


Synthesis of New Amino-Functionalized Porphyrins:Preliminary Study of Their Organophotocatalytic Activity


Abstract
1. Introduction
2. Results and Discussion
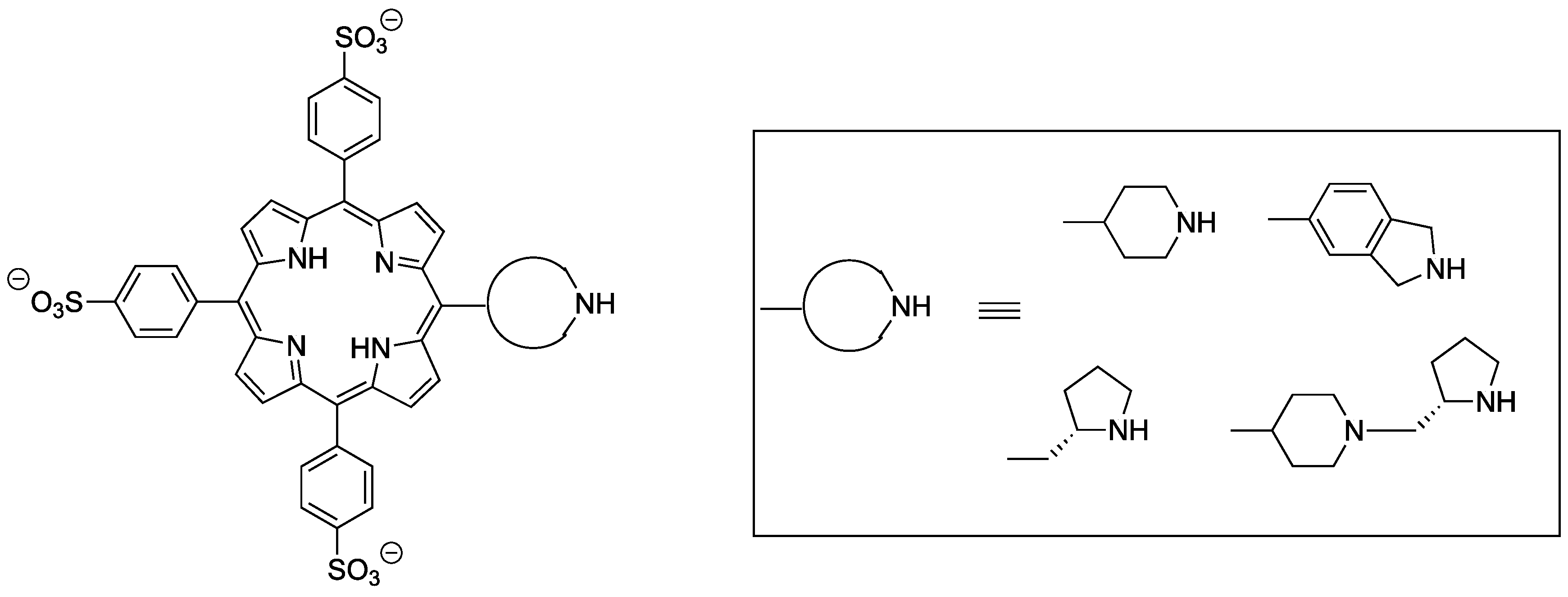
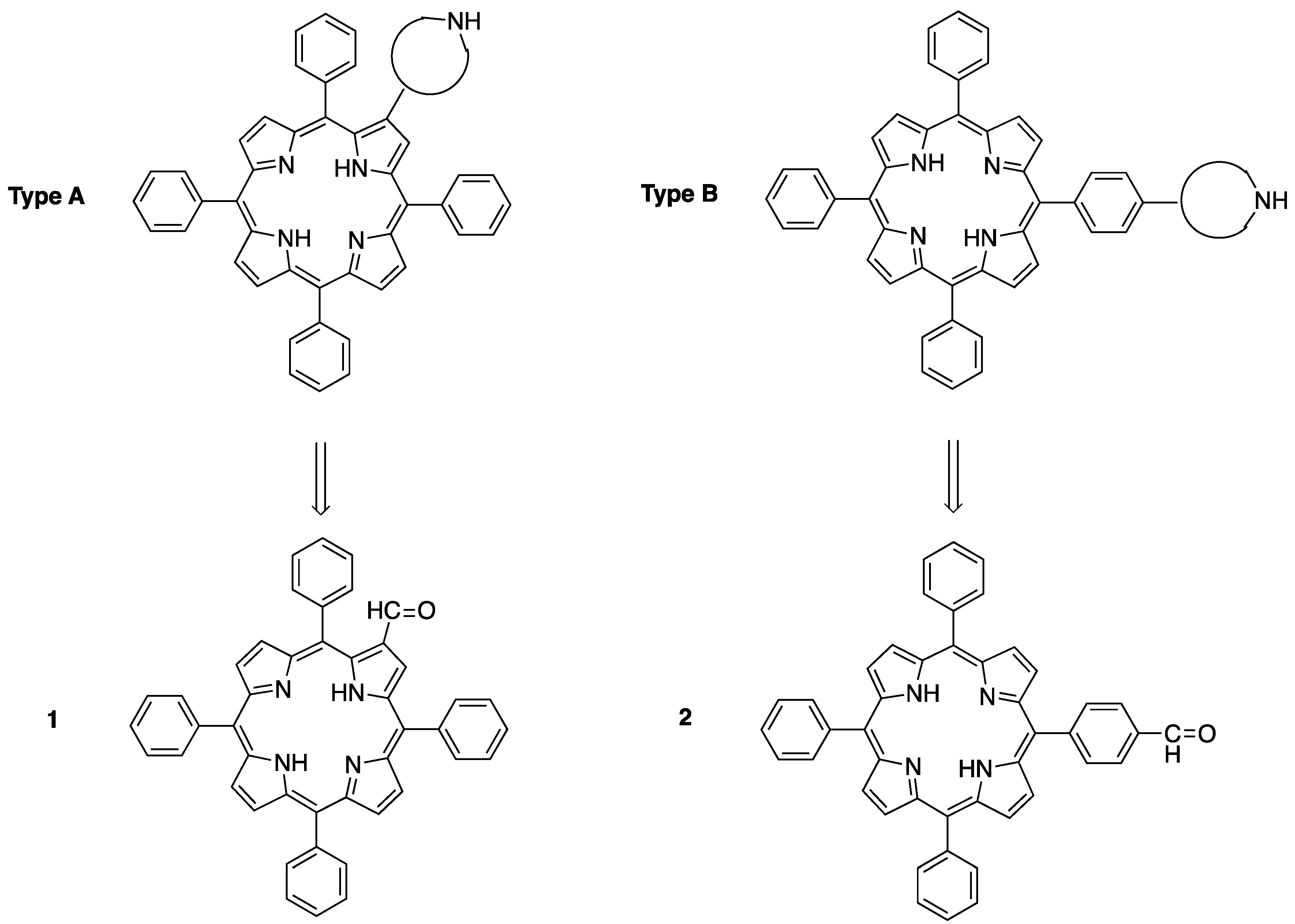
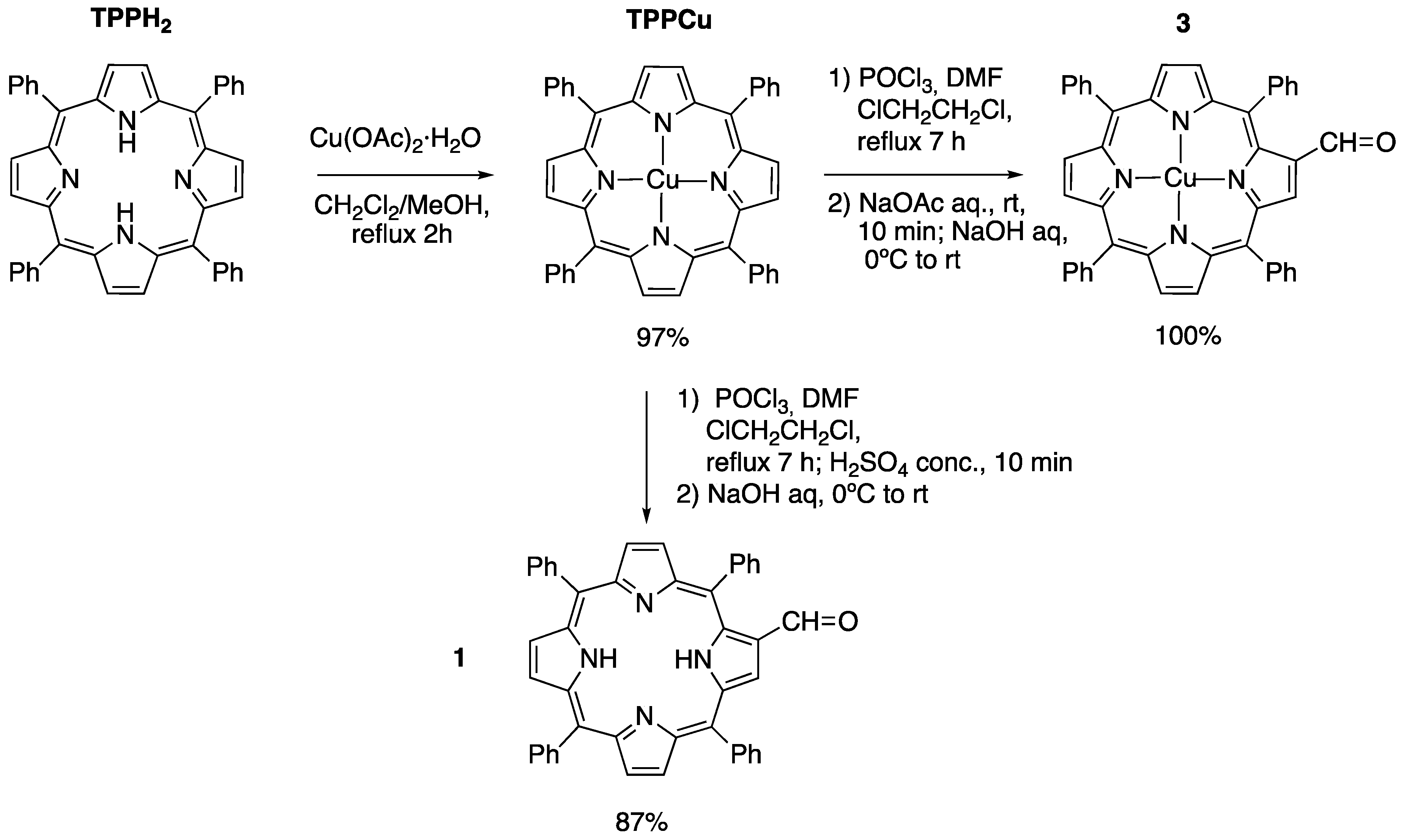
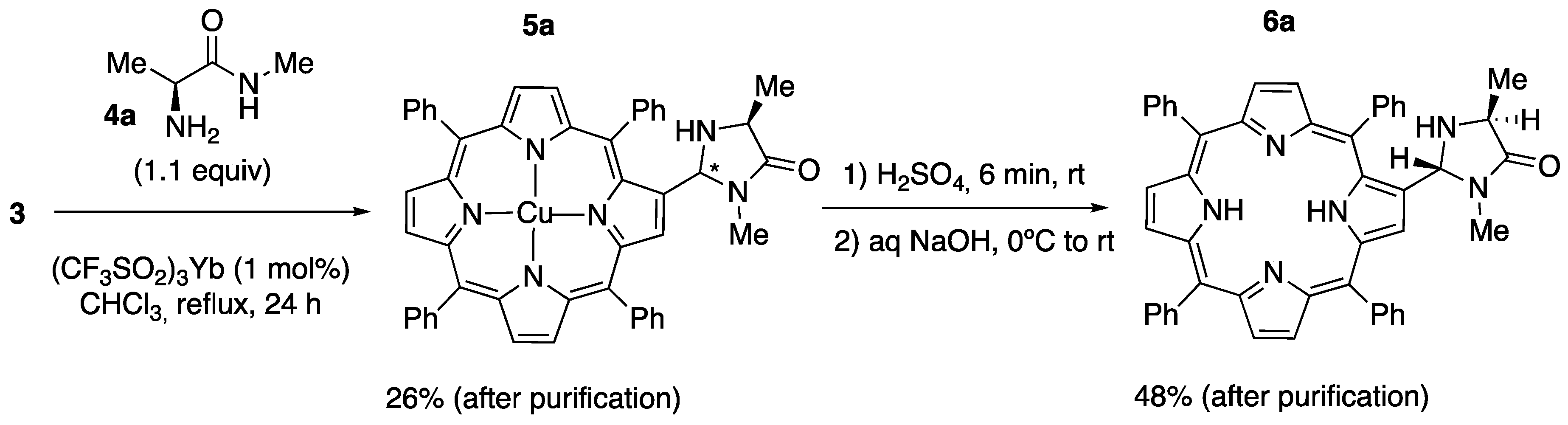
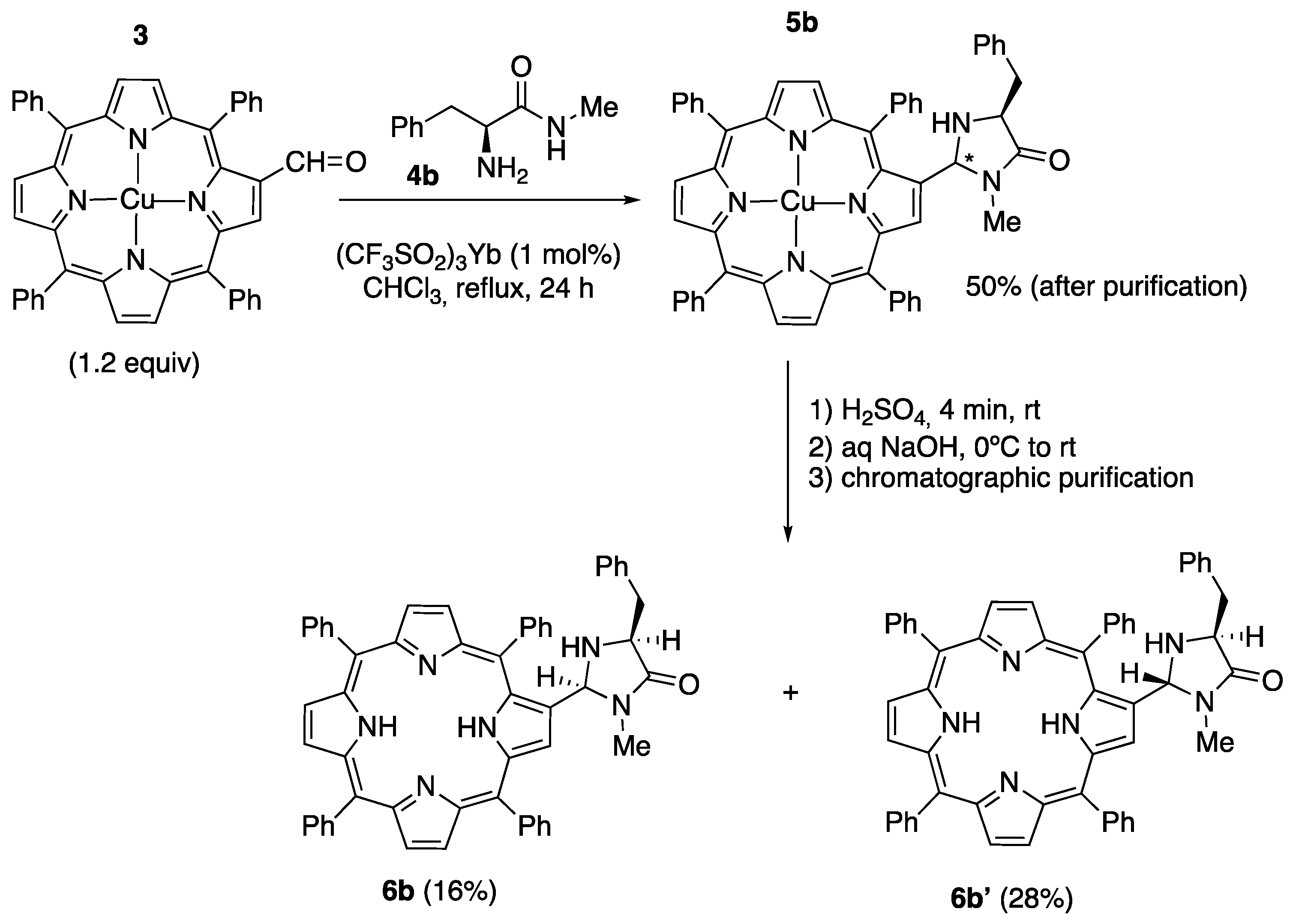
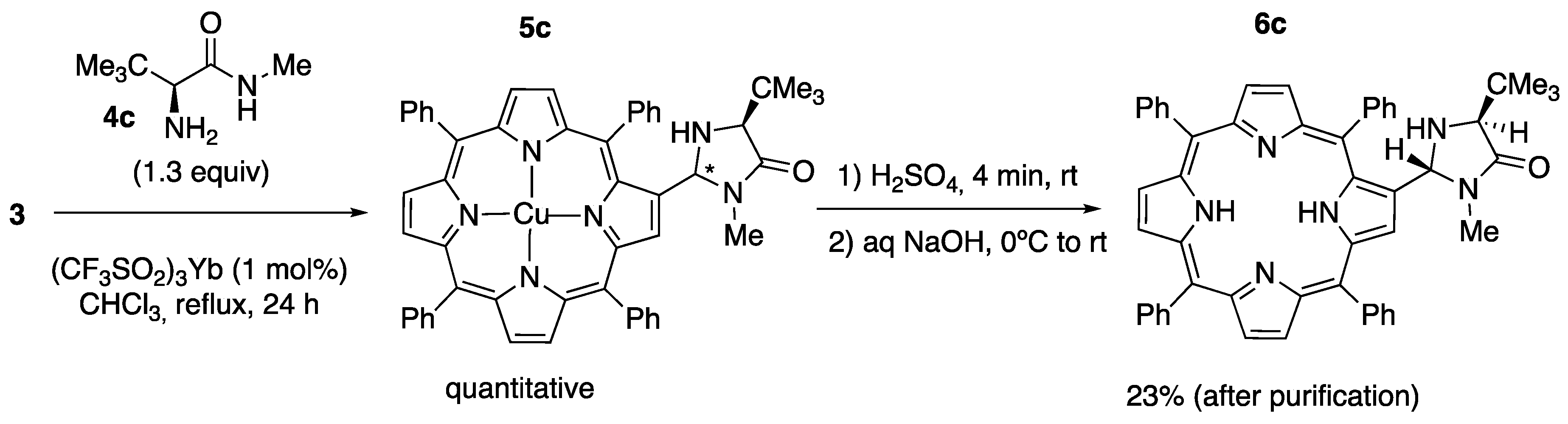
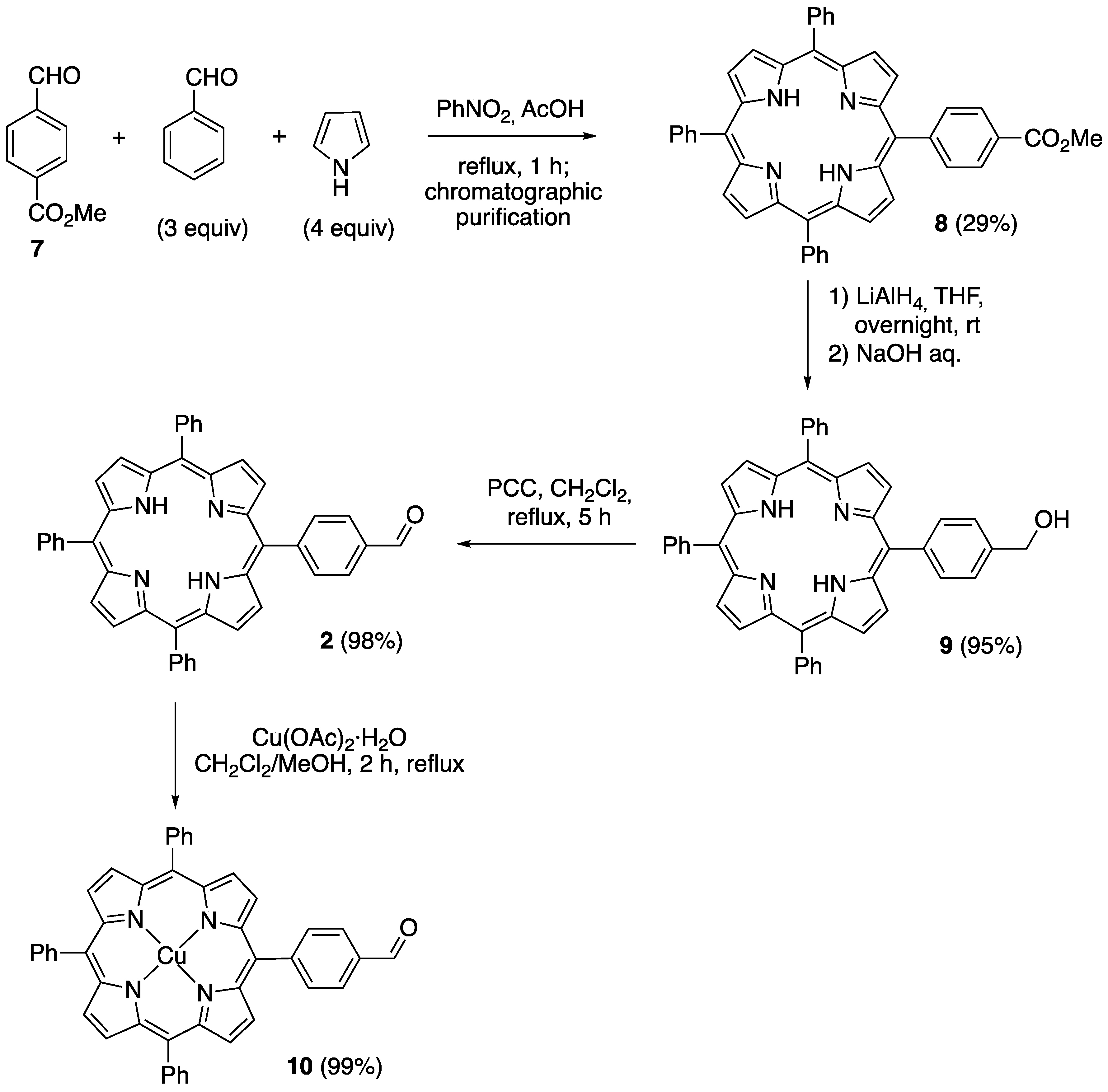
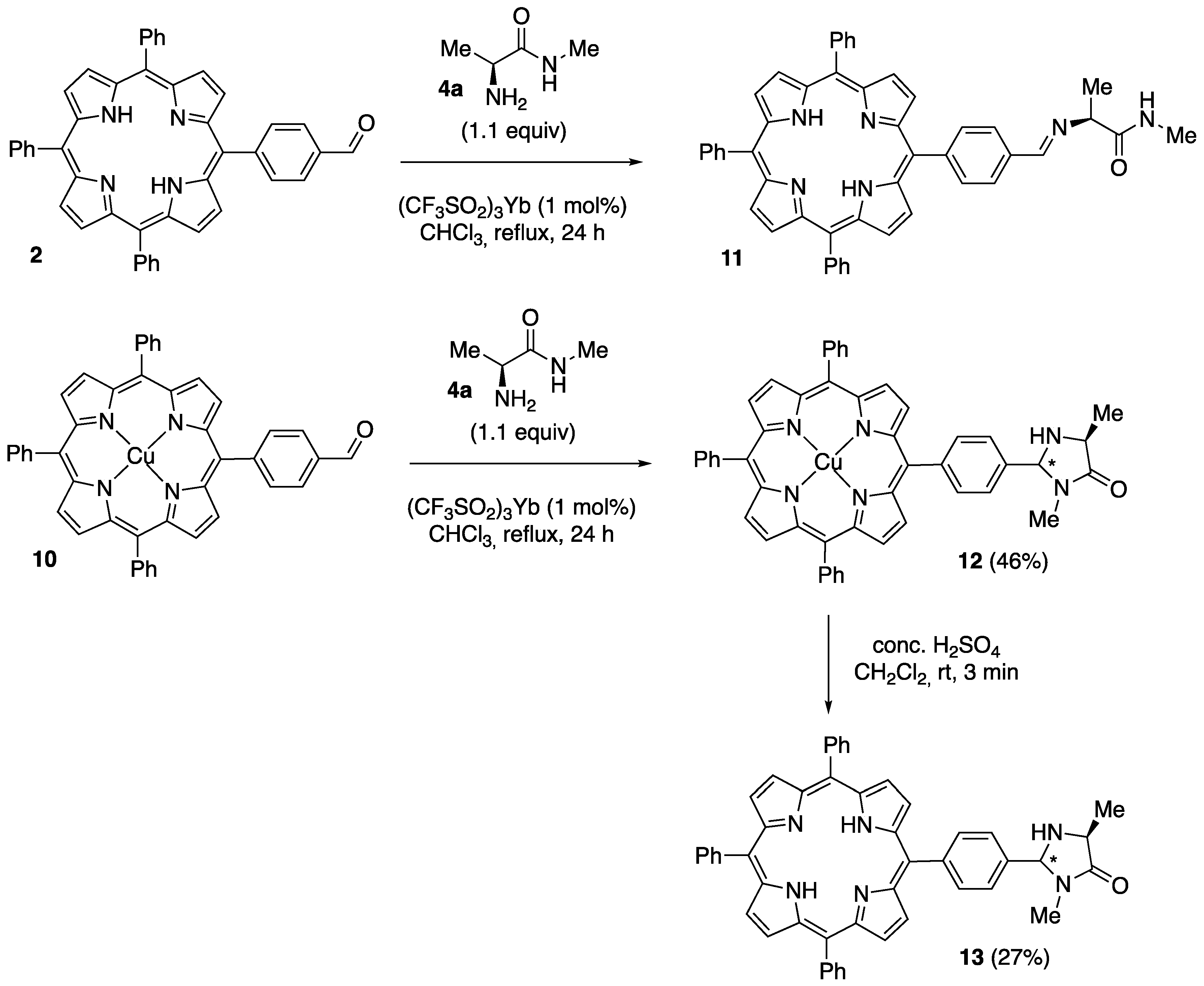
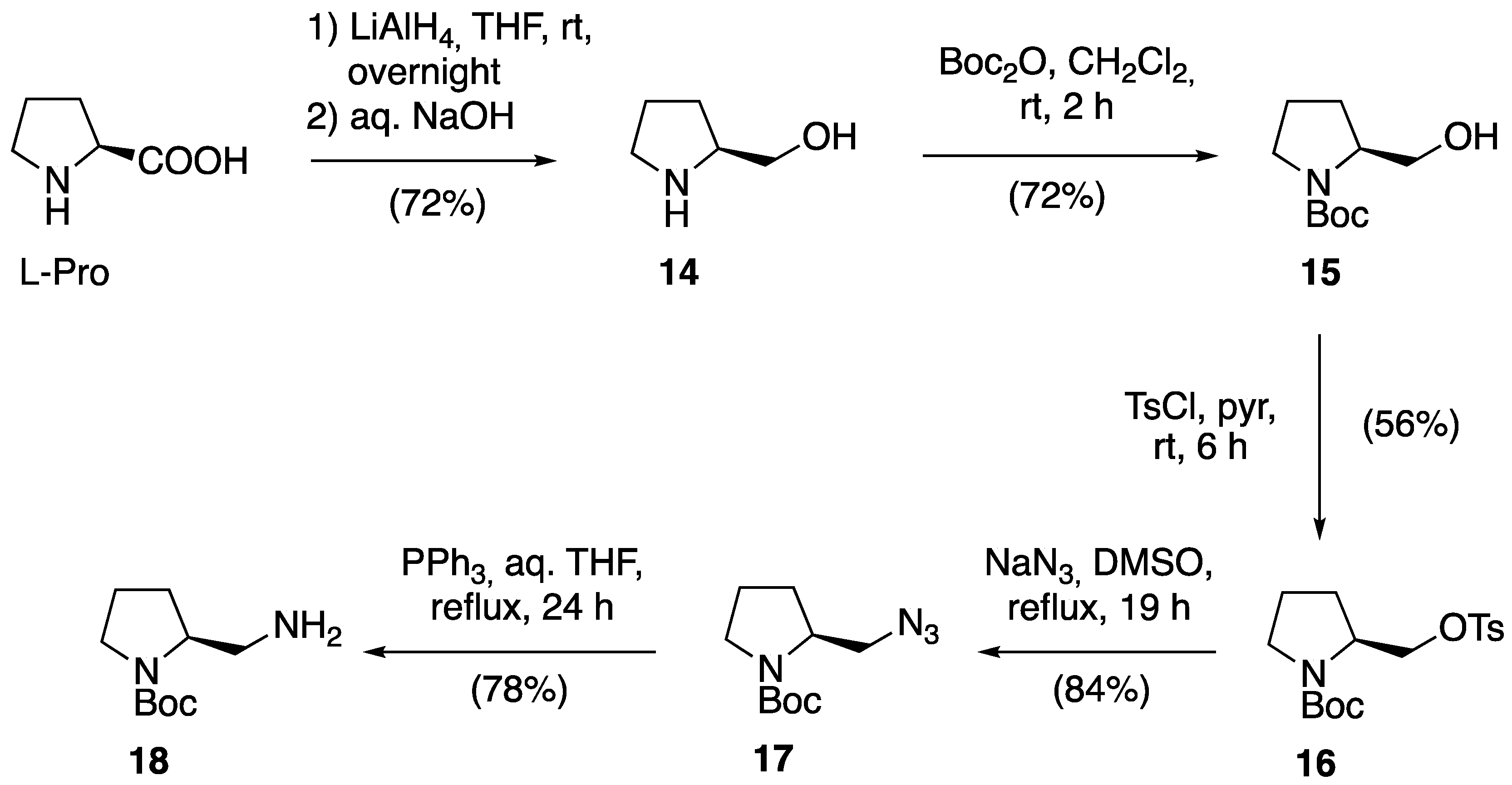
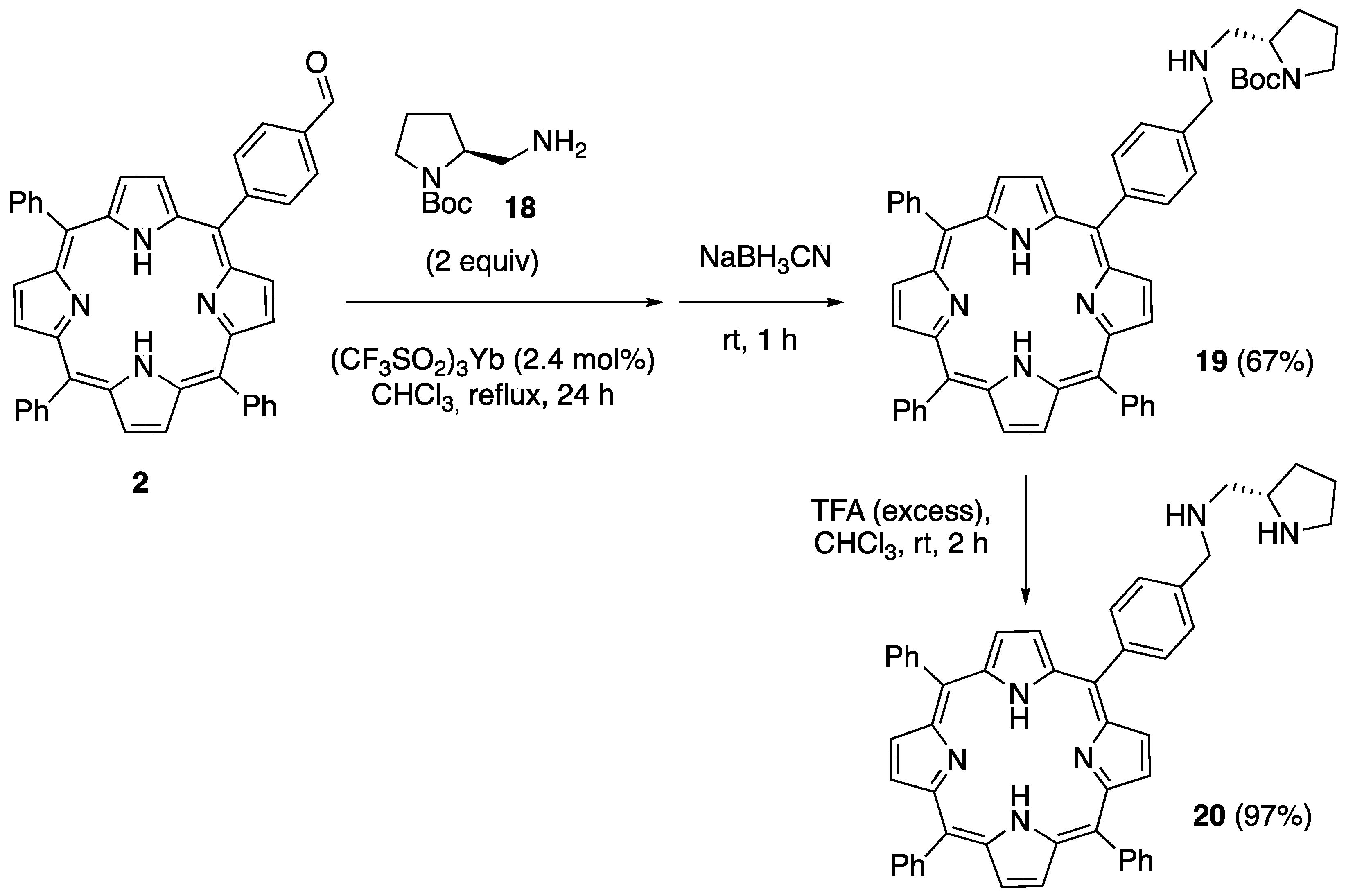
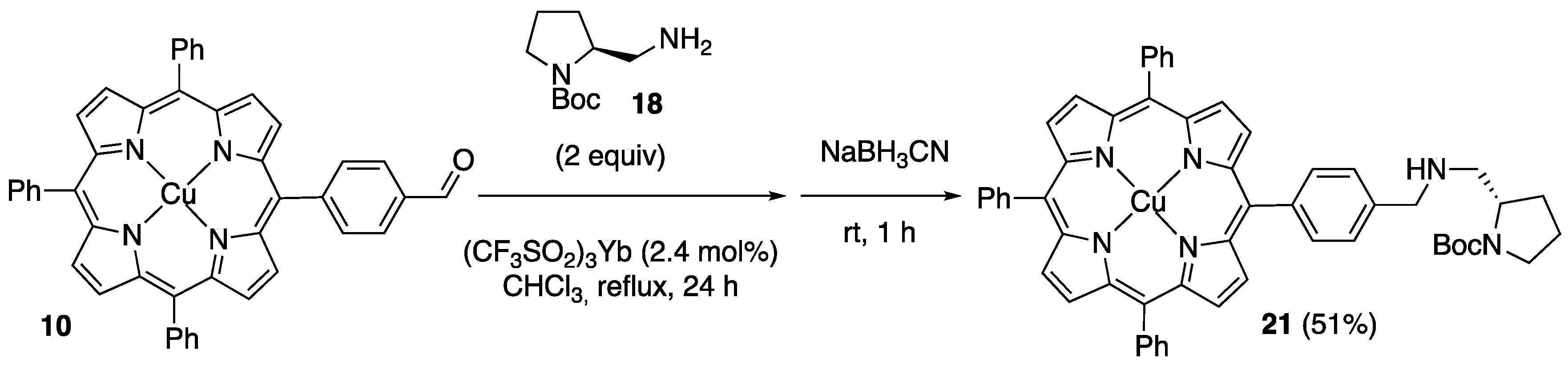
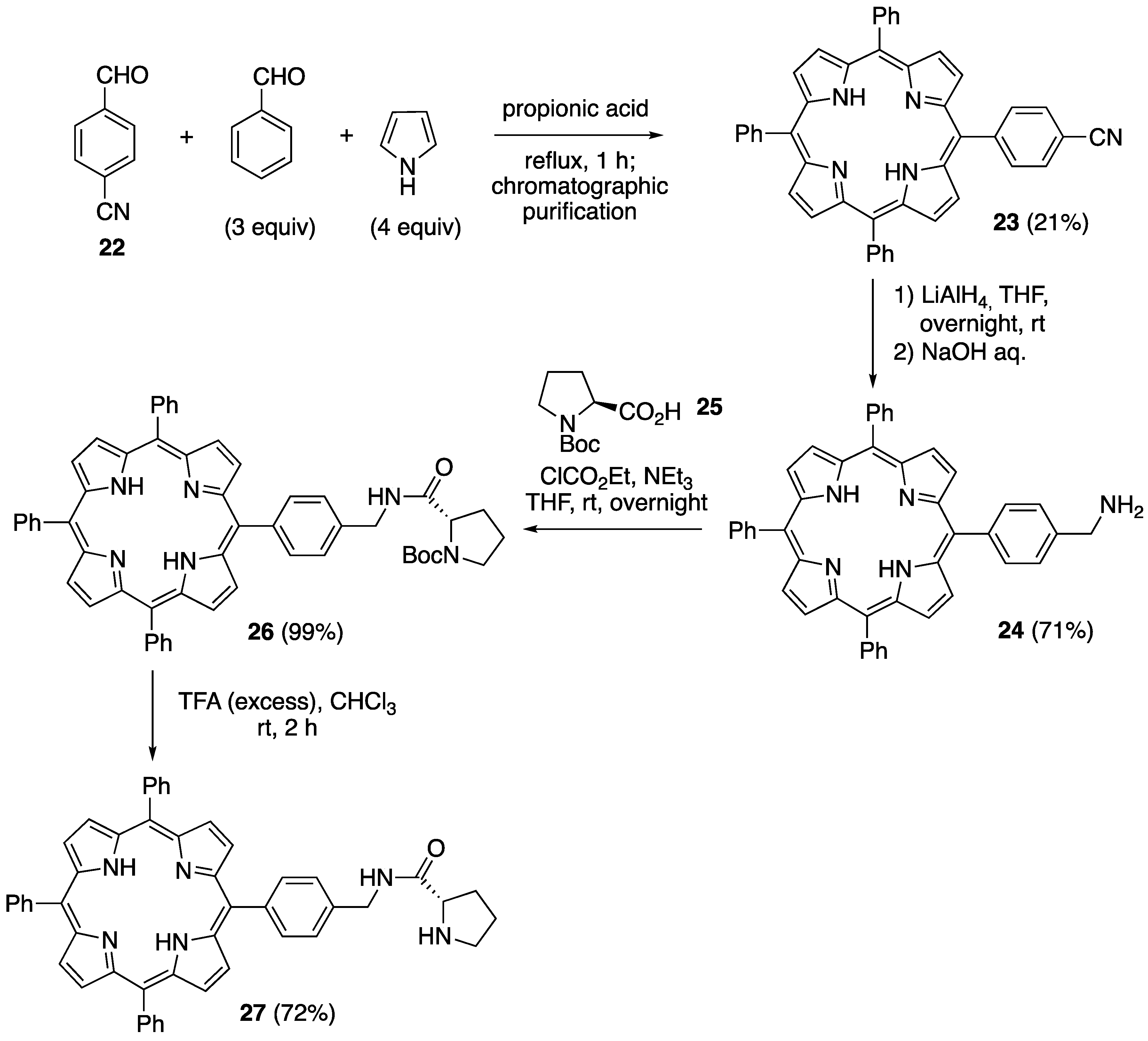
2.1. Synthesis of Novel Amino-Functionalized Porphyrins
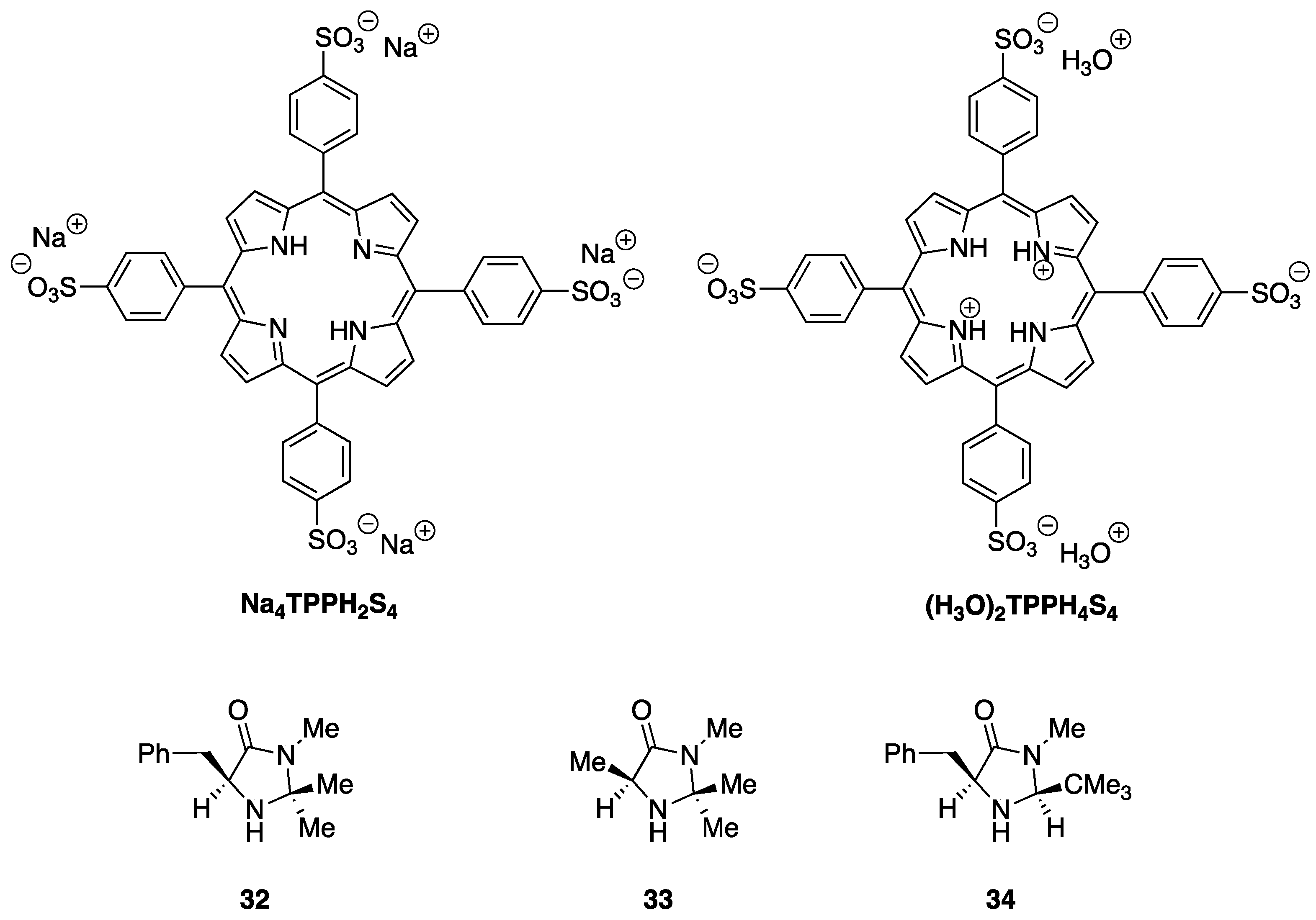
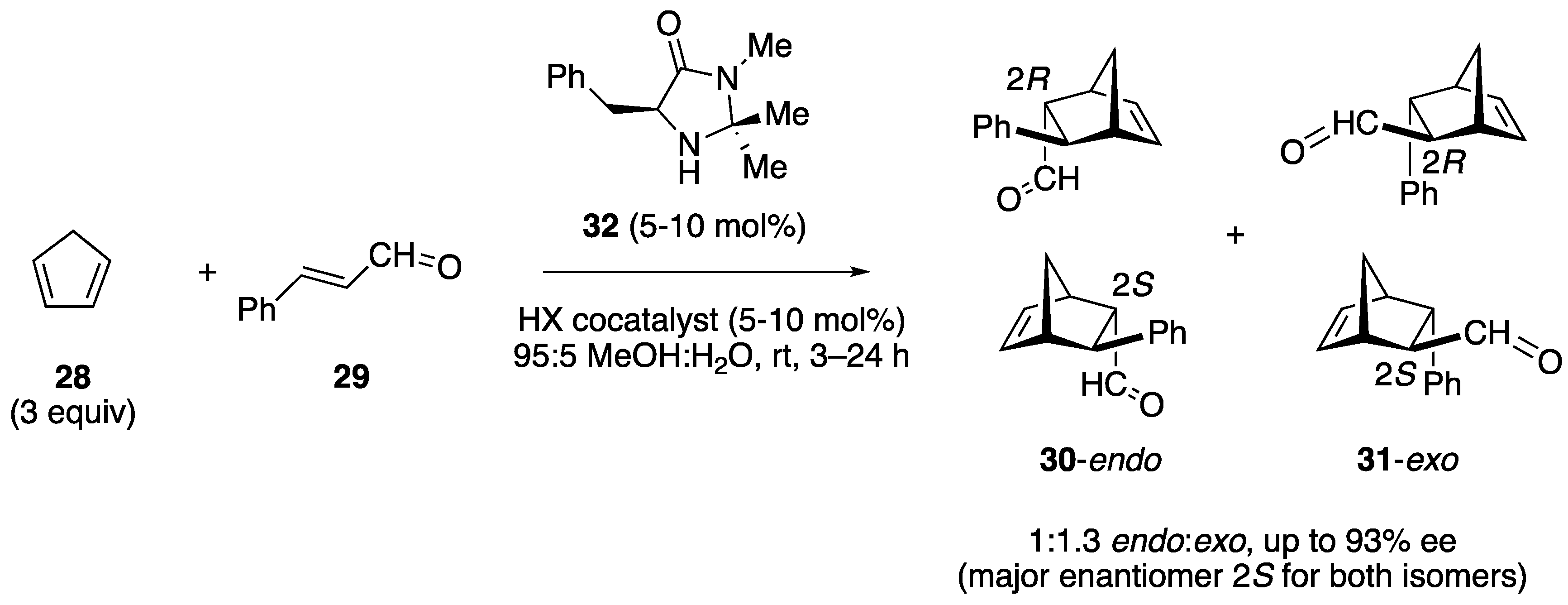
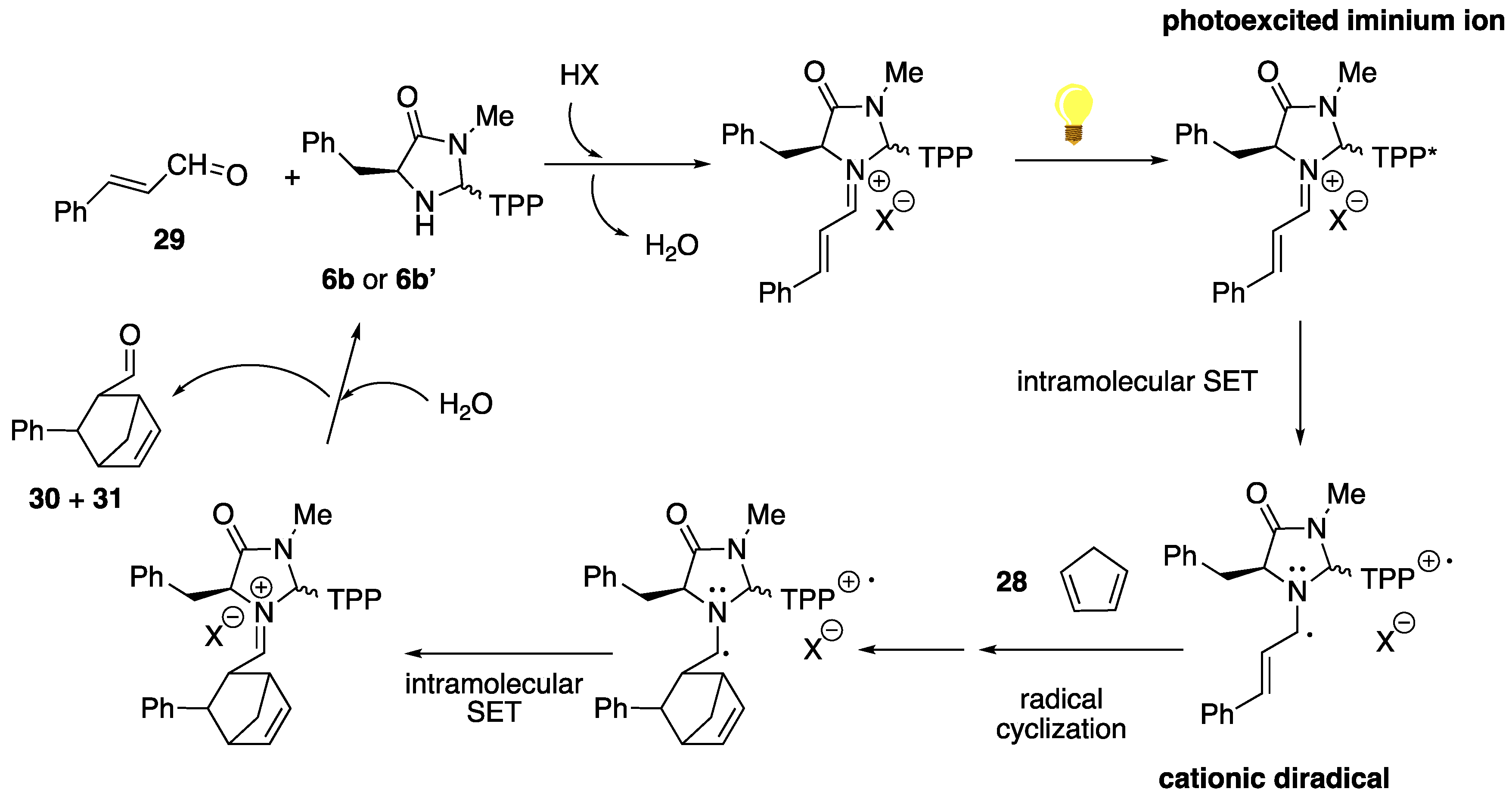
2.2. Study of the Catalytic Activity of Amine–Porphyrin Hybrids of Type A in the Organocatalyzed Diels–Alder Reaction
3. Materials and Methods
3.1. General Methods
3.2. Synthetic Procedures and Product Characterization
3.2.1. Synthesis of Porphyrin-Derived Imidazolidinones
3.2.2. Imidazolidinone-Catalyzed Diels–Alder Reactions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Della Sala, G.; Schettini, R. (Eds.) New Trends in Asymmetric Catalysis; MDPI: Basel, Switzerland, 2021. [Google Scholar]
- Akiyama, T.; Ojima, I. (Eds.) Catalytic Asymmetric Synthesis, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2022. [Google Scholar]
- Dalko, P.I. (Ed.) Comprehensive Enantioselective Organ Catalysis; Wiley-VCH: Weinheim, Germany, 2013; Volumes 1–3. [Google Scholar]
- Rios Torres, R. (Ed.) Stereoselective Organocatalysis. Bond Formation Methodologies and Activation Modes; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Strieth-Kalthoff, F.; James, M.J.; Teders, M.; Pitzer, L.; Glorius, F. Energy transfer catalysis mediated by visible light: Principles, applications, directions. Chem. Soc. Rev. 2018, 47, 7190–7202. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2013, 113, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Nicewicz, D.A.; MacMillan, D.W.C. Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Nagib, D.A.; Scott, M.E.; MacMillan, D.W.C. Enantioselective α-Trifluoromethylation of Aldehydes vis Photoredox Organocatalysis. J. Am. Chem. Soc. 2009, 131, 10875–10877. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Sahoo, B.; Li, J.-L.; Glorius, F. Dual Catalysis Sees the Light: Combining Photoredox with Organo-, Acid, and Transition-Metal Catalysis. Chem.—Eur. J. 2014, 20, 3874–3886. [Google Scholar] [CrossRef]
- Skubi, K.L.; Blum, T.R.; Yoon, T.P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. [Google Scholar] [CrossRef]
- Zou, Y.-Q.; Hörmann, F.M.; Bach, T. Iminium and Enamine Catalysis in Enantioselective Photochemical Reactions. Chem. Soc. Rev. 2018, 47, 278–290. [Google Scholar] [CrossRef]
- Yin, Y.; Zhao, X.; Qiao, B.; Jiang, Z. Cooperative photoredox and chiral hydrogen-bonding catalysis. Org. Chem. Front. 2020, 7, 1283–1296. [Google Scholar] [CrossRef]
- Shibasaki, M.; Kanai, M.; Matsunaga, S.; Kumagai, N. Recent Progress in Asymmetric Bifunctional Catalysis Using Multimetallic Systems. Acc. Chem. Res. 2009, 42, 1117–1127. [Google Scholar] [CrossRef]
- Paull, D.H.; Abraham, C.J.; Scerba, M.T.; Alden-Danforth, E.; Lectka, T. Bifunctional Asymmetric Catalysis: Cooperative Lewis Acid/base Systems. Acc. Chem. Res. 2008, 41, 655–663. [Google Scholar] [CrossRef]
- Bauer, A.; Westkamper, F.; Grimme, S.; Bach, T. Catalytic enantioselective reactions driven by photoinduced electron transfer. Nature 2005, 436, 1139–1140. [Google Scholar] [CrossRef]
- Arceo, E.; Jurberg, I.D.; Álvarez-Fernández, A.; Melchiorre, P. Photochemical activity of a key donor acceptor complex that can drive stereoselective catalytic α-alkylations of aldehydes. Nat. Chem. 2013, 5, 750–756. [Google Scholar] [CrossRef]
- Rigotti, T.; Casado-Sánchez, A.; Cabrera, S.; Pérez-Ruiz, R.; Liras, M.; de la Peña O’Shea, V.A.; Alemán, J. A Bifunctional Photoaminocatalyst for the Alkylation of Aldehydes: Design, Analysis, and Mechanistic Studies. ACS Catal. 2018, 8, 5928–5940. [Google Scholar] [CrossRef]
- Wu, Y.; Kim, D.; Teets, T.S. Photophysical Properties and Redox Potentials of Photosensitizers for Organic Photoredox Transformations. Synlett 2022, 33, 1154–1179. [Google Scholar]
- Costa e Silva, R.; da Silva, L.O.; de Andrade, A.B.; Brocksom, T.J.; de Oliveira, K.T. Recent applications of porphyrins as photocatalysts in organic synthesis: Batch and continuous flow approaches. Beilstein J. Org. Chem. 2020, 16, 917–955. [Google Scholar] [CrossRef]
- Rybika-Jasinska, K.; Shan, W.; Zawada, K.; Kadish, K.M.; Gryko, D. Porphyrins as Photoredox Catalysts: Experimental and Theoretical Studies. J. Am. Chem. Soc. 2016, 138, 15451–15458. [Google Scholar] [CrossRef]
- Rybika-Jasinska, K.; König, B.; Gryko, D. Porphyrin-Catalyzed Photochemical C–H Arylation of Heteroarenes. Eur. J. Org. Chem. 2017, 2017, 2104–2107. [Google Scholar] [CrossRef]
- De Souza, A.A.N.; Silva, N.S.; Müller, A.V.; Polo, A.S.; Brocksom, T.J.; de Oliveira, K.T. Porphyrins as Photoredox Catalysts in Csp2–H Arylations: Batch and Continuous Flow Approaches. J. Org. Chem. 2018, 83, 10577–15086. [Google Scholar] [CrossRef]
- Arlegui, A.; El-Hachemi, Z.; Crusats, J.; Moyano, A. 5-Phenyl-10,15,20-Tris(4-sulfonatophenyl)porphyrin: Synthesis, Catalysis, and Structural Studies. Molecules 2018, 23, 3363. [Google Scholar] [CrossRef]
- Arlegui, A.; Soler, B.; Galindo, A.; Orteaga, O.; Canillas, A.; Ribó, J.M.; El-Hachemi, Z.; Crusats, J.; Moyano, A. Spontaneous mirror-symmetry breaking coupled to top-bottom chirality transfer: From porphyrin self-assembly to scalemic Diels–Alder adducts. Chem. Commun. 2019, 55, 12219–12222. [Google Scholar] [CrossRef]
- Crusats, J.; Moyano, A. Absolute Asymmetric Catalysis, from Concept to Experiment: A Narrative. Synlett 2021, 32, 2013–2035. [Google Scholar] [CrossRef]
- Arlegui, A.; Torres, P.; Cuesta, V.; Crusats, J.; Moyano, A. A pH-Switchable Aqueous Organocatalysis with Amphiphilic Secondary Amine–Porphyrin Hybrids. Eur. J. Org. Chem. 2020, 2020, 4399–4407. [Google Scholar] [CrossRef]
- Arlegui, A.; Torres, P.; Cuesta, V.; Crusats, J.; Moyano, A. Chiral Amphiphilic Secondary Amine-Porphyrin Hybrids for Aqueous Organocatalysis. Molecules 2020, 25, 3420. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, J.S. Synthesis of meso-Substituted Porphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 1, pp. 45–118. [Google Scholar]
- Bonfantini, E.E.; Burrell, A.K.; Campbell, W.M.; Crossley, M.J.; Gosper, J.J.; Harding, M.M.; Officer, D.L.; Reid, D.C.W. Efficient synthesis of free-base 2-formyl-5,10,15,20-tetraarylporphyrins, their reduction and conversion to [(porphyrin-2-yl)methyl]phosphonium salts. J. Porphyr. Phthalocyanines 2002, 6, 708–719. [Google Scholar] [CrossRef]
- Liu, G.; Khlobystov, A.N.; Charalambidis, G.; Coutsolelos, A.G.; Briggs, G.A.D.; Porfyrakis, K. N@C60-Porphyrin: A Dyad of Two Radical Centers. J. Am. Chem. Soc. 2012, 134, 1938–1941. [Google Scholar] [CrossRef]
- Drovetskaya, T.; Reed, C.A.; Boyd, P. A Fullerene Porphyrin Conjugate. Tetrahedron Lett. 1995, 36, 7971–7974. [Google Scholar] [CrossRef]
- Silva, A.M.G.; Tomé, A.C.; Neves, M.G.P.M.S.; Silva, A.M.S.; Cavaleiro, J.A.S. Synthesis of New β-Substituted meso-Tetraphenylporphyrins via 1,3-Dipolar Cycloaddition Reactions. J. Org. Chem. 2002, 67, 726–732. [Google Scholar] [CrossRef]
- Liu, X.-G.; Feng, Y.-Q.; Tan, C.-J.; Chen, H.-L. Cycloaddition of Porphyrins to Isatin: Synthesis of Novel Spiro Porphyrin Derivatives. Synth. Commun. 2006, 36, 2655–2659. [Google Scholar] [CrossRef]
- Ladeira, B.M.F.; Dias, C.J.; Gomes, A.T.P.C.; Tomé, A.C.; Neves, M.G.P.M.S.; Moura, N.M.M.; Almeida, A.; Faustino, M.A.F. Cationic Pyrrolidine/Pyrroline-Substituted Porphyrins as Efficient Photosensitizers against E. coli. Molecules 2021, 26, 464. [Google Scholar] [CrossRef]
- Moura, N.M.M.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Almeida Paz, F.A.; Silva, A.M.S.; Tomé, A.C.; Cavaleiro, J.A.S. A new synthetic approach to benzoporphyrins and Kröhnke-type porphyrin-2-ylpyridines. Chem. Commun. 2012, 48, 6142–6144. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Nath, M. Novel 5-benzazolyl-10,15,20-triphenylporphyrins and β,meso-benzoxazolyl- bridged porphyrin dyads: Synthesis, characterization and photophysical properties. Dyes Pigments 2012, 92, 1241–1249. [Google Scholar] [CrossRef]
- Moura, N.M.M.; Ramos, C.I.V.; Linhares, I.; Santos, M.S.; Faustino, M.A.F.; Almeida, A.; Cavaleiro, J.A.S.; Amado, F.M.L.; Lodeiro, C.; Neves, M.G.P.M.S. Synthesis, characterization and biological evaluation of cationic porphyrin–terpyridine derivatives. RSC Adv. 2016, 6, 110674. [Google Scholar] [CrossRef]
- Moreira, X.; Santos, P.; Faustino, M.A.F.; Raposo, M.M.M.; Costa, S.P.G.; Moura, N.M.M.; Gomes, A.T.P.C.; Almeida, A.; Neves, M.G.P.M.S. An insight into the synthesis of cationic porphyrin-imidazole derivatives and their photodynamic inactivation efficiency against Escherichia coli. Dyes Pigments 2020, 178, 108330. [Google Scholar] [CrossRef]
- Richeter, S.; Jeandon, C.; Gisselbrecht, J.-P.; Ruppert, R.; Callot, H. Synthesis and Optical and Electrochemical Properties of Porphyrin Dimers Linked by Metal Ions. J. Am. Chem. Soc. 2002, 124, 6168–6179. [Google Scholar] [CrossRef]
- Samulis, L.; Tomkinson, N.C.O. Preparation of the MacMillan Imidazolidinones. Tetrahedron 2011, 67, 4263–4267. [Google Scholar] [CrossRef]
- Li, R.; Yao, S.; Cui, J.; Zhu, Y.; Ge, Z.; Cheng, T. Tripetide Boronic Acid or Boronic Ester, Preparative Method and Use Thereof. U.S. Patent 2012/0135921 A1, 31 May 2012. [Google Scholar]
- Welin, E.R.; Warkentin, A.A.; Conrad, J.C.; MacMillan, D.W.C. Enantioselective α-Alkylation of Aldehydes by Photoredox Organocatalysis: Rapid Access to Pharmacophore Fragments from β-Cyanoaldehydes. Angew. Chem. Int. Ed. 2015, 54, 9668–9672. [Google Scholar] [CrossRef]
- Tomé, J.P.C.; Neves, M.G.P.M.S.; Tomé, A.C.; Cavaleiro, J.A.S.; Mendonça, A.F.; Pegado, I.N.; Duarte, R.; Valdeira, M.L. Synthesis of glycoporphyrin derivatives and their antiviral activity against herpes simplex virus types 1 and 2. Bioorg. Med. Chem. 2005, 13, 3878–3888. [Google Scholar] [CrossRef]
- Tang, X.; Feng, S.; Wang, Y.; Yang, F.; Zheng, Z.; Zhao, J.; Wu, Y.; Yin, H.; Liu, G.; Meng, Q. Bifunctional metal-free photo-organocatalysts for enantioselective aerobic oxidation of β−dicarbonyl compounds. Tetrahedron 2018, 74, 3624–3633. [Google Scholar] [CrossRef]
- Liu, J.; Li, P.; Zhang, Y.; Ren, K.; Wang, L.; Wang, G. Recyclable Merrifield Resin-Supported Organocatalysts Containing Pyrrolidine Unit through A3-Coupling Reaction Linkage for Asymmetric Michael Addition. Chirality 2010, 22, 432–441. [Google Scholar] [CrossRef]
- Bryden, F.; Boyle, R.W. A Mild, Facile, One-Pot Synthesis of Zinc Azido Porphyrins as Substrates for Use in Click Chemistry. Synlett 2013, 24, 1978–1982. [Google Scholar]
- Huy, P.; Neudörfl, J.-M.; Schmalz, H.-G. A Practical Synthesis of Trans-3-Substituted Proline Derivatives through 1,4-Addition. Org. Lett. 2011, 13, 216–219. [Google Scholar] [CrossRef]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels–Alder Reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Holland, M.C.; Metternich, J.B.; Daniliuc, C.; Schweizer, W.B.; Gilmour, R. Aromatic Interactions in Organocatalyst Design: Augmenting Selectivity reversal in Iminium Ion Activation. Chem. Eur. J. 2015, 6, 10031–10038. [Google Scholar] [CrossRef]
- Hayashi, Y.; Samana, S.; Gotoh, H.; Ishikawa, H. Asymmetric Diels–Alder Reactions of α,β-Unsaturated Aldehydes Catalyzed by a Diarylprolinol Silyl ether Salt in the Presence of Water. Angew. Chem. Int. Ed. 2008, 47, 11616–11617. [Google Scholar] [CrossRef]
- Adler, A.D.; Longo, F.R.; Finarelli, J.D.; Goldmacher, J.; Assour, J.; Korsaloff, L.A. A Simplified Synthesis for meso-Tetraphenylporphin. J. Org. Chem. 1966, 32, 476. [Google Scholar] [CrossRef]
- López, M.C.; Royal, G.; Philouze, C.; Chavant, P.Y.; Blandin, V. Imidazolidinone nitroxides as catalysts in the aerobic oxidation of alcohols, en route to atroposelective oxidative desymmetrization. Eur. J. Org. Chem. 2014, 2014, 4884–4896. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Organocatalyst | Solvent | % Yield b (dr) c | % ee (30/31) d |
|---|---|---|---|---|
| 1 | 32 | MeOH/H2O e | 48 (53:47) | 71/81 |
| 2 | 5a | MeOH/H2O e | 17 (33:67) | 24/17 |
| 3 | 5a | Toluene | 30 (75:25) | 4/1 |
| 4 | 5b | MeOH/H2O e | 36 (29:71) | 37/43 |
| 5 | 6a | MeOH/H2O e | 0 (–) | – |
| 6 | 6b | Toluene | 9 (75:25) | 3/1 |
| 7 | 6b’ | Toluene | 27 (68:32) | 3/49 |
| 8 | 6c | Toluene | 0 (–) | – |
| Entry | Organocatalyst | Photocatalyst | Solvent | % Yield b (dr) c | % ee (30/31) d |
|---|---|---|---|---|---|
| 1 | 32 | TPPH2 | MeOH | 99 (53:47) | 69/76 |
| 2 | 32 | TPPH2 | Toluene | 0 (–) | – |
| 3 | 32 | (H3O)2TPPH4S4 | MeOH | 96 (55:45) | 60/72 |
| 4 e | 32 | Na4TPPH2S4 | MeOH/H2O f | 34 (56:44) | 37/24 |
| 5 | 33 | TPPH2 | MeOH | 90 (52:48) | 61/67 |
| 6 | 34 | TPPH2 | Toluene | 0 (–) | – |
| 7 | 34 | TPPH2 | MeOH | 0 (–) | – |
| Entry | Organophotocatalyst | Solvent | % Yield b (dr) c | % ee (30/31) d |
|---|---|---|---|---|
| 1 | 6b | Toluene | 12 (88:12) | 1/4 |
| 2 | 6b’ | Toluene | 21 (85:15) | 2/2 |
| 3 | 6c | Toluene | 0 (–) | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, P.; Guillén, M.; Escribà, M.; Crusats, J.; Moyano, A. Synthesis of New Amino-Functionalized Porphyrins:Preliminary Study of Their Organophotocatalytic Activity. Molecules 2023, 28, 1997. https://doi.org/10.3390/molecules28041997
Torres P, Guillén M, Escribà M, Crusats J, Moyano A. Synthesis of New Amino-Functionalized Porphyrins:Preliminary Study of Their Organophotocatalytic Activity. Molecules. 2023; 28(4):1997. https://doi.org/10.3390/molecules28041997
Chicago/Turabian StyleTorres, Pol, Marian Guillén, Marc Escribà, Joaquim Crusats, and Albert Moyano. 2023. "Synthesis of New Amino-Functionalized Porphyrins:Preliminary Study of Their Organophotocatalytic Activity" Molecules 28, no. 4: 1997. https://doi.org/10.3390/molecules28041997
APA StyleTorres, P., Guillén, M., Escribà, M., Crusats, J., & Moyano, A. (2023). Synthesis of New Amino-Functionalized Porphyrins:Preliminary Study of Their Organophotocatalytic Activity. Molecules, 28(4), 1997. https://doi.org/10.3390/molecules28041997