procedures: Method C
A solution of 1-amino-2-indanol (1.0 mmol) in ethanol was added to a solution of the corresponding salicylaldehyde (2.0 mmol) in ethanol. The mixture was stirred at room temperature for 24 h. Then, it was stored at 5 °C for one week, evaporated to dryness, and the resulting oil crystallized from diethyl ether.
(1R,2S)-1-[(2-Hydroxy-5-methoxybenzylidene)amino]-2-indanol (24). Following procedure A and from 5-methoxysalicylaldehyde, compound 24 was obtained (75%); m.p. 103–105 °C; [α]D20 − 16.4°; [α]57820 − 14.5°; [α]54620 − 4.2° (c 0.5, DMSO); IR (KBr) max/cm−1 3362 (OH), 1647 (C=N), 1623, 1588 (C=C), 1531, 1490 (arom); Raman max/cm−1 1639 (C=N), 1619 (C=C); 1H NMR (500 MHz, DMSO-d6) δ 13.05 (s, 1H, OH), 8.67 (s, 1H, CH=N), 7.31 (d, J = 7.5 Hz, 1H, H-arom), 7.26 (dt, J = 1.5 Hz, J = 8.0 Hz, 1H, H-arom), 7.24 (m, 2H, H-arom), 7.10 (d, J = 3.5 Hz, 1H, H-arom), 6.95 (dd, J = 3.0 Hz, J = 9.0 Hz, 1H, H-arom), 6.79 (d, J = 9 Hz, 1H, H-arom), 5.18 (d, J2,OH = 5.0 Hz, 1H, C2-OH), 4.74 (d, J1,2 = 5.5 Hz, 1H, H-1), 4.54 (q, J = 6.0 Hz, 1H, H-2), 3.73 (s, 3H, CH3), 3.10 (dd, J2,3 = 6.5 Hz, J3,3′ = 15.5 Hz, 1H, H-3), 2.93 (dd, J2,3′ = 6.0 Hz, J3,3′ = 15.5 Hz, 1H, H-3′); 13C NMR (125 MHz, DMSO-d6) δ 165.19 (CH=N), 154.62 (C-OH), 151.15, 141.82, 140.89, 127.76, 126.39, 124.85, 124.43, 119.11, 118.35, 116.99, 114.65 (C-arom), 74.06, 73.83 (C1, C2), 55.30 (CH3), 39.54 (C3). Anal. Calculated for C17H17NO3: C, 72.07; H, 6.05; N, 4.94. Found: C, 71.87; H, 6.15; N, 4.82.
(1R,2S)-1-[(5-Bromo-2-hydroxybenzylidene)amino]-2-indanol (25). Using procedure A and from 5-bromosalicylaldehyde: (51%); m.p. 148–150 °C; [α]D22 − 20.8°; [α]57822 − 18.1°; [α]54622 − 5.6° (c 0.5, DMSO); IR (KBr) max/cm−1 3330 (OH), 1648 (C=N), 1627, 1605 (C=C), 1514, 1499, 1477 (arom); Raman max/cm−1 1638 (C=N), 1625 (C=C); 1H NMR (500 MHz, DMSO-d6) δ 13.92 (bs, 1H, OH), 8.68 (s, 1H, CH=N), 7.70 (d, J = 3.0 Hz, 1H, H-arom), 7.44 (dd, J = 2.5 Hz, J = 8.5 Hz, 1H, H-arom), 7.31 (d, J = 7.0 Hz, 1H, H-arom), 7.26 (dt, J =1.5 Hz, J = 6.5 Hz, 1H, H-arom), 7.19 (m, 2H, H-arom), 6.79 (d, J = 8.5 Hz, 1H, H-arom), 5.28 (d, J2,OH = 5.0 Hz, 1H, C2-OH), 4.79 (d, J1,2 = 5.5 Hz, 1H, H-1), 4.55 (q, J = 5.5 Hz, 1H, H-2), 3.11 (dd, J2,3 = 6.0 Hz, J3,3′ = 16.0 Hz, 1H, H-3), 2.91 (dd, J2,3′ = 6.0 Hz, J3,3′ = 15.5 Hz, 1H, H-3′); 13C NMR (125 MHz, DMSO-d6) δ 164.54 (CH=N), 161.26 (C-OH), 141.48, 141.06, 134.83, 133.61, 128.05, 126.60, 125.05, 124.58, 120.13, 119.39, 108.32 (C-arom), 73.79, 73.47 (C1, C2), 39.02 (C3). Anal. Calculated for C16H14BrNO2: C, 57.85; H, 4.25; N, 4.22. Found: C, 57.66; H, 4.27; N, 4.11.
(1S,2R)-1-[(2-Hydroxy-5-methoxybenzylidene)amino]-2-indanol (31). Using procedure A and from 5-methoxysalicylaldehyde: (59%); m.p. 100–102 °C; [α]D22 + 13.1°; [α]57822 + 11.3°; [α]54622 + 1.4° (c 0.5, DMSO); IR (KBr) max/cm−1 3332 (OH), 1645 (C=N), 1624, 1588 (C=C), 1523, 1492, 1475 (arom); Raman max/cm−1 1639 (C=N), 1619 (C=C); 1H NMR (500 MHz, DMSO-d6) δ 13.05 (s, 1H, OH), 8.67 (s, 1H, CH=N), 7.31 (d, J = 7.5 Hz, 1H, H-arom), 7.26 (dt, J = 1.5 Hz, J = 7.0 Hz, 1H, H-arom), 7.24 (m, 2H, H-arom), 7.10 (d, J = 3.0 Hz, 1H, H-arom), 6.95 (dd, J = 3.5 Hz, J = 9.0 Hz, 1H, H-arom), 6.78 (d, J = 9.0 Hz, 1H, H-arom), 5.18 (d, J2,OH = 5.0 Hz, 1H, C2-OH), 4.74 (d, J1,2 = 5.0 Hz, 1H, H-1), 4.54 (q, J = 5.5 Hz, 1H, H-2), 3.73 (3H, s, CH3), 3.10 (dd, J2,3 = 6.0 Hz, J3,3′ = 15.5 Hz, 1H, H-3), 2.93 (dd, J2,3′ = 6.0 Hz, J3,3′ = 15.5 Hz, 1H, H-3′); 13C NMR (125 MHz, DMSO-d6) δ 165.53 (CH=N), 154.95 (C-OH), 151.48, 142.15, 141.23, 128.09, 126.73, 125.18, 124.76, 119.44, 118.68, 117.32, 114.98 (C-arom), 74.39, 73.17 (C1, C2), 55.63 (CH3) 39.19 (C3); 1H NMR (500 MHz, CDCl3) δ 12.42 (sa, 1H, OH), 8.55 (s, 1H, CH=N), 7.30 (m, 2H, H-arom), 7.23 (dt, J = 1.5 Hz, J = 7.5 Hz, 1H, H-arom), 7.24 (d, J = 7.0 Hz, 1H, H-arom), 6.95 (dd, J = 3.5 Hz, J = 9.0 Hz, 1H, H-arom), 6.89 (d, J = 9.0 Hz, 1H, H-arom), 6.84 (d, J = 3.0 Hz, 1H, H-arom), 4.80 (d, J1,2 = 5.5 Hz, 1H, H-1), 4.69 (c, J = 5.5 Hz, 1H, H-2), 3.79 (s, 3H, CH3), 3.24 (dd, J2,3 = 6.0 Hz, J3,3′ = 16.0 Hz, 1H, H-3), 3.09 (dd, J2,3′ = 5.5 Hz, J3,3′ = 16.0 Hz, 1H, H-3′); 13C NMR (125 MHz, CDCl3) δ 166.60 (CH=N), 155.13 (C-OH), 152.15, 140.71, 140.65, 128.62, 127.06, 125.48, 124.84, 120.06, 118.29, 117.91, 115.13 (C-arom), 75.66, 75.27 (C1, C2), 55.94 (CH3), 39.60 (C3).
(1S,2R)-1-[(5-Bromo-2-hydroxybenzylidene)amino]-2-indanol (32). Using procedure A and from 5-bromosalicylaldehyde: (59%); m.p. 147–149 °C; [α]D25 + 17.2°; [α]57825 + 14.4°; [α]54625 + 2.8° (c 0.5, DMSO); IR (KBr) max/cm−1 3329 (OH), 1648 (C=N), 1627, 1605 (C=C), 1515, 1477 (arom); Raman max/cm−1 1638 (C=N), 1625 (C=C); 1H NMR (500 MHz, DMSO-d6) δ 13.91 (1H, s, OH), 8.69 (s, 1H, CH=N), 7.72 (d, J = 2.5 Hz, 1H, H-arom), 7.44 (dd, J = 2.5 Hz, J = 9.0 Hz, 1H, H-arom), 7.31 (d, J = 7.5 Hz, 1H, H-arom), 7.26 (dt, J = 2.0 Hz, J = 8.5 Hz, 1H, H-arom), 7.19 (m, 2H, H-arom), 6.79 (d, J = 9.0 Hz, 1H, H-arom), 5.24 (d, J2,OH = 5.0 Hz, 1H, C2-OH), 4.79 (d, J1,2 = 5.5 Hz, 1H, H-1), 4.55 (q, J = 5.5 Hz, 1H, H-2), 3.11 (dd, J2,3 = 6.0 Hz, J3,3′ = 15.5 Hz, 1H, H-3), 2.91 (dd, J2,3′ = 5.5 Hz, J3,3′ = 15.5 Hz, 1H, H-3′); 13C NMR (125 MHz, DMSO-d6) δ 164.06 (CH=N), 160.77 (C-OH), 140.98, 140.57, 134.34, 133.09, 127.56, 126.11, 124.56, 124.09, 119.64, 118.91, 107.83 (C-arom), 73.29, 72.97 (C1, C2), 38.52 (C3). Anal. Calculated for C16H14BrNO2: C, 57.85; H, 4.25; N, 4.22. Found: C, 57.56; H, 4.31; N, 3.93.
(1R,2R)-1-[(3,5-Di-tert-butyl-2-hydroxybenzylidene)amino]-2-indanol (37). Using procedure A and from 3,5-di-tert-butylsalicylaldehyde: (78%); m.p. 69–71 °C; [α]D25 − 48.5°; [α]57825 − 49.5°; [α]54625 + 50.9° (c 0.5, DMSO); IR (KBr) max/cm−1 3339 (OH), 1627 (C=N), 1596, 1475 (arom); Raman max/cm−1 1622 (C=N), 1591 (C=C); 1H NMR (500 MHz, DMSO-d6) δ 13.96 (s, 1H, OH), 8.75 (s, 1H, CH=N), 7.39 (d, J = 2.0 Hz, 1H, H-arom),7.34 (s, 1H, H-arom), 7.23 (m, 3H, H-arom), 7.08 (d, J = 7.5 Hz, 1H, H-arom), 5.53 (d, J2,OH = 5.5 Hz, 1H, C2-OH), 4.63 (d, J1,2 = 6.0 Hz, 1H, H-1), 4.39 (q, J1,2 = J2,3 = J2,OH = 6.5 Hz, 1H, H-2), 3.23 (dd, J2,3 = 7.0 Hz, J3,3′ = 15.5 Hz, 1H, H-3), 2.84 (dd, J2,3′ = 8.0 Hz, J3,3′ = 15.5 Hz, 1H, H-3′), 1.37 (s, 9H, CH3), 1.29 (s, 9H, CH3); 13C NMR (125 MHz, DMSO-d6) δ 167.79 (CH=N), 157.37 (C-OH), 141.50, 139.66, 139.53, 135.45, 127.85, 126.72, 126.52, 126.14, 124.68, 123.78, 117.78 (C-arom), 79.17, 78.94 (C1,C2), 38.92 (C3), 34.43, 33.76 (C-(CH3)3), 31.19, 29.14 (CH3). Anal. Calculated for C28H33NO3: C, 76.62; H, 8.12; N, 3.44. Found: C, 76.31; H, 7.95; N, 3.33.
(6Z)-6-[(1R,2S)-(2-hydroxyindan-1-yl)amino]methylen]-4-nitrocyclohexa-2,4-dienone (38). Using procedure A and from 5-nitrosalicylaldehyde, compound 38 was obtained: (91%); m.p. 212–214 °C; [α]D20 + 35.2°; [α]57820 + 40.9°; [α]54620 + 63.9° (c 0.5, DMSO); IR (KBr) max/cm−1 3174 (OH), 1655 (C=O), 1611 (C=C), 1544 (NO2), 1515, 1441 (arom), 1330 (NO2); Raman max/cm−1 1655 (C=O), 1603 (C=C); 1H NMR (500 MHz, DMSO-d6) δ 14.49 (bs, 1H, NH), 8.94 (s, 1H, CH-N), 8.53 (d, J =3.0 Hz, 1H, H-cyclo), 8.04 (dd, J = 3.0 Hz, J =9.5 Hz, 1H, H-cyclo), 7.33 (m, 3H, H-arom), 7.27 (m, 1H, H-arom), 6.58 (d, J = 9.5 Hz, 1H, H-cyclo), 5.74 (d, J2,OH = 2.5 Hz, 1H, C2-OH), 5.20 (d, J1,2 = 4.5 Hz, 1H, H-1), 4.65 (bs, 1H, H-2), 3.18 (dd, J2,3 = 5.5 Hz, J3,3′ = 16.0 Hz, 1H, H-3), 2.91 (dd, J2,3′ = 2.5 Hz, J3,3′ = 16.0 Hz, 1H, H-3′); 13C NMR (125 MHz, DMSO-d6) δ 178.06 (C=O), 166.86 (C-NH), 140.99, 138.77, 133.45, 132.74, 129.12, 128.55, 126.70, 125.19, 124.36, 122.75, 113.30 (C-arom), 72.04, 68.09 (C1, C2), 39.93 (C3). Anal. Calculated for C18H14N2O4: C, 64.42; H, 4.73; N, 9.39. Found: C, 64.66; H, 4.81; N, 9.64.
(6Z)-6-[(1R,2S)-(2-hydroxyindan-1-yl)amino]methylen]-5-bromocyclohexa-2,4-dienone (39). Using procedure A and from 5-bromosalicylaldehyde: (62%); m.p. 179–181 °C; [α]D20 + 109.01°; [α]57820 + 121.10°; [α]54620 + 170.11° (c 0.5, DMSO); IR (KBr) max/cm−1 3174 (OH), 1642 (C=N), 1597, 1502, 1454 (arom); Raman max/cm−1 1641 (C=N);1H NMR (500 MHz, DMSO-d6) δ 14.32 (bs, 1H, OH), 8.69 (s, 1H, CH=N), 7.39 (d, J = 8.0 Hz, 1H, H-arom), 7.32 (d, J = 7.0 Hz, 1H, H-arom), 7.28 (m, 1H, H-arom), 7.21 (m, 2H, H-arom), 6.97 (d, J = 1.5 Hz, 1H, H-arom), 6.92 (dd, J = 2.0 Hz, J = 8.5 Hz, 1H, H-arom), 5.33 (d, J2,OH = 5.0 Hz, 1H, C2-OH), 4.87 (d, J1,2 = 5.0 Hz, 1H, H-1), 4.56 (q, J = 5.5 Hz, 1H, H-2), 3.12 (dd, J2,3 = 6.0 Hz, J3,3′ = 16.0 Hz, 1H, H-3), 2.91 (dd, J2,3′ = 5.0 Hz, J3,3′ = 15.5 Hz, 1H, H-3′); 13C NMR (125 MHz, DMSO-d6) δ 165.89 (C-OH), 165.23 (CH=N), 141.03, 133.75, 128.15, 126.64, 125.11, 124.53, 120.88, 119.60, 116.78 (C-arom), 73.39, 71.99 (C1, C2), 39.09 (C3). Anal. Calculated for C16H14BrNO2: C, 57.85; H, 4.25; N, 4.22. Found: C, 57.73; H, 4.29; N, 4.19.
(6Z)-6-[(1R,2S)-(2-hydroxyindan-1-yl)amino]methylen]-5-methoxycyclohexa-2,4-dienone(40). Using procedure A and from 4-methoxysalicylaldehyde: (86%); m.p. 169–171 °C; [α]D22 + 157.9°; [α]57822 + 172.5°; [α]54622 + 228.4° (c 0.5, DMSO); IR (KBr) max/cm−1 3182 (OH), 1644 (C=O), 1616 (C=C), 1514, 1484 (arom); Raman max/cm−1 1638 (C=O), 1608 (C=C); 1H NMR (500 MHz, DMSO-d6) δ 14.01 (s, 1H, OH), 8.49 (s, 1H, CH=N), 7.31 (d, J = 7.5 Hz, 1H, H-cyclo), 7.26 (m, 2H, H-arom), 7.21 (m, 2H, H-arom), 6.30 (dd, J = 2.5 Hz, J = 8.5 Hz, 1H, H-cyclo), 6.20 (d, J = 2.5 Hz, 1H, H-cyclo), 5.30 (d, J2,OH = 4.5 Hz, 1H, C2-OH), 4.81 (d, J1,2 = 5.5 Hz, 1H, H-1), 4.54 (q, J = 4.5 Hz, 1H, H-2), 3.74 (s, 3H, CH3), 3.11 (dd, J2,3 = 6.0 Hz, J3,3′ = 16.0 Hz, 1H, H-3), 2.90 (dd, J2,3′ = 4.5 Hz, J3,3′ = 15.5 Hz, 1H, H-3′); 13C NMR (125 MHz, DMSO-d6) δ 168.92 (C-OH), 164.03 (CH=N), 163.93, 141.48, 140.94, 133.56, 127.99, 126.59, 125.07, 124.40, 111.68, 105.49, 101.24 (C-arom), 73.33, 71.26 (C1, C2), 55.07 (CH3) 39.35 (C3). Anal. Calculated for C17H17NO3: C, 72.07; H, 6.05; N, 4.94. Found: C, 71.83; H, 6.05; N, 4.91.
(6Z)-6-[(1R,2S)-(2-hydroxyindan-1-yl)amino]methylen]-4,6-dinitrocyclohexa-2,4-dienone(41). Using procedure A and from 3,5-dinitrosalicylaldehyde: (80%); m.p. 145–147 °C; [α]D25 + 211.5°; [α]57825 + 234.34°; [α]54625 + 334.9°; (c 0.5, DMSO); IR (KBr) max/cm−1 3407, 3350 (OH), 1650 (C=O), 1616 (C=C), 1556 (NO2) 1531 (arom); Raman max/cm−1 1666 (C=O); 1H NMR (500 MHz, DMSO-d6) δ 13.86 (bs, 1H, NH), 9.10 (d, J = 14.0 Hz, 1H, CH-N), 8.82 (d, J = 2.5 Hz, 1H, H-cyclo), 8.76 (d, J = 3.0 Hz, 1H, H-cyclo), 7.36 (m, 3H, H-arom), 7.28 (dt, J = 2.5 Hz, J = 6.5 Hz, 1H, H-arom), 5.94 (d, J2,OH = 4.5 Hz, 1H, C2-OH), 4.37 (t, J1,2 = JNH,1 = 7.0 Hz, 1H, H-1), 4.70 (m, 1H, H-2), 3.22 (dd, J2,3 = 5.5 Hz, J3,3′ = 16.5 Hz, 1H, H-3), 2.94 (dd, J2,3′ = 5.0 Hz, J3,3′ = 16.5 Hz, 1H, H-3′); 13C NMR (125 MHz, DMSO-d6) δ 169.99 (C=O), 167.97 (CH-N), 141.36,140.83, 137.67, 137.31, 129.88, 129.04, 127.00, 126.96, 125.46, 124.70, 117.06 (C-arom), 71.79, 67.51 (C1, C2), 39.68 (C3). Anal. Calculated for C16H13N3O6: C, 55.98; H, 3.82; N, 12.24. Found: C, 55.98; H, 3.85; N, 12.25.
(6Z)-6-[(1R,2S)-(2-hydroxyindan-1-yl)amino]methylen]cyclohexa-2,4-dienone(42). Using procedure B and from salicylaldehyde: (28%); m.p. 113–115 °C; [α]D22 + 16.3°; [α]57822 + 19.4°; [α]54622 + 36.3 (c 0.5, DMSO); IR (KBr) max/cm−1 3015 (OH), 1634 (C=O), 1607 (C=C), 1513, 1473 (arom); Raman max/cm−1 1635 (C=O); 1H NMR (500 MHz, DMSO-d6) δ 13.73 (s, 1H, OH), 8.71 (s, 1H, CH=N), 7.50 (dd, J = 2.0 Hz, J = 8.0 Hz, 1H, H-arom), 7.33 (dd, J = 2.0 Hz, J = 8.5 Hz, 2H, H-arom), 7.27 (m, 1H, H-arom), 7.20 (m, 2H, H-arom), 6.87 (dt, J = 1.5 Hz, J = 7.5 Hz, 1H, H-arom), 6.84 (d, J = 8.0 Hz, 1H, H-arom), 5.19 (d, J2,OH = 5.5 Hz, 1H, C2-OH), 4.78 (d, J1,2 = 5.5 Hz, 1H, H-1), 4.56 (q, J = 6.0 Hz, 1H, H-2), 3.13 (dd, J2,3 = 6.0 Hz, J3,3′ = 15.5 Hz, 1H, H-3), 2.94 (dd, J2,3′ = 6.0 Hz, J3,3′ = 15.5 Hz, 1H, H-3′); 13C NMR (125 MHz, DMSO-d6) δ 166.24 (CH=N), 161.80 (C-OH), 142.48, 141.60, 132.81, 132.28, 128.50, 127.12, 125.58, 125.15, 119.26, 118.64, 117.14 (C-arom), 74.50, 74, 45 (C1,C2), 39.85 (C3); 1H NMR (500 MHz, CD3OD) δ 8.61 (s, 1H, CH=N), 7.38 (dd, J = 1.5 Hz, J = 3.0 Hz, 1H, H-arom), 7.31 (m, 3H, H-arom), 7.21 (m, 2H, H-arom), 6.82 (m, 2H, H-arom), 4.85 (d, J1,2 = 5.0 Hz, 1H, H-1), 4.64 (q, J1,2 = J2,3 = 5.5 Hz, 1H, H-2), 3.21 (dd, J2,3 = 6.0 Hz, J3,3′ = 16 Hz, 1H, H-3), 3.06 (dd, J2,3′ = 5.0 Hz, J3,3′ = 15.5 Hz, 1H, H-3′); 13C NMR (125 MHz, CD3OD) δ 167.33 (CH=N), 162.49 (C-OH), 142.22, 142.10, 134.53, 129.56, 128.05, 126.33, 125.74, 122.62, 120.91, 119.04, 118.75 (C-arom), 75.76, 74.76 (C1,C2), 40.09 (C3). Anal. Calculated for C16H15NO2: C, 75.87; H, 5.97; N, 5.53. Found: C, 75.98; H, 5.98; N, 5.71.
(6Z)-6-[(1S,2R)-(2-hydroxyindan-1-yl)amino]methylen]-4-nitrocyclohexa-2,4-dienone(43). Using procedure A and from 5-nitrosalicylaldehyde, compound 43 was obtained: (85%); m.p. 219–221 °C; [α]D22 − 45.5°; [α]57822 − 52.5°; [α]54622 − 80.4°; [α]43622 43.7° (c 0.5, DMSO); IR (KBr) max/cm−1 3209 (OH), 1656 (C=O), 1610 (arom), 1543 (NO2), 1514, 1444 (arom), 1330 (NO2); Raman max/cm−1 1655 (C=O), 1603 (C=C); 1H NMR (500 MHz, DMSO-d6) δ 14.50 (bs, 1H, NH), 8.95 (s, 1H, CH-N), 8.54 (d, J = 3.0 Hz, 1H, H-cyclo), 8.05 (dd, J = 3.0 Hz, J = 9.5 Hz, 1H, H-cyclo), 7.33 (m, 3H, H-arom), 7.27 (m, 1H, H-arom), 6.59 (d, J = 10.0 Hz, 1H, H-cyclo), 5.75 (d, J2,OH = 3.0 Hz, 1H, C2-OH), 5.20 (d, J1,2 = 5.0 Hz, 1H, H-1), 4.66 (sa, 1H, H-2), 3.19 (dd, J2,3 = 5.5 Hz, J3,3′ = 16.5 Hz, 1H, H-3), 2.91 (dd, J2,3′ = 3.0 Hz, J3,3′ = 16.5 Hz, 1H, H-3′); 13C NMR (125 MHz, DMSO-d6) δ 178.10 (C=O), 166.88 (C-NH), 140.99, 138.78, 133.45, 132.76, 129.14, 128.56, 126.71, 125.20, 124.37, 122.79, 113.30 (C-arom), 72.05, 68.10 (C1, C2), 39.96 (C3).
(6Z)-6-[(1S,2R)-(2-hydroxyindan-1-yl)amino]methylen]-5-methoxycyclohexa-2,4-dienone(44). Using procedure A and from 4-methoxysalicylaldehyde: (88%); m.p. 170–172 °C; [α]D22 − 167.6°; [α]57822 − 182.5°; [α]54622 − 241.1° (c 0.5, DMSO); IR (KBr) max/cm−1 3186 (OH), 1644 (C=O), 1615 (C=C), 1514, 1484, 1456 (arom); Raman max/cm−1 1638 (C=O), 1608 (C=C);1H NMR (500 MHz, DMSO-d6) δ 14.01 (s, 1H, OH), 8.49 (s, 1H, CH=N), 7.31 (d, J = 7.0 Hz, 1H, H-cyclo), 7.26 (m, 2H, H-arom), 7.20 (m, 2H, H-arom), 6.30 (dd, J = 2.0 Hz, J = 8.5 Hz, 1H, H-cyclo), 6.20 (d, J = 2.0 Hz, 1H, H-cyclo), 5.29 (d, J2,OH = 4.0 Hz, 1H, C2-OH), 4.81 (d, J1,2 = 5.0 Hz, 1H, H-1), 4.54 (q, J = 4.0 Hz, 1H, H-2), 3.74 (s, 3H, CH3), 3.10 (dd, J2,3 = 6.0 Hz, J3,3′ = 16.0 Hz, 1H, H-3), 2.89 (dd, J2,3′ = 5.0 Hz, J3,3′ = 16.0 Hz, 1H, H-3′); 13C NMR (125 MHz, DMSO-d6) δ 168.88 (C-OH), 164.03 (CH=N), 163.92, 141.48, 140.94, 133.54, 127.98, 126.58, 125.06, 124.39, 111.68, 105.48, 101.23 (C-arom), 73.32, 71.26 (C1, C2), 55.06 (CH3) 36.68 (C3). Anal. Calculated for C17H17NO3: C, 72.07; H, 6.05; N, 4.94. Found: C, 71.83; H, 6.05; N, 4.91.
(6Z)-6-[(1S,2R)-(2-hydroxyindan-1-yl)amino]methylen]cyclohexa-2,4-dienone(45). Using procedure B and from salicylaldehyde: (35%); m.p. 110–112 °C; [α]D18 − 15.2°; [α]57818 − 17.9°; [α]54618 − 33.6 (c 0.5, DMSO); IR (KBr) max/cm−1 3119 (OH), 1639 (C=O), 1611 (C=C), 1513, 1490 (arom); Raman max/cm−1 1635 (C=O); 1H NMR (500 MHz, DMSO-d6) δ 13.74 (s, 1H, OH), 8.71 (s, 1H, CH=N), 7.49 (dd, J = 1.0 Hz, J = 7.5 Hz, 1H, H-arom), 7.33 (m, 2H, H-arom), 7.27 (m, 1H, H-arom), 7.20 (m, 2H, H-arom), 6.90 (t, J = 7.5 Hz, 1H, H-arom), 6.84 (d, J = 8.0 Hz, 1H, H-arom), 5.21 (d, J2,OH = 5.0 Hz, 1H, C2-OH), 4.78 (d, J1,2 = 5.5 Hz, 1H, H-1), 4.56 (q, J = 5.5 Hz, 1H, H-2), 3.12 (dd, J2,3 = 6.5 Hz, J3,3′ = 16.0 Hz, 1H, H-3), 2.94 (dd, J2,3′ = 5.5 Hz, J3,3′ = 15.5 Hz, 1H, H-3′); 13C NMR (125 MHz, DMSO-d6) δ 165.87 (CH=N), 161.41 (C-OH), 141.22, 132.44, 131.89, 128.12, 126.74, 125.20, 124.77, 118.26, 116.76 (C-arom), 74.11, 74.08 (C1,C2), 39.27 (C3). Anal. Calculated for C16H15NO2: C, 75.87; H, 5.97; N, 5.53. Found: C, 75.47; H, 5.90; N, 5.35.
Reaction of (1R,2S)-1-amino-2-indanol with salicylaldehyde. Using procedure C and from salicylaldehyde, compound 47 was obtained: (88%); m.p. 157–158 °C; [α]D18 + 26.1°; [α]57818 + 27.0°; [α]54618 + 31.7°; [α]43618 + 66.0° (c 0.5, CHCl3); IR (KBr) max/cm−1 3187 (OH), 1619, 1589 (arom), 1253, 1221 (C-O-C), 1027 (C-O); 1H NMR (500 MHz, DMSO-d6) δ 9.91 (s, 1H, OH), 7.81 (dd, J = 1.5 Hz, J = 7.5 Hz, 1H, H-arom), 7.32 (m, 2H, H-arom), 7.27 (m, 1H, H-arom), 7.24 (m, 2H, H-arom), 7.20 (m, 2H, H-arom), 7.05 (dt, J = 1.0 Hz, J = 8.0 Hz, 1H, H-arom), 6.99 (m, 2H, H-arom), 6.93 (d, J = 8.0 Hz, 1H, H-arom), 5.75 (s, 1H, H-2 oxazine), 4.92 (s, 1H, H-2 oxazolidine), 4.83 (dt, J2,3′ = 1.0 Hz, J1,2 = J2,3 = 6.0 Hz, 1H, H-2 indanol), 4.61 (d, J1,2 = 5.5 Hz, 1H, H-1 indanol), 3.23 (dd, J2,3 = 6.5 Hz, J3,3′ = 18.0 Hz, 1H, H-3 indanol), 3.14 (d, J3,3′ = 17.5 Hz, 1H, H-3′ indanol); 13C NMR (125 MHz, DMSO-d6) δ 156.66 (C-OH), 153.85, 142.60, 140.05, 130.94, 130.42, 130.04, 129.57, 128.89, 127.48, 125.73, 125.20, 123.88, 121.72, 120.07, 119.73, 116.57, 116.12 (C-arom), 86.72 (C2 oxazolidine), 81.12 (C2 oxazine), 77.17 (C2 indanol), 70.53 (C1 indanol), 40.17 (C3 indanol); 1H NMR (500 MHz, CD3OD) δ 7.69 (dd, J = 1.5 Hz, J = 7.5 Hz, 1H, H-arom), 7.32 (m, 6H, H-arom), 7.22 (m, 1H, H-arom), 7.24 (m, 2H, H-arom), 7.04 (m, 2H, H-arom), 6.94 (t, J = 8.5 Hz, 2H, H-arom), 5.73 (s, 1H, H-2 oxazine), 5.02 (s, 1H, H-2 oxazolidine), 4.77 (d, J1,2 = J2,3 = 6.0 Hz, 1H, H-2 indanol), 4.62 (s, 1H, H-1 indanol), 3.25 (d, J2,3 = 5.5 Hz, 2H, H-3, H-3′ indanol); 13C NMR (125 MHz, CD3OD) δ 157.76(C-OH), 155.11, 143.55, 140.64, 132.00, 131.26, 130.98, 130.58, 129.83, 128.42, 126.82, 125.85, 124.38, 122.69, 121.07, 120.49, 117.63, 117,08 (C-arom), 87.94 (C2 oxazolidine) 84.73 (C2 oxazine) 78.70 (C2 indanol), 71.97 (C1 indanol), 40.19 (C3 indanol). HRMS-CI (C23H19NO3 [M+H]+): calculated 358.1457; found 358.1443.
Reaction of (1S,2R)-1-amino-2-indanol with salicylaldehyde. Using procedure C and from salicylaldehyde, compound 48 was obtained: (20%); m.p. 152–154 °C; [α]D18 − 23.9°; [α]57818 − 25.2°; [α]54618 − 29.3°; [α]43618 − 61.0° (c 0.5, CHCl3); IR (KBr) max/cm−1 3201 (OH), 1617, 1587 (arom), 1250, 1225 (C-O-C), 1025 (C-O); 1H NMR (500 MHz, DMSO-d6) δ 9.91 (s, 1H, OH), 7.81 (dd, J = 1.5 Hz, J = 8.0 Hz, 1H, H-arom), 7.31 (m, 2H, H-arom), 7.27 (m, 1H, H-arom), 7.23 (m, 2H, H-arom), 7.20 (m, 2H, H-arom), 7.05 (t, J = 7.5 Hz, 1H, H-arom), 6.98 (m, 2H, H-arom), 6.93 (d, J = 8.5 Hz, 1H, H-arom), 5.75 (s, 1H, H-2 oxazine), 4.91 (s, 1H, H-2 oxazolidine), 4.83 (t, J1,2 = J2,3 = 6.0 Hz, 1H, H-2), 4.60 (d, J1,2 = 6.0 Hz, 1H, H-1), 3.22 (dd, J2,3 = 6.0 Hz, J3,3′ = 18.0 Hz, 1H, H-3), 3.13 (d, J3,3′ = 18.0 Hz, 1H, H-3′); 13C NMR (125 MHz, DMSO-d6) δ 156.65 (C-OH), 153.84, 142.60, 140.03, 130.94, 130.42, 130.04, 129.57, 128.89, 127.48, 125.72, 125.20, 123.86, 121.72, 120.07, 119.72, 116.57, 116.10 (C-arom), 86.71 (C2 oxazolidine), 81.10 (C2 oxazine), 77.16 (C2 indanol), 70.53 (C1 indanol), 39.30 (C3 indanol).
3.7. General Acetylation Procedure
For a solution of the corresponding imine (1.0 mmol) in pyridine (3.0 mL), acetic anhydride (2.0 mL) was added. The mixture was kept at room temperature for 24 h and then was poured into ice water. The resulting white solid was filtered and washed with water. The mixture containing imine and oxazolidine was purified by fractional crystallization from ethanol. The first crystalline material corresponds to acetylated imine, while the mother liquors contain the oxazolidine derivative.
(1R,2S)-2-O-Acetyl-1-[(3,5-di-tert-butyl-2-hydroxybenzylidene)amino]-2-indanol(52). Following the above procedure and 35: 45%; m.p. 144–146 °C; [α]D20 − 100.0°; [α]57820 − 103.1°; [α]54620 − 114.2° (c 0.5, CHCl3); IR (KBr) max/cm−1 1739 (C=O), 1627 (C=N), 1594, 1473 (arom); 1H NMR (400 MHz, CDCl3) δ 13.64 (s, 1H, OH), 8.52 (s, 1H, CH=N), 7.41 (d, J = 2.4 Hz, 1H, H-arom), 7.34 (m, 2H, H-arom), 7.27 (m, 1H, H-arom), 7.20 (d, J = 7.6 Hz, 1H, H-arom), 7.15 (d, J = 2.8 Hz, 1H, H-arom), 5.68 (1H, c, J1,2 = J2,3 = J2,3′ 5.6 Hz, H-2), 5.04 (d, J1,2 = 6.0 Hz, 1H, H-1), 3.35 (dd, J2,3 = 6.8 Hz, J3,3′ = 16.4 Hz, 1H, H-3), 2.28 (dd, J2,3′ = 5.2 Hz, J3,3′ = 16.4 Hz, 1H, H-3′), 2.07 (s, 3H, OCH3), 1.44 (s, 9H, CH3), 1.34 (s, 9H, CH3); 13C NMR (100 MHz, CDCl3) δ 171.01 (C=O), 167.00 (CH=N), 158.28 (C-OH), 140.79, 140.04, 139.85, 136.76, 128.66, 127.35,127.30, 126.27,125.07, 124.95, 117.79 (C-arom), 76.06, 73.17 (C1, C2), 36.84 (C3), 35.05, 34.16 (C-(CH3)3), 31.52, 29.38 (CH3), 20.97 (OCH3). 1H NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H, OH), 8.79 (s, 1H, CH=N), 7.36 (m, 4H, H-arom), 7.25 (t, J = 6.4 Hz, 1H, H-arom), 7.18 (d, J = 7.2 Hz, 1H, H-arom), 5.64 (sa, 1H, H-2), 5.10 (d, J1,2 = 4.8 Hz, 1H, H-1), 3.36 (m, 1H, H-3), 3.11 (dd, J2,3′ = 2.4 Hz, J3,3′ = 16.8 Hz, 1H, H-3′), 1.95 (s, 3H, OCH3), 1.36 (s, 9H, CH3), 1.29 (s, 9H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 170.29 (C=O), 168.99 (CH=N), 158.35 (C-OH), 141.56, 140.37, 140.11, 136.18, 128.82, 127.57, 127.11, 127.08, 125.42, 124.89, 118.20 (C-arom), 76.39, 72.59 (C1,C2), 37.11 (C3), 35.04, 34.34 (C-(CH3)3), 31.76, 29.68 (CH3), 21.05 (OCH3). Anal. Calculated for C26H33NO3: C, 76.62; H, 8.16; N, 3.44. Found: C, 76.35; H, 8.07; N, 3.35.
N-Acetyl-3,5-di-tert-butyl-2-{(2R,3aR,8aS)-3,3a,8,8a-tetrahydro-2H-indeno [1,2-d]oxazol-2-yl}phenol(53). Following the general procedure from compound 35: 11%; m.p. 164–166 °C; [α]D20 − 208.8°; [α]57820 − 217.8°; [α]54620 − 250.3°; [α]43620 − 449.7° (c 0.5, CHCl3); IR (KBr) max/cm−1 3118 (OH), 1622 (C=O), 1602, 1480 (arom); 1H NMR (400 MHz, CDCl3) δ 9.15 (s, 1H, OH), 7.46 (d, J = 7.6 Hz, 1H, H-arom), 7.40 (t, J = 7.2 Hz, 1H, H-arom), 7.33 (d, J = 7.6 Hz, 1H, H-arom), 7.27 (m, 1H, H-arom), 7.16 (d, J = 2.4 Hz, 1H, H-arom), 6.63 (s, 1H, CH), 6.23 (d, J = 2.4 Hz, 1H, H-arom), 5.50 (d, J1,2 = 4.8 Hz, 1H, H-1), 5.06 (t, J1,2 = J2,3′ = 4.4 Hz, 1H, H-2), 3.43 (d, J3,3′ = 17.2 Hz, 1H, H-3), 3.28 (dd, J2,3′ = 4.4 Hz, J3,3′ = 17.2 Hz, 1H, H-3′), 2.44 (s, 3H, OCH3), 1.41 (s, 9H, CH3), 0.91 (s, 9H, CH3); 13C NMR (100 MHz, CDCl3) δ 170.14 (C=O), 151.95 (C-OH), 141.66, 141.17, 139.91, 138.03, 129.36, 127.77, 126.05, 125.26, 124.93, 124.46, 121.25 (C-arom), 86.57 (CH) 82.34 (C2), 66.61 (C1), 37.09 (C3), 35.04, 33.97 (C-(CH3)3), 31.20, 29.82 (CH3), 23.22 (OCH3). Anal. Calculated for C26H33NO3: C, 76.62; H, 8.16; N, 3.44. Found: C, 76.37; H, 8.08; N, 3.45.
(1S,2R)-2-O-Acetyl-1-[(3,5-di-tert-butyl-2-hydroxybenzylidene)amino]-2-indanol(54). Following the general procedure from compound 36: 44%; m.p. 145–147 °C; [α]D20 + 115.2°; [α]57820 + 117.9°; [α]54620 + 132.4° (c 0.5, CHCl3); IR (KBr) max/cm−1 1739 (C=O), 1627 (C=N), 1595, 1473 (arom); 1H NMR (400 MHz, CDCl3) δ 13.62 (s, 1H, OH), 8.51 (s, 1H, CH=N), 7.42 (d, J = 2.4 Hz, 1H, H-arom), 7.34 (m, 2H, H-arom), 7.28 (m, 1H, H-arom), 7.21 (d, J = 7.2 Hz, 1H, H-arom), 7.16 (d, J = 2.4 Hz, 1H, H-arom), 5.69 (c, J1,2 = J2,3 = J2,3′ = 5.6 Hz, 1H, H-2), 5.04 (d, J1,2 = 6.0 Hz, 1H, H-1), 3.36 (dd, J2,3 = 6.4 Hz, J3,3′ = 16.4 Hz, 1H, H-3), 2.28 (dd, J2,3′ = 5.2 Hz, J3,3′ = 16.4 Hz, 1H, H-3′), 2.06 (s, 3H, OCH3), 1.44 (s, 9H, CH3), 1.35 (s, 9H, CH3); 13C NMR (100 MHz, CDCl3) δ 170.97 (C=O), 166.98 (CH=N), 158.29 (C-OH), 140.79, 140.05, 139.85, 136.77, 128.66, 127.34, 127.30, 126.26, 125.06, 124.95, 117.81 (C-arom), 76.05, 73.15 (C1, C2), 36.83 (C3), 35.05, 34.16 (C-(CH3)3), 31.51, 29.39 (CH3), 20.94 (OCH3). Anal. Calculated for C26H33NO3: C, 76.62; H, 8.16; N, 3.44. Found: C, 76.39; H, 8.09; N, 3.34.
N-Acetyl-3,5-di-tert-butyl-2-{(2S,3aS,8aR)-3,3a,8,8a-tetrahydro-2H-indeno [1,2-d]oxazol-2-yl}phenol(55). Following the general procedure from compound 36: 10%; m.p. 166–168 °C; [α]D20 + 242.8°; [α]57820 + 255.1°; [α]54620 + 292.2°; [α]43620 + 523.5° (c 0.5, CHCl3); IR (KBr) max/cm−1 3117 (OH), 1623 (C=O), 1602, 1480 (arom); 1H NMR (500 MHz, CDCl3) δ 9.15 (s, 1H, OH), 7.46 (d, J = 7.5 Hz, 1H, H-arom), 7.40 (t, J = 7.0 Hz, 1H, H-arom), 7.33 (d, J = 7.5 Hz, 1H, H-arom), 7.27 (m, 1H, H-arom), 7.16 (d, J = 2.0 Hz, 1H, H-arom), 6.63 (s, 1H, CH), 6.22 (d, J = 2.0 Hz, 1H, H-arom), 5.50 (d, J1,2 = 4.5 Hz, 1H, H-1), 5.07 (t, J1,2 = J2,3′ = 4.5 Hz, 1H, H-2), 3.43 (d, J3,3′ =17.5 Hz, 1H, H-3), 3.29 (dd, J2,3′ = 4.0 Hz, J3,3′ = 17.5 Hz, 1H, H-3′), 2.45 (s, 3H, OCH3), 1.41 (s, 9H, CH3), 0.91 (s, 9H, CH3); 13C NMR (125 MHz, CDCl3) δ 170.26 (C=O), 151.96 (C-OH), 141.65, 141.17, 139.92, 138.03, 129.35, 127.77, 126.04, 125.26, 124.93, 124.44, 121.26 (C-arom), 86.59 (CH) 82.34 (C2), 66.63 (C1), 37.10 (C3), 35.04, 33.97 (C-(CH3)3), 31.21, 29.83 (CH3), 23.21 (OCH3).
(1R,2R)-2-O-Acetyl-1-[(3,5-di-tert-butyl-2-hydroxybenzylidene)amino]-2-indanol(56). Following the general procedure from compound 37: 64%; m.p. 106–108 °C; [α]D25 − 48.1°; [α]57825 − 48.5°; [α]54625 − 51.3°; [α]43625 − 28.7° (c 0.5, CHCl3); IR (KBr) max/cm−1 1737 (C=O), 1625 (C=N), 1598, 1460 (arom); 1H NMR (500 MHz, CDCl3) δ 13.25 (sa, 1H, OH), 8.55 (s, 1H, CH=N), 7.42 (d, J = 2.0 Hz, 1H, H-arom), 7.27 (m, 3H, H-arom), 7.16 (m, 2H, H-arom), 5.45 (c, J1,2 = J2,3 = J2,3′ = 6.0 Hz, 1H, H-2), 4.88 (d, J1,2 = 5.5 Hz, 1H, H-1), 3.63 (dd, J2,3 = 7.0 Hz, J3,3′ = 16.0 Hz, 1H, H-3), 2.93 (dd, J2,3′ = 6.0 Hz, J3,3′ = 16.5 Hz, 1H, H-3′), 2.08 (s, 3H, OCH3), 1.44 (s, 9H, CH3), 1.35 (s, 9H, CH3); 13C NMR (125 MHz, CDCl3) δ 170.81 (C=O), 167.31 (CH=N), 158.08 (C-OH), 140.37, 140.29, 139.49, 136.90, 128.67, 127.50, 127.32, 126.32, 125.04, 124.55, 117.68 (C-arom), 81.19, 77.62 (C1, C2), 37.14 (C3), 35.06, 34.19 (C-(CH3)3), 31.52, 29.2 (CH3), 21.17 (OCH3).
4. Conclusions
In conclusion, a series of Schiff bases from chiral and conformationally rigid 1-amino-2-indanol enantiomers have been obtained and fully characterized by spectroscopic methods. Crystal data reveal the tautomeric preference and electronic delocalization in the solid phase, as does NMR spectroscopy for structures in solution, which were further assessed by computational analyses. Good correlations have been obtained between tautomeric equilibria and chemical shifts of phenolimine or ketoamine signals, thus revealing the influence of substitution patterns. Reaction of cis-amino indanols and salicylaldehyde exhibit a high level of stereocontrol, as disclosed by the formation of a single diastereomeric adduct arising from two aldehyde molecules. All configurations of the stereogenic elements through the oxazolidine–oxazine fragment created could be determined by single-crystal diffraction analysis with structures computed by DFT methods. The geometrical and energy features of intramolecular H-bonding present in such structures have also been discussed. Finally, the mechanism of 1,3-oxazolidine formation and the evaluation of imine/oxazolidine mixtures under acetylating conditions have been investigated by NMR spectroscopy and corroborated by theoretical calculations as well. All data point to interplay between kinetic and thermodynamic control, even if imines constitute the thermodynamically generated structures. It is worth mentioning that cyclization of imines under acylating conditions leads to oxazolidines having a 1,4-trans arrangement.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28041670/s1; Figures and Tables of spectral and computational data as stated through the entire manuscript.
Author Contributions
E.M. and J.C.P. conceived the conceptual ideas and manuscript outline. J.C.P. and P.C. drafted the manuscript. M.E.L. performed crystallographic and structural studies. E.M., P.C. and J.C.P. reviewed and critically edited the final content. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Junta de Extremadura and Fondo Europeo de Desarrollo Regional (Grant GR21039).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Crystallographic data have been deposited with the Cambridge Structural Database (CSD) and can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by email at: data_request@ccdc.cam.ac.uk, or by contacting the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK.
Acknowledgments
We gratefully acknowledge the Servicio de Apoyo a la Investigación (SAIUEX) at the University of Extremadura for analytical and spectroscopic resources, and the computational facilities at the LUSITANIA Supercomputing Centre supported by Cenits and Computaex Foundation. Last, but not least, this manuscript is dedicated to the memory of María Dolores Méndez.
Conflicts of Interest
The authors declare no conflict of interest (financial or ethical) regarding this submission.
Sample Availability
Samples of the new compounds synthesized could be available by contacting the authors.
References
- Martínez, R.F.; Ávalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.L.; Light, M.E.; Palacios, J.C.; Pérez, E.M.S. An anomeric effect drives the regiospecific ring-opening of 1,3-oxazolidines under acetylating conditions. Eur. J. Org. Chem. 2010, 2010, 5263–5273. [Google Scholar] [CrossRef]
- Martínez, R.F.; Ávalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.L.; Light, M.E.; Palacios, J.C.; Pérez, E.M.S. Schiff Bases from TRIS and formylpyridines: Structure and mechanistic rationale aided by DFT calculations. Eur. J. Org. Chem. 2010, 2010, 6224–6232. [Google Scholar] [CrossRef]
- Neelakantan, L.; Molin-Case, J.A. Crystal and molecular structure of 2-p-bromophenyl-3,4-dimethyl-5-phenyloxazolidine. J. Org. Chem. 1971, 36, 2261–2262. [Google Scholar] [CrossRef] [PubMed]
- Just, G.; Potvin, P.; Uggowitzer, P. Configuration at the 2-position of oxazolidines derived from l-ephedrine and p-bromobenzaldehyde. An x-ray structure redetermination. J. Org. Chem. 1983, 48, 2923–2924. [Google Scholar] [CrossRef]
- Andrés, C.; González, A.; Pedrosa, R.; Pérez-Encabo, A.; García-Granda, S.; Salvado, M.A.; Gómez-Beltran, F. A new chiral glycine synthon. Synthesis, x-ray structure of (−).(2S,4R)-2-ethoxycarbonyl-4-phenyl-1,3-oxazolidine and diastereoselective nucleophilic ring opening to (R)-ethyl α-amino carboxylates. Tetrahedron Lett. 1992, 33, 4743–4746. [Google Scholar] [CrossRef]
- O’Brien, P.; Warren, S. Asymmetric synthesis with diphenylphosphine oxides: Bicyclic aminals and oxazolidines as chiral auxiliaries. Tetrahedron Asymmetry 1996, 7, 3431–3444. [Google Scholar] [CrossRef]
- Agami, C.; Couty, F.; Lequesne, C. Asymmetric synthesis of homochiral 1,2-diols via N-boc oxazolidines. Tetrahedron Lett. 1994, 35, 3309–3312. [Google Scholar] [CrossRef]
- Santes, V.; Ortiz, A.; Santillan, R.; Gutiérrez, A.; Farfan, N. Syntheses of Bisoxazolidines and Morpholones. Synth. Commun. 1999, 29, 1277–1286. [Google Scholar] [CrossRef]
- Agami, C.; Rizk, T. Stereochemistry-60: Kinetic control of asymmetric induction during oxazolidine formation from (-)-ephedrine and aromatic aldehydes. Tetrahedron 1985, 41, 537–540. [Google Scholar] [CrossRef]
- Ávalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.L.; Light, M.E.; Palacios, J.C.; Pérez, E.M. Chiral N-Acyloxazolidines: Synthesis, Structure, and Mechanistic Insights. J. Org. Chem. 2008, 73, 661–672. [Google Scholar] [CrossRef]
- Martínez, R.F.; Avalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.L.; Light, M.E.; Palacios, J.C.; Pérez, E.M.S. An efficient and highly diastereoselective synthesis of C-glycosylated 1,3-oxazolidines from N-methyl-D-glucamine. Tetrahedron 2008, 64, 6377–6386. [Google Scholar] [CrossRef]
- Lindeman, S.V.; Andrianov, V.G.; Kravcheni, S.G.; Potapov, V.M.; Potekhin, K.A.; Struchkov, Y.T. Crystal and molecular structures of the two crystalline modifications of N-salicylidene-pentafluoroaniline. J. Struct. Chem. 1981, 22, 578–585. [Google Scholar] [CrossRef]
- Filipenko, O.S.; Ponomarev, V.I.; Bolotin, B.M.; Atovmyan, L.O. Crystal and molecular structure of a red modification of N-salicylidene-p-dimethylaminoaniline. Kristallografiya 1983, 28, 889–895. [Google Scholar]
- Aldoshin, S.M.; Atovmyan, L.O.; Ponomarev, V.I. Structure, spectral-luminescent and thermochromic properties of “yellow” N-salicylidene-p-dimethylaminoaniline crystals. Khim. Fiz. Sov. 1984, 3, 787–791. [Google Scholar]
- Bregman, J.; Leiserowitz, L.; Osaki, K.; Topochemistry, X. The crystal and molecular structures of 2-chloro-N-salicylideneaniline. J. Chem. Soc. 1964, 2086–2100. [Google Scholar] [CrossRef]
- Aldoshin, S.M.; Knyazhanskii, M.I.; Tymyanskii, Y.R.; Atovmyan, L.O.; D’Yachenko, O.A. Effect of intermolecular interactions on photo- and thermochromic properties of crystalline salicylaldehyde arylimines. Khim. Fiz. 1982, 1015–1023. [Google Scholar]
- Obodovskaya, A.E.; Starikova, Z.A.; Bolotin, B.M.; Safonova, T.N.; Etingen, N.B. X-Ray diffraction study of two crystalline modifications of N-(4-ethoxysalicylidene)-4-methylaniline. J. Struct. Chem. 1985, 26, 92–99. [Google Scholar] [CrossRef]
- Filipenko, O.S.; Atovmyan, L.O.; Tarnopol’skii, B.L.; Safina, Z.S. Crystal structures of nematogenic p-ethoxy- and p-propoxysalicylidene-p’-butylanilines. J. Struct. Chem. 1979, 20, 60–66. [Google Scholar] [CrossRef]
- Ondracek, J.; Kovarova, Z.; Maixner, J.; Jursik, F. Structure of o-(salicylideneamino)phenol hydrochloride. Acta Crystallogr. 1993, C49, 1948–1949. [Google Scholar]
- Mansilla-Koblavi, F.; Toure, S.; Lapasset, J.; Carles, M.; Bodot, H. N-Salicylidene trimethyl-2,4,6 aniline. Acta Crystallogr. 1989, C45, 451–453. [Google Scholar] [CrossRef]
- Inabe, T.; Hoshino, N.; Mitani, T.; Maruyama, Y. Structure and optical properties of a thermochromic Schiff Base. Low-temperature structural studies of the N,N′-disalicylidene-p-phenylenediamine and N,N′-disalicylidene-1,6-pyrenediamine crystals. Bull. Chem. Soc. Jpn. 1989, 62, 2245–2251. [Google Scholar] [CrossRef]
- Moloney, G.P.; Gable, R.W.; Iskander, M.N.; Craik, D.J.; Mackay, M.F. Anomalies in the reduction of the Schiff bases 5-(diethylamino)-2-(phenyliminomethyl)phenol and 2-[(4-diethylaminophenyl)iminomethyl]-phenol and their crystal Structures. Aust. J. Chem. 1990, 43, 99–107. [Google Scholar] [CrossRef]
- Inabe, T.; Gautier-Luneau, I.; Hoshino, N.; Okaniwa, K.; Okamoto, H.; Mitani, T.; Nagashima, U.; Maruyama, Y. Structure and optical properties of themochromic schiff bases. Charge transfer interaction and proton transfer in the N-tetrachlorosalicylideneaniline and N-tetrachlorosalicylidene-1-pyrenylamine crystals. Bull. Chem. Soc. Jpn. 1991, 64, 801–810. [Google Scholar] [CrossRef]
- Sergienko, V.S.; Mistryuko, A.E.; Litvino, V.V.; Knyazhanski, M.I.; Garnovskii, A.D.; Porai-Koshits, M.A. Preparation and crystal structure of 2-hydroxy-1-naphthylmethyleneaniline and its zinc chloride complex. Koord. Khim. 1990, 16, 168–176. [Google Scholar]
- Yeap, G.-Y.; Gan, C.-L.; Fun, H.-K.; Shawkataly, O.B.; Teoh, S.-G. Structure of 2-[(3-nitrophenylimino)methyl]phenol. Acta Crystallogr. 1992, C48, 1143–1144. [Google Scholar] [CrossRef]
- Wozniak, K.; He, H.; Klinowski, J.; Jones, W.; Dziembowska, T.; Grech, E. Intramolecular hydrogen bonding in N-salicylideneanilines. X-ray diffraction and solid-state NMR studies. J. Chem. Soc. Faraday Trans. 1995, 91, 77–85. [Google Scholar] [CrossRef]
- Kwiatkowski, E.; Olechnowicz, A.; Kosciuszko-Panek, B.; Ho, D.M. Crystal and molecular structure of N-salicylidene-1,2-diaminobenzene. Pol. J. Chem. 1994, 68, 85–92. [Google Scholar]
- Mansilla-Koblavi, F.; Tenon, J.A.; Toure, S.; Ebby, N.; Lapasset, J.; Carles, M. Une serie de N-(2,3-dihydroxybenzilidene)amines: Manifestation d’equilibres tautomères. Acta Crystallogr. 1995, C51, 1595–1602. [Google Scholar] [CrossRef]
- Fernández-G., J.M.; Rodríguez-Romero, A.; Pannerselvam, K.; Soriano-García, M. Two 2,3-naphthalenic Schiff bases. Acta Crystallogr. 1995, C51, 1643–1646. [Google Scholar] [CrossRef]
- Elerman, Y.; Elmali, A.; Atakol, O.; Svoboda, I. N-(2-Hydroxyphenyl)salicylaldimine. Acta Crystallogr. 1995, C51, 2344–2346. [Google Scholar] [CrossRef]
- Tenon, J.A.; Carles, M.; Aycard, J.-P. N-(5-Hydroxysalicylidène)-2,4,6-triméthylaniline. Acta Crystallogr. 1995, C51, 2603–2606. [Google Scholar] [CrossRef]
- Tafeenko, V.A.; Popov, S.I.; Medvedev, S.V. X-Ray study of the base of blue-black alizarine B dye at room and low (−150 °C) temperatures. Zh. Strukt. Khim. 1991, 32, 106–109. [Google Scholar]
- Tafeenko, V.A.; Bogdan, T.V.; Medvedev, S.V.; Kozyrev, A.A.; Popov, S.I. Crystal and molecular structure of the dibutyl derivative of alizarin blue-black B. Zh. Strukt. Khim 1991, 32, 169–171. [Google Scholar]
- Puranik, V.-G.; Tavale, S.S.; Kumbhar, A.S.; Yerande, R.G.; Padhye, S.B.; Butche, R.J. Crystal and molecular structure of an anchored catechol ligand: 2,3-dihydroxy benzenemethanimine α-(2-hydroxymethyl)phenyl. J. Cryst. Spectrosc. Res. 1992, 22, 725–731. [Google Scholar] [CrossRef]
- Schilf, W.; Kamiénski, B.; Szady-Chelmieniecka, A.; Grech, E. The 15N and 13C solid state NMR study of intramolecular hydrogen bond in some Schiff bases. J. Mol. Struct. 2004, 700, 105–108. [Google Scholar] [CrossRef]
- Schilf, W.; Kamiénski, B.; Kolodziej, B.; Grech, E. The NMR study of hydrogen bond formation in some tris(((-salicylidene)amino)ethyl)amine derivatives in solution and in the solid state. J. Mol. Struct. 2004, 708, 33–38. [Google Scholar] [CrossRef]
- Wojciechowski, G.; Przybylski, P.; Schilf, W.; Kamiénski, B.; Brzezinski, B. Spectroscopic studies of Schiff bases of 2,2′-dihydroxybiphenyl-3-carbaldehyde and para substituted anilines. J. Mol. Struct. 2003, 649, 197–205. [Google Scholar] [CrossRef]
- Wojciechowski, G.; Ratajczak-Sitarz, M.; Katrusiak, A.; Schilf, W.; Przybylski, P.; Brzezinski, B. Crystal structure of Schiff base derivative of 2,2′-dihydroxybiphenyl-3-carbaldehyde with n-butylamine. J. Mol. Struct. 2003, 650, 191–199. [Google Scholar] [CrossRef]
- Kolodziej, B.; Dominiak, P.M.; Koscielecka, A.; Schilf, W.; Wozniak, K. Neutral and ionic multiple hydrogen bonded moieties in crystal structure of a one tripodal Schiff base. J. Mol. Struct. 2004, 691, 133–139. [Google Scholar] [CrossRef]
- Montalvo-González, R.; Ariza-Castolo, A. Molecular structure of di-aryl-aldimines by multinuclear magnetic resonance and X-ray diffraction. J. Mol. Struct. 2003, 655, 375–389. [Google Scholar] [CrossRef]
- Berger, S.; Braun, S.; Kalinowski, H.-O. NMR Spectroscopy of the Non-Metallic Elements; John Wiley & Sons: Hoboken, NJ, USA, 1996. [Google Scholar]
- Alarcón, S.H.; Olivieri, A.C.; Sanz, D.; Claramunt, R.M.; Elguero, J. Substituent and solvent effects on the proton transfer equilibrium in anils and azo derivatives of naphthol. Multinuclear NMR study and theoretical calculations. J. Mol. Struct. 2004, 705, 1–9. [Google Scholar] [CrossRef]
- Schilf, W.; Bloxsidge, J.P.; Jones, J.R.; Lu, S.-Y. Investigations of intramolecular hydrogen bonding in three types of Schiff bases by 2H and 3H NMR isotope effects. Magn. Reson. Chem. 2004, 42, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Schilf, W.; Kamieński, B.; Szady-Chelmieniecka, A.; Grech, E. The intramolecular hydrogen bonds in some Schiff bases derived from cyclopropyl-, cyclobutyl- and cyclopentylamine. J. Mol. Struct. 2005, 743, 237–241. [Google Scholar] [CrossRef]
- Schilf, W.; Kamieński, B.; Szady-Chelmieniecka, A.; Grech, E.; Makal, A.; Woźniak, K. NMR and X-ray studies of 2,6-bis (alkylimino) phenol Schiff bases. J. Mol. Struct. 2007, 844, 94–101. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Abanowski, J.K.; Andzelm, J.W. Density Functional Methods in Chemistry; Springer: New York, NY, USA, 1991. [Google Scholar]
- Andzelm, J.; Wimmer, E. Density functional Gaussian-type-orbital approach to molecular geometries, vibrations, and reaction energies. J. Chem. Phys. 1992, 96, 1280–1303. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- Gill, P.M.W.; Johnson, B.G.; Pople, J.A.; Frisch, M. The performance of the Becke-Lee-Yang-Parr (B-LYP) density functional theory with various basis sets. J. Chem. Phys. Lett. 1992, 197, 499–505. [Google Scholar] [CrossRef]
- Scuseria, G.E. Comparison of coupled-cluster results with a hybrid of Hartree–Fock and density functional theory. J. Chem. Phys. 1992, 97, 7528–7530. [Google Scholar] [CrossRef]
- Sosa, C.; Lee, C. Density functional description of transition structures using nonlocal corrections. Silylene insertion reactions into the hydrogen molecule. J. Chem. Phys. 1993, 98, 8004–8011. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Frisch, M.J.; Chabalowski, C.F. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 215–241. [Google Scholar]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Martínez, R.F.; Matamoros, E.; Cintas, P.; Palacios, J.C. Imine or Enamine? Insights and Predictive Guidelines from the Electronic Effect of Substituents in H-Bonded Salicylimines. J. Org. Chem. 2020, 85, 5838–5862. [Google Scholar] [CrossRef]
- Matamoros, E.; Cintas, P.; Light, M.E.; Palacios, J.C. Electronic effects in tautomeric equilibria: The case of chiral imines from D-glucamine and 2-hydroxyacetophenones. Org. Biomol. Chem. 2019, 17, 10209–10222. [Google Scholar] [CrossRef]
- Romero-Fernández, M.P.; Ávalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.L.; Light, M.E.; Palacios, J.C. Pseudo-cyclic structures of mono- and di-azaderivatives of malondialdehydes. Synthesis and conformational disentanglement by computational analyses. Org. Biomol. Chem. 2014, 12, 8997–9010. [Google Scholar] [CrossRef]
- Romero-Fernández, M.P.; Ávalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.L.; Palacios, J.C. Rethinking Aromaticity in H-Bonded Systems. Caveats for Transition Structures Involving Hydrogen Transfer and π-Delocalization. J. Phys. Chem. A 2015, 119, 525–534. [Google Scholar] [CrossRef]
- Matamoros, E.; Cintas, P.; Palacios, J.C. Tautomerism and stereodynamics in Schiff bases from gossypol and hemigossypol with N-aminoheterocycles. Org. Biomol. Chem. 2019, 17, 6229–6250. [Google Scholar] [CrossRef]
- Alarcón, S.; Pagani, D.; Bacigalupo, J.; Olivieri, A.C. Spectroscopic and Semi-Empirical MO Study of Substituent Effects on the Intramolecular Proton Transfer in Anils of 2-Hydroxybenzaldehydes. J. Mol. Struct. 1999, 475, 233–240. [Google Scholar] [CrossRef]
- Neuvonen, K.; Fülöp, F.; Neuvonen, H.; Koch, A.; Kleinpeter, E.; Pihlaja, K. Substituent Influences on the Stability of the Ring and Chain Tautomers in 1,3-O,N-Heterocyclic Systems: Characterization by 13C NMR Chemical Shifts, PM3 Charge Densities, and Isodesmic Reactions. J. Org. Chem. 2001, 66, 4132–4140. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, F.; Pihlaja, K. Ring-chain tautomerism of oxazolidines derived from serine esters. Tetrahedron 1993, 49, 6701–6706. [Google Scholar] [CrossRef]
- Fülöp, F.; Pihlaja, K.; Neuvonen, K.; Bernáth, G.; Argay, G.; Kálmán, A. Ring-chain tautomerism in oxazolidines. J. Org. Chem. 1993, 58, 1967–1969. [Google Scholar] [CrossRef]
- Maireanu, C.; Darabantu, M.; Plé, G.; Berghian, C.; Condamine, E.; Ramondenc, Y.; Silaghi-Dumitrescu, I.; Mager, S. Ring-chain tautomerism and other versatile behaviour of 1,4-diimino- and 1,2-phenylene derivatives of some C-substituted serinols. Tetrahedron 2002, 58, 2681–2693. [Google Scholar] [CrossRef]
- Matamoros, E.; Cintas, P.; Palacios, J.C. Amphipathic 1,3-oxazolidines from N-alkyl glucamines and benzaldehydes: Stereochemical and mechanistic studies. N. J. Chem. 2021, 45, 4365–4386. [Google Scholar] [CrossRef]
- Stoddart, J.F. On the nomenclature of the five- or six-membered ring conformations. In Stereochemistry of Carbohydrates; John Wiley: New York, NY, USA, 1971; Chapter 3; pp. 55–58. [Google Scholar]
- Radhakrishnan, T.P.; Agranat, I. Measures of pyramidalization. Struct. Chem. 1991, 2, 107–115. [Google Scholar] [CrossRef]
- Eliel, E.L.; Wilen, S.H.; Mander, L.N. Stereochemistry of Organic Compounds; Wiley-Interscience: New York, NY, USA, 1994. [Google Scholar]
- Oki, M. The Chemistry of Rotational Isomers. In Reactivity and Structure Concepts in Organic Chemistry; Hafner, K., Lehn, J.-M., Rees, C.W., Ragué Schleyer, P.V., Trost, B.M., Zahradník, R., Eds.; Springer: Berlin/Heidelberg, Germany, 1993; Volume 30. [Google Scholar]
- Schaefer, T. Relation between hydroxyl proton chemical shifts and torsional frequencies in some ortho-substituted phenol derivatives. J. Phys. Chem. 1975, 79, 1888–1890. [Google Scholar] [CrossRef]
- Musin, R.N.; Mariam, Y.H. An integrated approach to the study of intramolecular hydrogen bonds in malonaldehyde enol derivatives and naphthazarin: Trend in energetic versus geometrical consequences. J. Phys. Org. Chem. 2006, 19, 425–444. [Google Scholar] [CrossRef]
- Raiford, L.C.; Taft, R.; Lankelman, H.P. Steric relations in the acylation of aromatic amines and aminophenols. J. Am. Chem. Soc. 1924, 46, 2051–2057. [Google Scholar] [CrossRef]
- Auwers, K.; Bondy, R. Mixed observations over acylations. Ber 1904, 37, 3905–3915. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).