
Improved Bioproduction of the Nylon 12 Monomer by Combining the Directed Evolution of P450 and Enhancing Heme Synthesis


Abstract
:1. Introduction
2. Results and Discussion
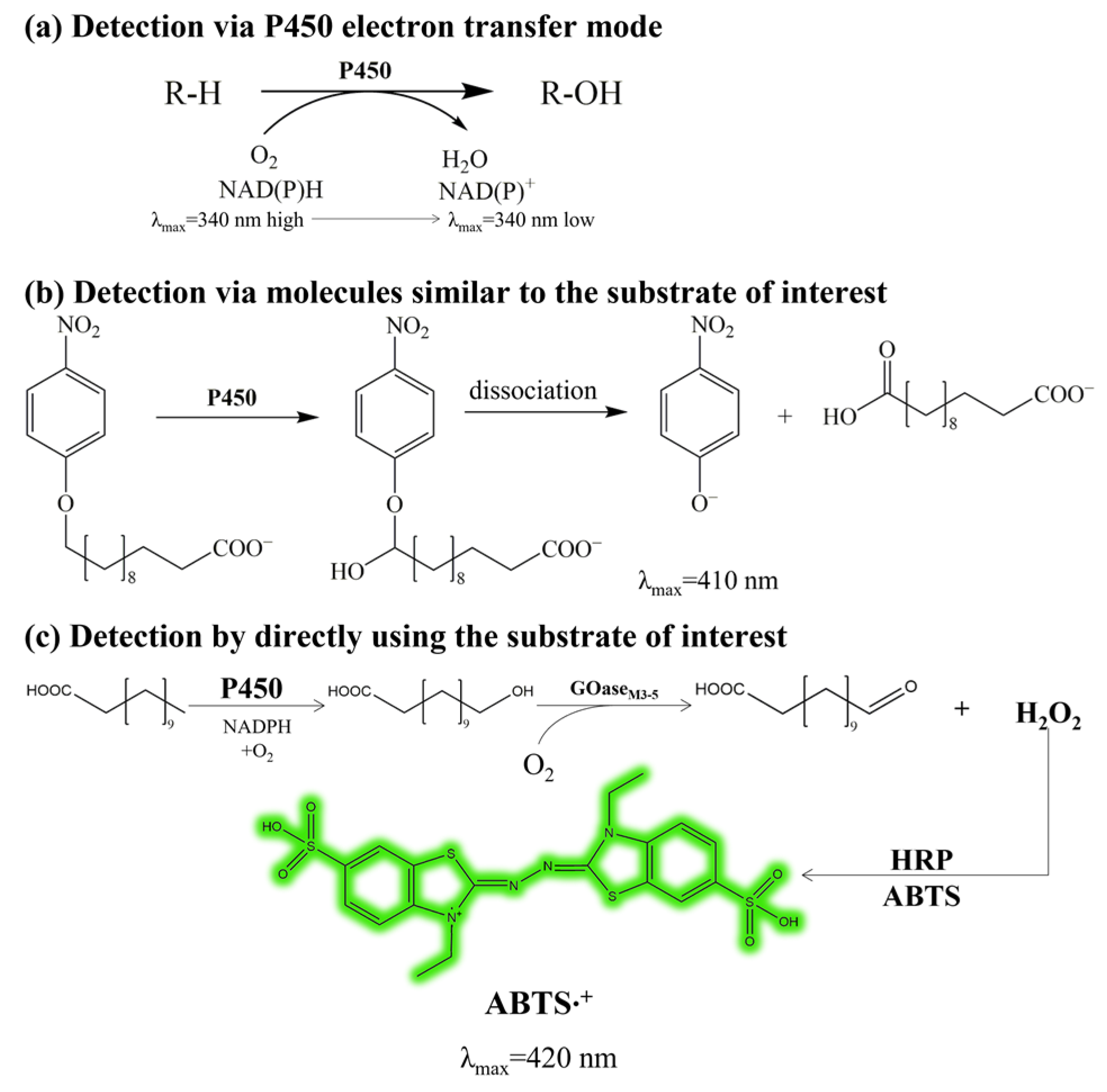
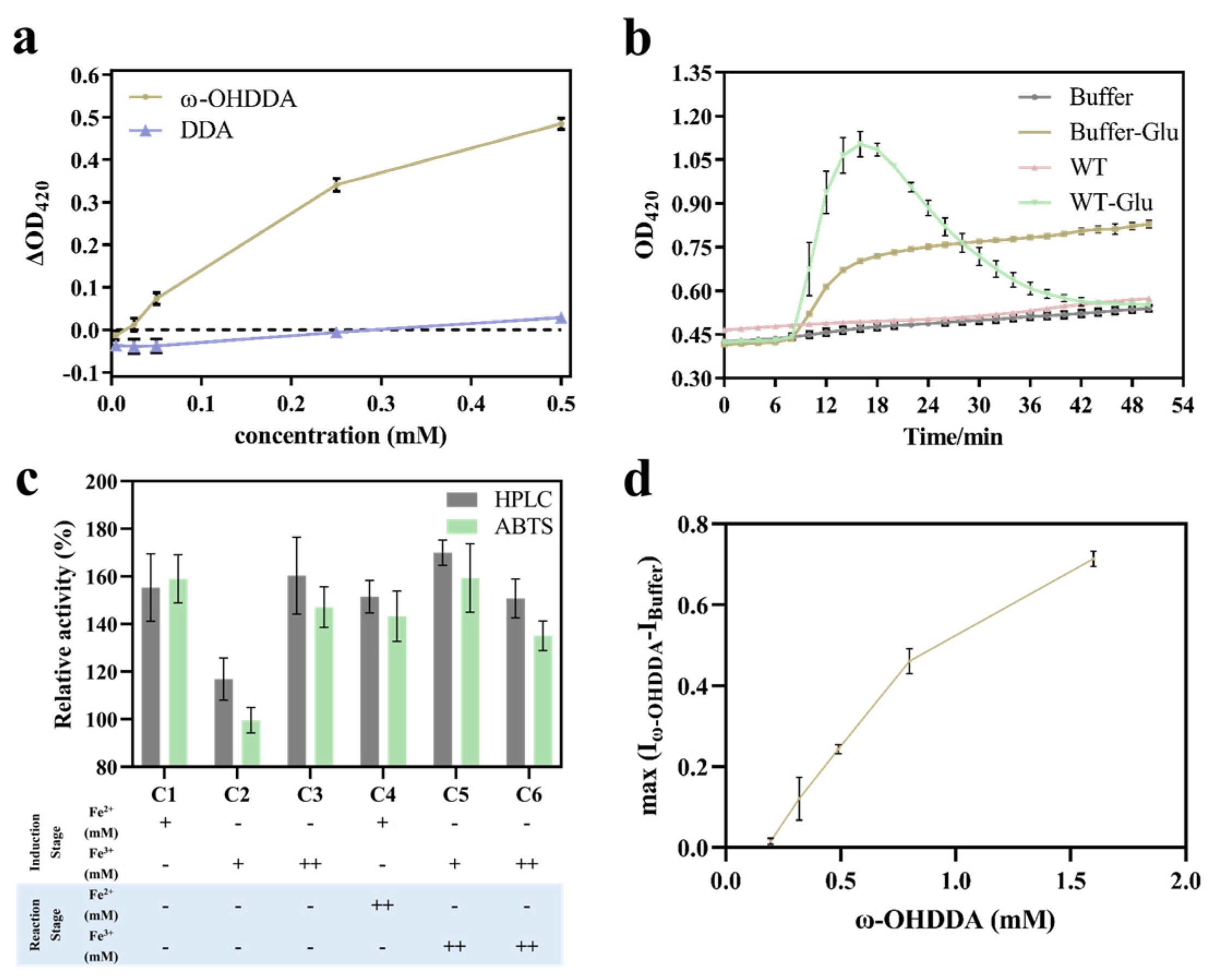
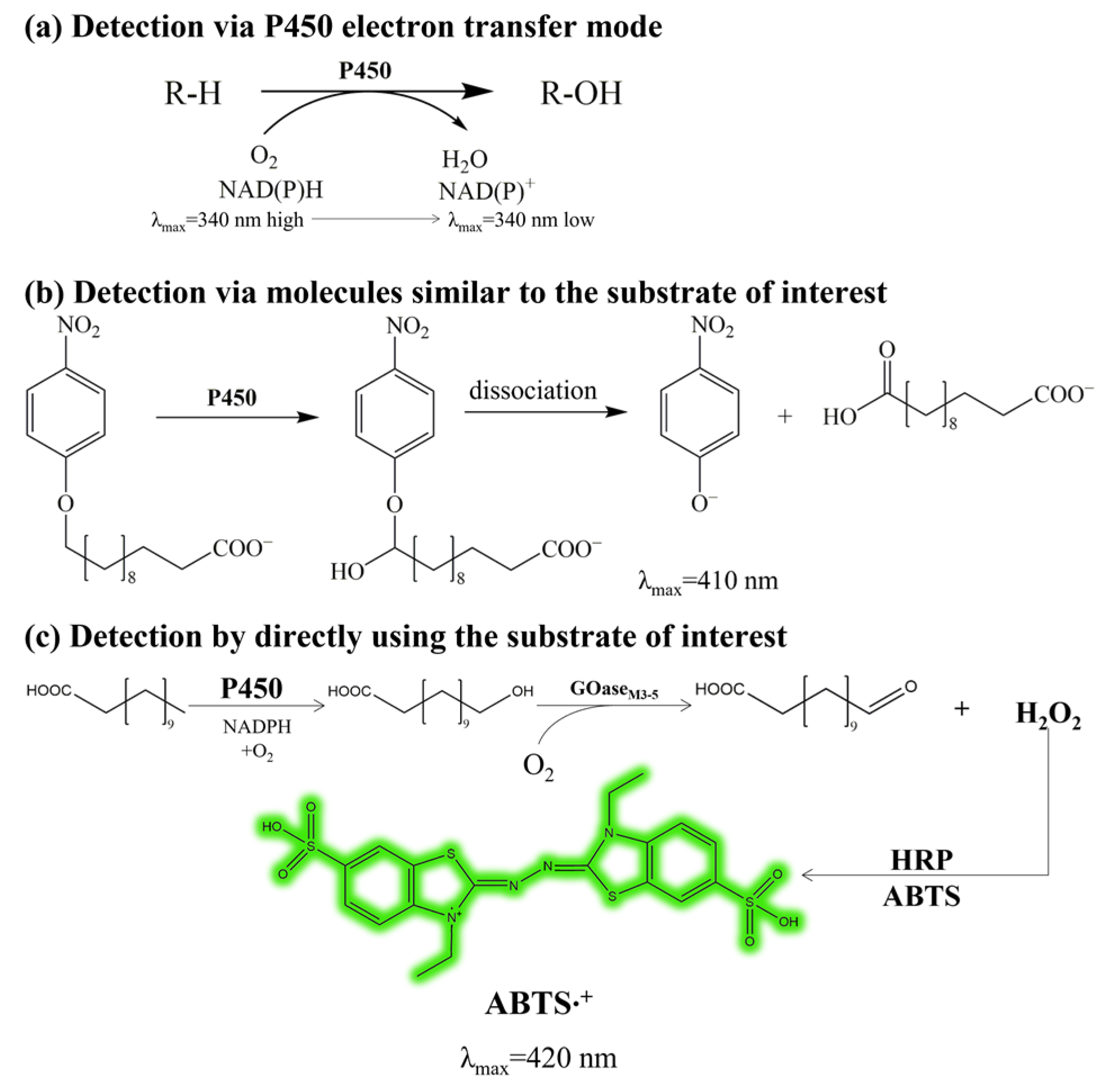
2.1. Establishment of the High-Throughput Screening Method
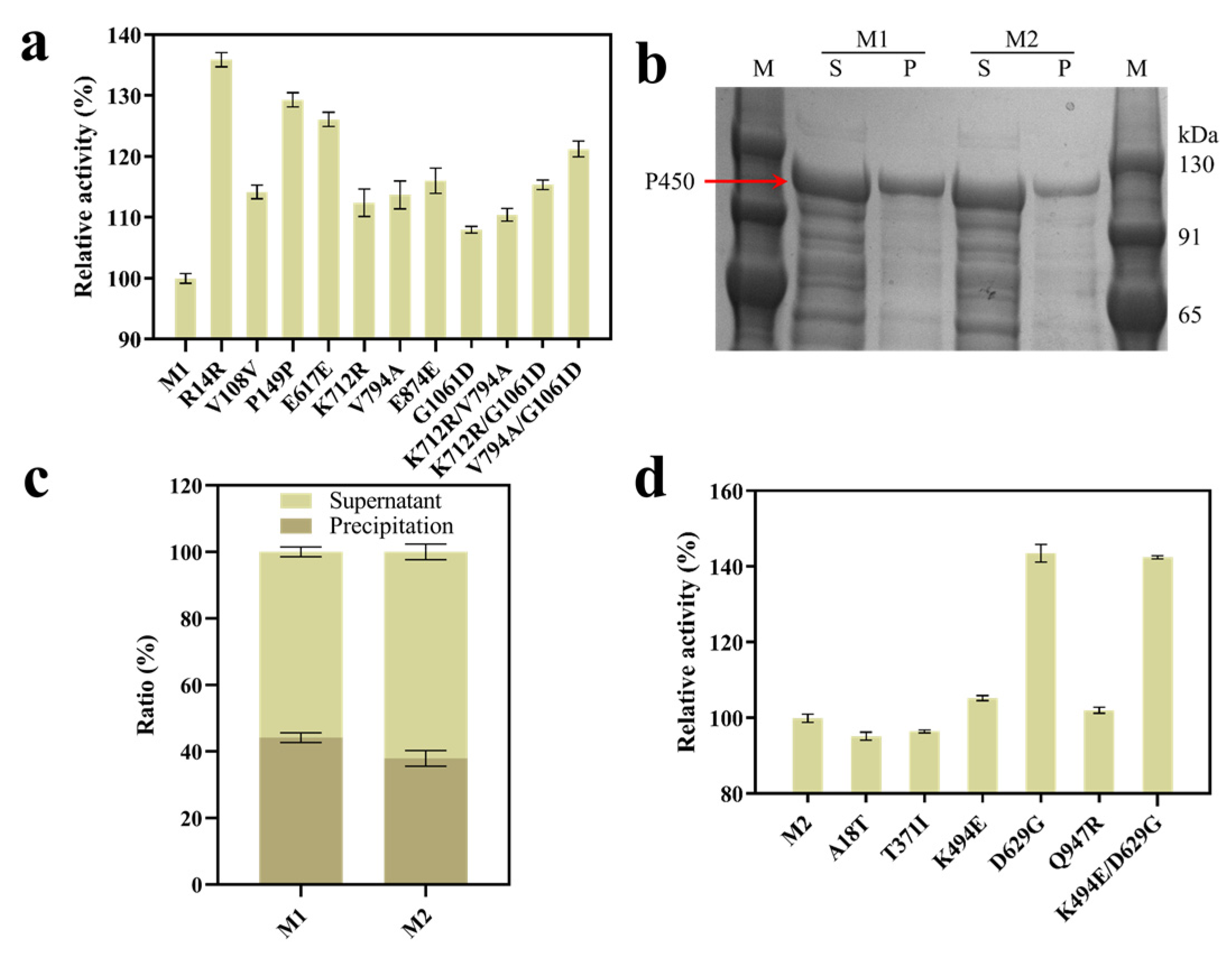
2.2. Directed Evolution of cyp153a-ncpG307A (M1)
2.3. Molecular Simulation Analysis
2.4. Enhancement of the Heme Synthesis Pathway
2.5. Construction of the ω-AmDDA Synthesis Pathway
3. Materials and Methods
3.1. Strains and Plasmids
3.2. Chemicals and Enzymes
3.3. Expression, Purification and SDS-PAGE Analysis of Galactose Oxidase
3.4. Cloning, Expression and Purification of P450
3.5. Whole-Cell Biotransformation
3.6. High-Throughput Screening of P450
3.7. HPLC and GC/MS Analysis
3.8. Kinetic Analysis of P450
3.9. Molecular Simulation Analysis of P450
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Winnacker, M. Polyamides and their functionalization: Recent concepts for their applications as biomaterials. Biomater. Sci. 2017, 5, 1230–1235. [Google Scholar] [CrossRef]
- Carole, T.M.; Pellegrino, J.; Paster, M.D. Opportunities in the industrial biobased products industry. Appl. Biochem. Biotechnol. 2004, 113, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Dachs, K.; Schwartz, E. Pyrrolidon, capryllactam und laurinlactam als neue grundstoffe für polyamidfasern. Angew. Chem. Int. Ed. 1962, 74, 540–545. [Google Scholar] [CrossRef]
- Ladkau, N.; Assmann, M.; Schrewe, M.; Julsing, M.K.; Schmid, A.; Buhler, B. Efficient production of the Nylon 12 monomer omega-aminododecanoic acid methyl ester from renewable dodecanoic acid methyl ester with engineered Escherichia coli. Metab. Eng. 2016, 36, 1–9. [Google Scholar] [CrossRef]
- Ahsan, M.M.; Jeon, H.; Nadarajan, S.; Chung, T.; Yoo, H.W.; Kim, B.G.; Patil, M.D.; Yun, H. Biosynthesis of the nylon 12 monomer, ω-aminododecanoic acid with novel CYP153A, AlkJ, and ω-TA enzymes. Biotechnol. J. 2018, 13, 1700562. [Google Scholar] [CrossRef]
- Ge, J.W.; Yang, X.H.; Yu, H.W.; Ye, L.D. High-yield whole cell biosynthesis of Nylon 12 monomer with self-sufficient supply of multiple cofactors. Metab. Eng. 2020, 62, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Notonier, S.; Gricman, L.; Pleiss, J.; Hauer, B. Semirational protein engineering of CYP153A(M.aq.)-CPRBM3 for efficient terminal hydroxylation of short- to long-chain fatty acids. ChemBioChem 2016, 17, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Ba, L.; Gao, J.W.; Gao, X.X.; Zhu, D.M.; de Jong, R.M.; Mink, D.; Kaluzna, I.; Lin, Z.L. Semi-rational engineering of cytochrome CYP153A from Marinobacter aquaeolei for improved omega-hydroxylation activity towards oleic acid. Appl. Microbiol. Biotechnol. 2016, 100, 8779–8788. [Google Scholar] [CrossRef]
- Malca, S.H.; Scheps, D.; Kuhnel, L.; Venegas-Venegas, E.; Seifert, A.; Nestl, B.M.; Hauer, B. Bacterial CYP153A monooxygenases for the synthesis of omega-hydroxylated fatty acids. Chem. Commun. 2012, 48, 5115–5117. [Google Scholar] [CrossRef]
- Zhang, L.L.; Xie, Z.Z.; Liu, Z.W.; Zhou, S.Y.; Ma, L.X.; Liu, W.D.; Huang, J.W.; Ko, T.P.; Li, X.Q.; Hu, Y.C.; et al. Structural insight into the electron transfer pathway of a self-sufficient P450 monooxygenase. Nat. Commun. 2020, 11, 2676. [Google Scholar] [CrossRef]
- Olsen, M.; Iverson, B.; Georgiou, G. High-throughput screening of enzyme libraries. Curr. Opin. Biotechnol. 2000, 11, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Weissenborn, M.J.; Notonier, S.; Lang, S.L.; Otte, K.B.; Herter, S.; Turner, N.J.; Flitsch, S.L.; Hauer, B. Whole-cell microtiter plate screening assay for terminal hydroxylation of fatty acids by P450s. Chem. Commun. 2016, 52, 6158–6161. [Google Scholar] [CrossRef] [PubMed]
- Escalettes, F.; Turner, N.J. Directed evolution of galactose oxidase: Generation of enantioselective secondary alcohol oxidases. ChemBioChem 2008, 9, 857–860. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Li, H.Y.; Zhang, H.; Peterson, J.A.; Poulos, T.L. Structure of a cytochrome P450-redox partner electron-transfer complex. Proc. Natl. Acad. Sci. USA 1999, 96, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.M.; Danesh-Azari, H.R.; Spandolf, C.; Weissenborn, M.J.; Grogan, G.; Hauer, B. Structure-guided redesign of CYP153A(M.aq) for the improved terminal hydroxylation of fatty acids. ChemCatChem 2016, 8, 3234–3239. [Google Scholar] [CrossRef]
- Green, M.T. C-H bond activation in heme proteins: The role of thiolate ligation in cytochrome P450. Curr. Opin. Chem. Biol. 2009, 13, 84–88. [Google Scholar] [CrossRef]
- Ge, B.S.; Chen, Y.; Yu, Q.; Lin, X.J.; Li, J.Q.; Qin, S. Regulation of the heme biosynthetic pathway for combinational biosynthesis of phycocyanobilin in Escherichia coli. Process Biochem. 2018, 71, 23–30. [Google Scholar] [CrossRef]
- Zhao, X.R.; Choi, K.R.; Lee, S.Y. Metabolic engineering of Escherichia coli for secretory production of free haem. Nat. Catal. 2018, 1, 720–728. [Google Scholar] [CrossRef]
- Sathesh-Prabu, C.; Lee, S.K. Production of long-chain alpha,omega-dicarboxylic acids by engineered Escherichia coli from renewable fatty acids and plant oils. J. Agric. Food Chem. 2015, 63, 8199–8208. [Google Scholar] [CrossRef]
- Munro, A.W.; Leys, D.G.; McLean, K.J.; Marshall, K.R.; Ost, T.W.B.; Daff, S.; Miles, C.S.; Chapman, S.K.; Lysek, D.A.; Moser, C.C.; et al. P450BM3: The very model of a modern flavocytochrome. Trends Biochem. Sci. 2002, 27, 250–257. [Google Scholar] [CrossRef]
- Tsotsou, G.E.; Cass, A.E.G.; Gilardi, G. High throughput assay for cytochrome P450BM3 for screening libraries of substrates and combinatorial mutants. Biosens. Bioelectron. 2002, 17, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.S.; Schwaneberg, U.; Fischer, M.; Schmitt, J.; Pleiss, J.; Lutz-Wahl, S.; Schmid, R.D. Rational evolution of a medium chain-specific cytochrome P-450 BM-3 variant. Biophys. Acta Protein Struct. Molec. Enzym. 2001, 1545, 114–121. [Google Scholar] [CrossRef]
- Murakami, K.; Tsubouchi, R.; Fukayama, M.; Ogawa, T.; Yoshino, M. Oxidative inactivation of reduced NADP-generating enzymes in E-coli: Iron-dependent inactivation with affinity cleavage of NADP-isocitrate dehydrogenase. Arch. Microbiol. 2006, 186, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, N.; Kotre, A.M.; Savageau, M.A. Glutamate-dehydrogenase from Escherichia coli: Purification and properties. J. Bacteriol. 1975, 124, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Decker, R.V.; Anderson, W.M.; Spivey, H.O. Substrate channeling of NADH and binding of dehydrogenases to complex-I. J. Biol. Chem. 1989, 264, 16483–16488. [Google Scholar] [CrossRef]
- Galkin, A.; Kulakova, L.; Tishkov, V.; Esaki, N.; Soda, K. Cloning of formate dehydrogenase gene from a methanol-utilizing bacterium Mycobacterium vaccae N10. Appl. Microbiol. Biotechnol. 1995, 44, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Jan, J.; Martinez, I.; Wang, Y.P.; Bennett, G.N.; San, K.Y. Metabolic engineering and transhydrogenase effects on NADPH availability in Escherichia coli. Biotechnol. Prog. 2013, 29, 1124–1130. [Google Scholar] [CrossRef]
- Ichinose, H.; Hatakeyama, M.; Yamauchi, Y. Sequence modifications and heterologous expression of eukaryotic cytochromes P450 in Escherichia coli. J. Biosci. Bioeng. 2015, 120, 268–274. [Google Scholar] [CrossRef]
- Hausjell, J.; Halbwirth, H.; Spadiut, O. Recombinant production of eukaryotic cytochrome P450s in microbial cell factories. Biosci. Rep. 2018, 38, BSR20171290. [Google Scholar] [CrossRef]
- Reis, A.C.; Salis, H.M. An automated model test system for systematic development and improvement of gene expression models. ACS Synth. Biol. 2020, 9, 3145–3156. [Google Scholar] [CrossRef]
- Salis, H.M.; Mirsky, E.A.; Voigt, C.A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009, 27, 946–952. [Google Scholar] [CrossRef]
- Joyce, M.G.; Ekanem, I.S.; Roitel, O.; Dunford, A.J.; Neeli, R.; Girvan, H.M.; Baker, G.J.; Curtis, R.A.; Munro, A.W.; Leys, D. The crystal structure of the FAD/NADPH-binding domain of flavocytochrome P450 BM3. FEBS J. 2012, 279, 1694–1706. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Roberts, D.L.; Paschke, R.; Shea, T.M.; Masters, B.S.; Kim, J.J. Three-dimensional structure of NADPH-cytochrome P450 reductase: Prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. USA 1997, 94, 8411–8416. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Sugishima, M.; Taira, J.; Sagara, T.; Nakao, R.; Sato, H.; Noguchi, M.; Fukuyama, K.; Yamamoto, K.; Yasunaga, T.; Sakamoto, H. Conformational equilibrium of NADPH-cytochrome P450 oxidoreductase is essential for heme oxygenase reaction. Antioxidants 2020, 9, 673. [Google Scholar] [CrossRef]
- Iijima, M.; Ohnuki, J.; Sato, T.; Sugishima, M.; Takano, M. Coupling of redox and structural states in cytochrome P450 reductase studied by molecular dynamics simulation. Sci. Rep. 2019, 9, 9341. [Google Scholar] [CrossRef]
- Weng, H.; Ding, W.; Shi, Y.; Li, J.; Kang, Z. Enhancement of heme synthesis pathway in Escherichia coli via a modular optimization strategy. J. Food Sci. Biotechnol. 2019, 38, 86–94. [Google Scholar]
- Zhang, J.L.; Kang, Z.; Chen, J.; Du, G.C. Optimization of the heme biosynthesis pathway for the production of 5-aminolevulinic acid in Escherichia coli. Sci. Rep. 2015, 5, 8584. [Google Scholar] [CrossRef]
- Zhou, Y.J.J.; Yang, W.; Wang, L.; Zhu, Z.W.; Zhang, S.F.; Zhao, Z.B.K. Engineering NAD(+) availability for Escherichia coli whole-cell biocatalysis: A case study for dihydroxyacetone production. Microb. Cell Fact. 2013, 12, 103. [Google Scholar] [CrossRef]
- Bao, T.; Zhang, X.; Zhao, X.J.; Rao, Z.M.; Yang, T.W.; Yang, S.T. Regulation of the NADH pool and NADH/NADPH ratio redistributes acetoin and 2,3-butanediol proportion in Bacillus subtilis. Biotechnol. J. 2015, 10, 1298–1306. [Google Scholar] [CrossRef]
- Sakoda, H.; Imanaka, T. Cloning and sequencing of the gene coding for alcohol dehydrogenase of Bacillus stearothermophilus and rational shift of the optimum pH. J. Bacteriol. 1992, 174, 1397–1402. [Google Scholar] [CrossRef]
- Vanbeilen, J.B.; Eggink, G.; Enequist, H.; Bos, R.; Witholt, B. DNA sequence determination and functional characterization of the OCT-plasmid-encoded alkJKL genes of Pseudomonas oleovorans. Mol. Microbiol. 1992, 6, 3121–3136. [Google Scholar]
- Liu, S.; Zhang, X.; Liu, F.; Xu, M.J.; Yang, T.W.; Long, M.F.; Zhou, J.P.; Osire, T.; Yang, S.T.; Rao, Z. Designing of a cofactor self-sufficient whole-cell biocatalyst system for production of 1,2-amino alcohols from epoxides. ACS Synth. Biol. 2019, 8, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, X.; Wang, Q.; Xu, J.; Dong, F.; Yang, S.; Yang, J.; Zhang, Z.; Qian, Y.; Chen, J.; et al. Multicopy chromosomal integration using CRISPR-associated transposases. ACS Synth. Biol. 2020, 9, 1998–2008. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.K.; Zhou, Y.; Wang, T.W.; Too, H.P.; Wang, D.I.C.; Li, Z. Highly regio- and enantioselective multiple oxy- and amino-functionalizations of alkenes by modular cascade biocatalysis. Nat. Commun. 2016, 7, 11917. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- HamediRad, M.; Weisberg, S.; Chao, R.; Lian, J.; Zhao, H. Highly efficient single-pot scarless Golden Gate assembly. ACS Synth. Biol. 2019, 8, 1047–1054. [Google Scholar] [CrossRef]
- Joo, H.; Arisawa, A.; Lin, Z.L.; Arnold, F.H. A high-throughput digital imaging screen for the discovery and directed evolution of oxygenases. Chem. Biol. 1999, 6, 699–706. [Google Scholar] [CrossRef]
- Omura, T.; Sato, R. Carbon monoxide-binding pigment of liver microsomes. J. Biol. Chem. 1964, 239, 2370–2378. [Google Scholar] [CrossRef]
- Panicco, P.; Astuti, Y.; Fantuzzi, A.; Durrant, J.R.; Gilardi, G. P450 versus P420: Correlation between cyclic voltammetry and visible absorption spectroscopy of the immobilized heme domain of cytochrome P450 BM3. J. Phys. Chem. B 2008, 112, 14063–14068. [Google Scholar] [CrossRef] [PubMed]
- Rontani, J.F.; Aubert, C. Trimethylsilyl transfer during electron ionization mass spectral fragmentation of some omega-hydroxycarboxylic and omega-dicarboxylic acid trimethylsilyl derivatives and the effect of chain length. Rapid Commun. Mass Spectrom. 2004, 18, 1889–1895. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinf. 2016, 54, 5–6. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2021, 2199, 239–255. [Google Scholar] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clones a | Amino Acid Substitutions (Synonymous Mutation) | Relative Activity b |
|---|---|---|
| M1 | G307A | 1 |
| A641 | G307A/E801K (R14R/V108V/P149P/E617E/E874E) | 1.32 ± 0.05 |
| A692 | G307A/I442V/N511S/G1061D (A208A/S473S/H500H) | 1.35 ± 0.03 |
| A735 | G307A/T55A/K190I/K712R (R701R) | 1.16 ± 0.02 |
| A1049 | G307A/ (L32L/Y459Y) | 1.25 ± 0.02 |
| A1067 | G307A/V794A (E951E) | 1.20 ± 0.07 |
| B375 | G307A/R14R/A18T/T371I/K494E/D629G/Q947R | 1.34 ± 0.04 |
| Enzyme Variant | Km (mM) a | kcat (Min−1) a | kcat/Km (Min−1·mM−1) a |
|---|---|---|---|
| M1 | 0.27 ± 0.01 | 10.92 ± 0.10 | 40.94 ± 0.39 |
| M3 | 0.21 ± 0.04 | 17.31 ± 1.13 | 81.69 ± 5.33 |
| Substrate a | Relative Activity b |
|---|---|
| C10:0 | 1.13 ± 0.05 |
| C13:0 | 1.65 ± 0.05 |
| C14:0 | 1.17 ± 0.04 |
| C16:0 | 1.93 ± 0.09 |
| Plasmids/Strains | Description | Source |
|---|---|---|
| Plasmids | ||
| E-M1-3 | pETDuet-1 derivative, rbs3-cyp153a-ncpG307A and Bacillus cereus gdh1; AmpR | [6] |
| C-M2-2 | pCDFDuet-1 derivative, Geobacillus stearothermophilus BsADHC257L, Chromobacterium violaceum DSM30191 cv2025 and Bacillus subtilis aladh2; SmR | [6] |
| pET-30a(+)-M3-5 | pET-30a(+) derivative, Dactylium dendroides GOaseM3-5; KmR | [13] |
| pACYCDuet-1 | Expression vector, p15A ori, double T7lac promoters; CmR | [46] |
| pRSFDuet-1 | Expression vector, RSF1030 ori, double T7lac promoters; KmR | [46] |
| pCDFDuet-1 | Expression vector, CDF ori, double T7lac promoters; SmR | [46] |
| E-3 | pETDuet-1 derivative, rbs3-cyp153a-ncp and Bacillus cereus gdh1; AmpR | This study |
| E-M2-3 | pETDuet-1 derivative, rbs3-cyp153a-ncpG307A/R14R and Bacillus cereus gdh1; AmpR | This study |
| E-M3-3 | pETDuet-1 derivative, rbs3-cyp153a-ncpG307A/R14R/D629G and Bacillus cereus gdh1; AmpR | This study |
| pAC-hemB | pACYCDuet-1 derivative, E. coli hemB; KmR | This study |
| pAC-hemE | pACYCDuet-1 derivative, E. coli hemE; SmR | This study |
| pAC-hemH | pACYCDuet-1 derivative, E. coli hemH; SmR | This study |
| pRS-hemB | pRSFDuet-1 derivative, E. coli hemB; KmR | This study |
| pRS-hemF | pRSFDuet-1 derivative, E. coli hemF; KmR | This study |
| pAC-hemBCDE | pACYCDuet-1 derivative, E. coli hemB, hemC, hemD and hemE; SmR | This study |
| pAC-hemEFGH | pACYCDuet-1 derivative, E. coli hemE, hemF, hemG and hemH; SmR | This study |
| pRS-hemBCD | pRSFDuet-1 derivative, E. coli hemB, hemC and hemD; KmR | This study |
| pRS-hemFGH | pRSFDuet-1 derivative, E. coli hemF, hemG and hemH; KmR | This study |
| E-M1-3-gdhA | pETDuet-1 derivative, rbs3-cyp153a-ncpG307A and E. coli gdhA; AmpR | This study |
| E-M1-3-icd | pETDuet-1 derivative, rbs3-cyp153a-ncpG307A and E. coli icd; AmpR | This study |
| E-M1-3-fdh1 | pETDuet-1 derivative, rbs3-cyp153a-ncpG307A and Burkholderia stabilis 15516 fdh1; AmpR | This study |
| pCD-pntAB | pCDFDuet-1 derivative, E. coli pntAB; SmR | This study |
| pCD-sthA | pCDFDuet-1 derivative, E. coli sthA; SmR | This study |
| pCD-pntAB-fdh2 | pCDFDuet-1 derivative, E. coli pntAB and Mycolicibacterium vaccae fdh2; SmR | This study |
| pCD-sthA-fdh2 | pCDFDuet-1 derivative, E. coli sthA and M. vaccae fdh2; SmR | This study |
| C-alkJ | pCDFDuet-1 derivative, Pseudomonas oleovorans alkJ, Chromobacterium violaceum DSM30191 cv2025 and Bacillus subtilis aladh2; SmR | This study |
| C-rbs2-alkJ | pCDFDuet-1 derivative, rbs2-alkJ, Chromobacterium violaceum DSM30191 cv2025 and Bacillus subtilis aladh2; SmR | This study |
| Strains | ||
| B1-1 | E. coli BL21(DE3), ΔfadD::PlacUV5-alkL | [6] |
| P1-1 | B1-1, ΔfadD::PT7-yaaDE | [6] |
| MG1655 | E. coli K-12 wild type | Lab stock |
| BL21+M3-5 | B1-1 strain harboring plasmid pET-30a(+)-M3-5; KmR | This study |
| B1-1-1 | B1-1 strain harboring the plasmids pAC-hemBCDE, pRS-hemFGH and E-M1-3; SmR, KmR and AmpR | This study |
| B1-1-2 | B1-1 strain harboring the plasmids pAC-hemB and E-M1-3; KmR and AmpR | This study |
| B1-1-3 | B1-1 strain harboring the plasmids pRS-hemBCD, pAC-hemEFGH and E-M1-3; SmR, KmR and AmpR | This study |
| B1-1-3+ΔyfeX | B1-1, ΔyfeX strain harboring the plasmids pRS-hemBCD, pAC-hemEFGH and E-M1-3; SmR, KmR and AmpR | This study |
| B2-1 | B1-1, T10::Plac-hemB-T1 | This study |
| B2-2 | B1-1, T10::Ptac-hemB-T1 | This study |
| B2-3 | B1-1, T10::PT7-hemB-T1 | This study |
| B3-1 | P1-1 strain harboring the plasmids E-M1-3 and C-alkJ; SmR and AmpR | This study |
| B3-2 | P1-1, ΔyfeX-T10::Ptac-hemB-T1 strain harboring the plasmids E-M3-3 and C-rbs2-alkJ; SmR and AmpR | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.; Ge, J.; Yu, H.; Ye, L. Improved Bioproduction of the Nylon 12 Monomer by Combining the Directed Evolution of P450 and Enhancing Heme Synthesis. Molecules 2023, 28, 1758. https://doi.org/10.3390/molecules28041758
Yu J, Ge J, Yu H, Ye L. Improved Bioproduction of the Nylon 12 Monomer by Combining the Directed Evolution of P450 and Enhancing Heme Synthesis. Molecules. 2023; 28(4):1758. https://doi.org/10.3390/molecules28041758
Chicago/Turabian StyleYu, Jiaming, Jiawei Ge, Hongwei Yu, and Lidan Ye. 2023. "Improved Bioproduction of the Nylon 12 Monomer by Combining the Directed Evolution of P450 and Enhancing Heme Synthesis" Molecules 28, no. 4: 1758. https://doi.org/10.3390/molecules28041758
APA StyleYu, J., Ge, J., Yu, H., & Ye, L. (2023). Improved Bioproduction of the Nylon 12 Monomer by Combining the Directed Evolution of P450 and Enhancing Heme Synthesis. Molecules, 28(4), 1758. https://doi.org/10.3390/molecules28041758