Abstract
An extremely small proportion of the X-ray crystal structures deposited in the Protein Data Bank are of RNA or RNA–protein complexes. This is due to three main obstacles to the successful determination of RNA structure: (1) low yields of pure, properly folded RNA; (2) difficulty creating crystal contacts due to low sequence diversity; and (3) limited methods for phasing. Various approaches have been developed to address these obstacles, such as native RNA purification, engineered crystallization modules, and incorporation of proteins to assist in phasing. In this review, we will discuss these strategies and provide examples of how they are used in practice.
1. Introduction
Of the over 190,000 macromolecular structures present in the Protein Data Bank, as of 2022, only 3678 represent X-ray structures of RNA or RNA–protein complexes. Of these, the majority are of small RNAs (≤50 nucleotides in size), making large RNA crystal structures even more of a minority. RNA crystallography is challenging for several reasons: (1) traditional RNA purification methods resulting in low yields of pure, properly folded RNA; (2) nucleic acids having less sequence diversity than proteins, making crystal contacts harder to come by; and (3) difficulties in solving the phase problem in RNA-only structures.
RNA crystallization is complicated by inherent difficulties, such as vulnerability to degradation by RNases and susceptibility to misfolding. A major bottleneck in RNA crystallography is often the production of sufficient amounts of high-quality, homogeneously folded RNA. This review is meant to be used as a reference for the crystallization of large-structured RNAs such as ribozymes and riboswitches. We will discuss the advantages and disadvantages of both traditional and native RNA purification methods. Furthermore, examples of crystallization strategies using both RNA-dependent and protein-driven modules will be discussed. Finally, strategies for phasing—including molecular replacement and general tools for heavy metal soaking to obtain anomalous diffraction data—will be presented.
2. RNA Purification and Folding
T7 RNA polymerase is commonly used to produce large quantities of RNA by in vitro transcription [1]. Despite the versatility of this common method, there are several important caveats to consider. T7 RNA polymerase is prone to non-templated additions of 1–3 nucleotides to the 3′ end of the RNA transcript. These non-templated additions can be circumvented by the incorporation of two sequential 2′-O-methyl substitutions in the last two nucleotides of the 5′ end of the DNA template strand [2]. Template slippage is also known to occur with T7 RNA polymerase when it encounters polyA sequences during transcription [3]. This polymerase also requires the 5′ end of the RNA sequence to contain at least two sequential guanosine residues for efficient promoter firing [1]. Careful sequence design can mitigate these known problems and enhance RNA yield [2]. Heterogeneity in transcript ends can also be overcome using ribozymes.
2.1. Producing Homogeneous Transcripts: Hammerhead Ribozyme
In vitro transcription produces a continuum of transcript lengths due to premature polymerase termination. In some cases, this heterogeneity can be problematic for crystallization. To overcome this, self-cleaving ribozymes, such as the hammerhead and glmS, can be engineered into the sequence to produce homogenous ends.
The hammerhead ribozyme is a small (~50 nucleotides), self-cleaving ribozyme that is found in plant pathogens [4,5] and in the satellite RNAs of newt mitochondria [6]. The use of the hammerhead ribozyme for directed transcript cleavage proved successful in the crystallization of multiple targets [7,8]. There are two forms of hammerhead ribozyme: minimal and full-length. Both contain the same conserved 13-nucleotide catalytic core, but they differ in catalytic efficiency [9]. Hammerhead ribozymes are optimally active in 10 mM Mg2+ at pH 7.5 [10]. Interestingly, the activity of the full-length ribozyme is increased over 100-fold compared to the minimal ribozyme [11]. Implementing hammerhead ribozymes to create homogenous transcript ends requires complementary base pairing with the target RNA (Figure 1A) [12,13]. The hammerhead ribozyme folds into three helices that flank conserved, single-stranded core nucleotides necessary for autocatalytic cleavage (Figure 1A). A conserved group of nucleotides bind Mg2+ metal ions required for folding of the ribozyme into its active conformation (Figure 1B) [14,15]. Cleavage occurs when bound Mg2+ activates the 2′-hydroxyl of C17 for nucleophilic attack on the adjacent phosphodiester bond between C17 and A1.1, producing a 2′,3′-cyclic phosphate and 5′-hydroxyl termini (Figure 1B) [10]. Use of the hammerhead ribozyme is complicated by sequence requirements immediately upstream of the cleavage site. Mutagenic studies have shown that this sequence usually needs to be an NUH trinucleotide (N = any nucleotide, H = not G), but that CAC, CGC, and AAC trinucleotide sequences also lead to efficient cleavage [16,17,18,19].
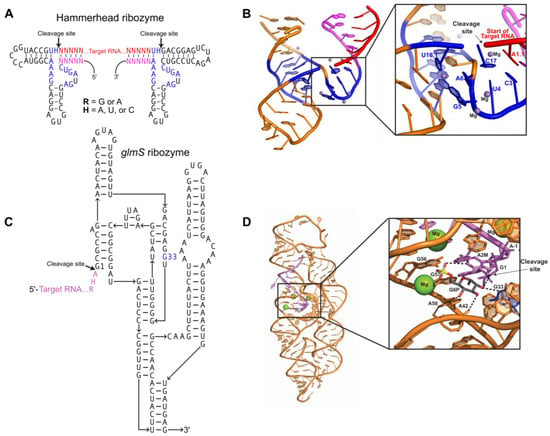
Figure 1.
Ribozymes for homogenous RNA end production: (A) Secondary structure of two hammerhead ribozymes connected by an RNA crystallization target. Conserved nucleotides necessary for catalysis (blue); short complements (magenta) to the 5′ and 3′ ends of the target RNA (red) must be engineered into the ribozyme sequence. Canonical base pairs in helical stems are denoted by dashes. R = G or A; H = A, U, C; N = A, U, C, G. (B) Tertiary structure of the minimal hammerhead ribozyme (PDB:300D). Color scheme matches Figure 1A. Inset shows the ribozyme active site turned upward, with catalytic residues, participating substrate residues, and metal ions labeled. (C) Secondary structure for a glmS ribozyme consisting of the target RNA (purple) and ribozyme strand (black). The G33 (blue) residue interacts with Glc6P and is required for catalysis. (D) Tertiary structure of the Bacillus anthracis glmS ribozyme (PDB:3L3C) bound to Glc6P (shown as sticks) before cleavage. Color scheme matches Figure 1C. Inset shows the ribozyme active site, with catalytic residues and participating substrate residues (shown as sticks) labeled. Hydrogen bonds between catalytic residues, substrate, and Glc6P indicated by black dashes; cleavage site indicated by arrow. Conserved G33 residue colored blue; bound Mg2+ shown as green spheres.
2.2. Producing Homogeneous Transcripts: glmS Ribozyme
The glmS ribozyme is a self-cleaving sequence found in the 5′ UTR of the glmS gene, encoding the protein glucosamine-6-phosphate aminotransferase. In vivo, this ribozyme serves as a riboswitch, regulating glucosamine-6-phosphate (GlcN6P) production by inactivating glmS mRNA through self-cleavage only when GlcN6P is bound in the active site [20,21]. The glmS ribozyme consists of a nested double-pseudoknot fold at its core, and another pseudoknot-containing domain peripheral to the core (Figure 1C) [20,21,22]. Pre-catalytic glmS ribozyme structures from Bacillus anthracis confirmed that effector binding immediately initiates the cleavage reaction through acid–base catalysis (Figure 1D) [20,22]. The glmS ribozyme requires both the GlcN6P effector and the G33 base for activity, and mutation of G33 disrupts catalytic activity even though it does not affect ribozyme folding [22].The self-cleavage activity of the glmS ribozyme can be utilized to produce homogeneous transcript ends through incorporation of the ribozyme at the 3′ end of the target RNA sequence. Addition of GlcN6P induces site-specific self-cleavage of the glmS ribozyme, producing the co-transcriptionally folded target RNA with homogenous 3′ ends [23].
2.3. Purification of Transcribed RNA
Properly folded RNA is critical, as conformational heterogeneity leads to poorly ordered crystals and an inability to diffract at a sufficient resolution. Purification of transcribed RNA has been accomplished by excising bands from denaturing PAGE gels, followed by a refolding protocol where the RNA is heat-denatured at 90 °C and allowed to refold by gentle cooling [23]. Unfortunately, temperature-based folding methods have proven unsuccessful for many RNAs due to misfolding. For example, Pereira et al. showed that heat refolding of the VS ribozyme after T7 transcription resulted in conformational heterogeneity and an inactive ribozyme. Urea titrations in conjunction with the same denaturing protocol can be used to mitigate this [24,25,26,27]. Moreover, the small amount of UV used to visualize RNA on a polyacrylamide gel can permanently damage the sample [28]. RNA itself has a free energy of folding of over 100 kcal/mol, so complete denaturation may never fully occur before refolding occurs [29]. Stacking interactions and hydrogen bonding between the small number of bases that comprise RNA are far more prevalent than niche long-range tertiary interactions, which create many different conformational folding possibilities rather than a select few unique folds. In addition, PAGE purification leads to residual acrylamide oligomer contamination that binds to the RNA and is impossible to eliminate, leading to deceptive increases in the molecular weight of the RNA and loss of significant amounts of transcriptional yield from the formation of irreversible aggregates caused by partial denaturation [30]. These issues can make obtaining the high yields needed for crystallography difficult.
Native folding of RNA during transcription can eliminate almost all of the aforementioned issues. Larger RNAs, such as group II self-splicing introns in both pre- and post-catalytic states [31,32], demonstrate a difficulty in maintaining an active fold. Using native gel analysis, Toor et al. showed that denaturing methods prevent refolding from occurring. To circumvent this, a large group of intron-variant sequences were screened to try to find one that had high splicing activity in low-magnesium and higher-temperature conditions [31]. Once the effective intron sequence was found, it was allowed to fold co-transcriptionally using T7 RNA polymerase. Next, the mixture was treated with DNase I to remove the residual DNA template, and then with Proteinase K to remove the DNase and T7 RNA polymerase. Finally, the Proteinase K (29 kDa) was removed by ultrafiltration using an Amicon 100 kDa cutoff filter. During this process, many washes with a simple buffer containing MgCl2 and sodium cacodylate (pH 6.5) were added, allowing buffer exchange and concentration of the RNA [31]. This process leaves fully folded and active catalytic RNA without contaminants.
RNAs natively fold when transcribed in cells and can possess unique post-transcriptional modifications that are important for understanding their structure and function. Modifications can include methylation [33], acetylation [34], glycosylation [35], and many others. In these cases, in vitro transcription is not feasible and other methods must be implemented. Overexpression of a vector containing the target RNA, an inducible promotor, and an affinity purification tag is one way to create modified RNA targets. The viral coat protein MS2 is a small RNA-binding protein [36] that has been widely used as a tool for purifying RNA. This protein binds specifically to a hairpin RNA motif called the MS2 aptamer. The aptamer can be engineered peripherally to the RNA of interest or within non-essential solvent-exposed structured RNA regions. By fusing the MS2 protein to a purification tag such as a FLAG-, HA-, or GST-tag, it is possible to selectively purify the RNA target using affinity chromatography [37]. Other RNA-binding proteins, such as Pseudomonas phage 7 (PP7) [38], can also be used and can function to purify RNA as well as RNA–protein complexes [39,40].
3. RNA-Driven Crystallization Modules
The crystallization of large RNAs can be a difficult task, as nucleic acids do not tend to crystallize as readily as proteins. The diversity of protein side chains offers more opportunities for crystal contacts due to properties such as differential charge, polarity, and hydrophobicity [25,41]. In contrast, nucleic acids present a continuous surface of negative charge that may serve to repel other molecules and inhibit crystal contact formation [25]. Luckily, strategies exist to engineer RNAs containing secondary structure domain modules that can promote crystallization.
Placement of engineered crystallization modules requires careful consideration to ensure solvent accessibility and to prevent the disruption of the native core RNA structure. A useful tool to help guide the modification of the RNA crystallization target is the use of phylogenetic sequence analysis to identify non-conserved variable regions within the RNA periphery. These variable regions are most likely non-essential to the function of the RNA and, thus, can be altered in order to promote crystallization. It is important to have an experimental method to assess the activity and proper folding of the modified RNA sequence following the addition of such modules. Information on phylogenetic variants can be obtained from RNA family databases such as Rfam [42]. Additionally, small changes in helical lengths and terminal loops can have a significant effect on the formation of lattice contacts [43]. Thus, several variations of the engineered molecule, such as the addition or subtraction of base pairs, should be screened for crystallization and diffraction. The formation of RNA’s tertiary structure relies primarily on interactions between secondary structure elements. Figure 2A shows variable regions identified through phylogenic analysis of group IIC introns that could be altered without disrupting folding or activity. Altering of the length of peripheral helices can be particularly helpful in changing how crystal contacts form between molecules and, thus, may aid in the formation of well-ordered crystals.
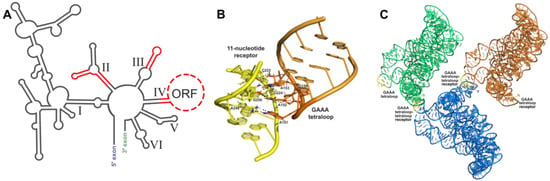
Figure 2.
Tetraloop–tetraloop receptor crystallization module: (A) Phylogenetic covariation in group IIC introns. Stems and loops in red are variable regions and are amenable areas to engineer crystallization modules. (B) Interaction between the GAAA tetraloop (orange) and the 11 nt tetraloop receptor (yellow) in the O.i. group II intron (PDB:3IGI); hydrogen bonding shown by black dashes. (C) Crystal symmetry present in one unit cell from the O.i. group II intron crystal structure (PDB:3IGI). Three RNA molecules pack into one unit cell, each one colored a different color. GAAA tetraloops are colored yellow to show where they form crystal contacts between RNA molecules.
3.1. Tetraloop Interactions as RNA Crystallization Modules
Crystallization of large RNAs often requires the insertion of a “crystallization module”, which promotes nucleation and enhances crystal growth. This method takes advantage of common RNA tertiary structural elements such as tetraloop (four-nucleotide loop sequence) [44] interactions and kissing loop interactions [45]. Tetraloops are common hairpin loop motifs found in RNAs [46], with GNRA (N = any nucleotide, R = A or G) tetraloops being the most prominent in naturally occurring folded RNAs [44]. The GAAA tetraloop is frequently used to enhance crystal contacts and has been used in the contexts of both random and specific tetraloop receptor binding [31,32,47,48,49,50,51]. Crystallization of the SAM-I riboswitch [52]—an mRNA element that binds S-adenosyl methionine to regulate gene expression in bacteria [53,54]—is one example of the GAAA tetraloop used for random binding in conjunction with peripheral helical length variation. GAAA tetraloops tend to bind tandem GC pairs in minor grooves of RNA helices [55,56] or sequences consisting of two Watson–Crick GC pairs, a reverse Hoogsteen AU pair, an adenosine platform, and a wobble GU pair [57]. However, biochemical studies have shown that both the GNRA tetraloops and the receptor sites can tolerate a high degree of variability without losing their binding affinity or specificity [58,59].
The interaction between a GAAA tetraloop and a specific engineered tetraloop receptor can also be used as a crystallization module. The GAAA tetraloop, in conjunction with the 11-nucleotide tetraloop receptor motif (Figure 2B), has been extensively studied and utilized for this purpose. Without affecting the structure biochemically, this module has been used to crystallize multiple large RNAs, including domains 5 and 6 of the group IIB intron ai5γ [47], the human hepatitis delta virus (HDV) ribozyme [47], a CUG RNA helix implicated in myotonic dystrophy type 1 [48], a bacterial ribonuclease P holoenzyme in complex with tRNA [49], and four separate group II intron structures (Figure 2C) [31,32,50,51]. This interaction was first identified by Murphy and Cech, who observed a tertiary contact between a GAAA tetraloop and a conserved bulge on a distal helix stabilizing the fold of the Tetrahymena thermophila ribozyme’s P4-P6 domain [60]. Costa and Michel later characterized the GAAA-tetraloop-specific 11-nucleotide receptor as a highly conserved asymmetric internal loop with the sequence 5′-UAUGG-3′:5′-CCUAAG-3′ [57]. Finally, the structure was obtained by Cate et al. [61]. This interaction is a good choice for enhancing crystallization because of its strength and specificity, acting as a thermodynamic clamp [60,62]. GAAA tetraloop–tetraloop receptor interactions have also been shown to positively affect the accuracy of ribozyme folding pathways [63,64,65,66,67] and, when disrupted by mutation, can cause destabilization of other tertiary interactions within the folded RNA structure [64].
3.2. Loop–Loop Interactions as RNA Crystallization Modules
As mentioned above, loop–loop or “kissing” loop interactions are another RNA tertiary motif that can be utilized as a module for crystallizing large RNAs [50,68]. Kissing loop complexes are formed by base pairing between the single-stranded residues of sequence-complementary loops [45]. Restricted forms of intramolecular kissing loop interactions were first identified between the D and T loops of tRNAs [69], and more extensive interactions were later found in the peripheral components of subgroup IC1 and ID introns [70]. Here, loop residues participate in intramolecular base pairing, creating a single composite, coaxially stacked helix composed of the two original hairpin loops and a new helix created by Watson–Crick base pairing of the nucleotides in the complimentary loops between the two original stems [71]. All nucleotides in each loop are stacked on the 3′ side of the main helix and are involved in pairing interactions [71]. Overall, the structure of the interaction resembles a bent RNA helix and requires magnesium ions to form [72]. Kissing loop interactions can also be intermolecular, which has been observed in the recognition of complementary anticodons between different tRNA pairs [73,74,75,76,77,78], the dimerization of genomic RNA of retroviruses [79,80], and in reverse transcription during HIV-1 replication [81].
The most notable example of a kissing loop interaction being used as an RNA crystallization module is in the human spliceosomal U1 snRNP structure [68]. The Nagai lab set out to crystallize the full U1 snRNP complex in 2009 and made many changes to the sequences of both the RNA and proteins to obtain constructs that would support crystallization. Previous studies had shown that the U1A binding site of the U1 snRNA is not crucial to U1 snRNP activity, making the region containing this sequence a reasonable place for the insertion of a kissing loop interaction in the crystallization construct [68]. After soaking with heavy metals (see below), the team obtained crystals that diffracted to 5.5 angstrom [68].
4. Protein-Assisted RNA Crystallography
Crystallization modules can also be composed of RNA-binding proteins or individual protein domains. Protein crystallization modules introduce surfaces that are chemically different from the negatively charged RNA surface, which help to position molecules in a repeating pattern and facilitate the growth of well-ordered crystals [41].
4.1. U1A Protein Module
The most widely used protein crystallization module has been the U1A protein—one of the components of the spliceosomal U1 small nuclear RNP (snRNP), which specifically recognizes a 10-nucleotide sequence in stem-loop II of the U1 snRNA [41]. This sequence can be engineered into a functionally unimportant stem-loop of the RNA target to facilitate binding to the RNA recognition motif (RRM) of the U1A protein and co-crystallization of the resulting RNP [41]. The crystal structure of the U1A protein bound to its cognate RNA shows that the RNA–protein interactions are confined to only seven nucleotides at the 5’ end of the 10-nucleotide loop and the closing base pair of the stem (Figure 3A) [82]. This makes insertion of a U1A binding site into an RNA target quite simple, requiring only a 12-nucleotide insertion to recapitulate the site [82]. Several RNA structures have been solved using the U1A crystallization module, including the hairpin ribozyme [83], the glmS ribozyme–riboswitch [84], the Azoarcus group I intron [85], and the HDV ribozyme [86]. This module has also been used for the crystallization of in vitro evolved ribozymes, aptamer domains, and artificial riboswitches [87,88,89,90].
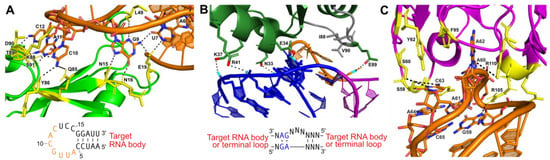
Figure 3.
Protein-assisted crystallization modules: (A) Interaction between U1A (green) and a 21-nucleotide (nt) hairpin (orange) (PDB:1URN). Several residues form hydrogen bonds (black dashes) with 7 nucleotides of the 12 nt stem-loop U1A binding site in the RNA hairpin. The interaction is further stabilized by base stacking between adjacent nucleotides in the stem-loop. Participating amino acid residues are colored yellow. The hairpin sequence for engineering is also shown with interacting nucleotides (orange). (B) Interaction between ribosomal protein L7Ae and a kink-turn (PDB:4BW0). The NC helix (blue) and C helix (magenta) are shown. The bulge (orange) forms hydrophobic interactions with several residues in L7Ae (green; hydrophobic interacting gray). The consensus sequence for engineering is also shown. (C) Interaction between the Fab BL3 antibody (purple) and the GAAACAC stem-loop binding site (orange) in an in vitro evolved RNA ligase (PDB:3IVK). Several base stacking interactions and hydrogen bonds (black dashes) form between nucleotides of the stem-loop and amino acids from the Fab. Base stacking between nucleotides of the stem-loop also occurs. Participating amino acid residues are colored yellow. No engineering sequence is shown, as antibody–RNA interactions are unique for each target obtained from the phage display pool.
4.2. Kink-Turn Module
Another protein crystallization module used to create crystal contacts is the kink-turn (k-turn) motif. This RNA motif, originally discovered in 2001 by Klein et al. in the 50S ribosomal subunit of Haloarcula marismortui, is ~15 nucleotides, containing two helices interrupted by a 3-nucleotide bulge [91]. The asymmetric bulge bends the helical axes 120°, leaving the three unpaired nucleotides free to interact with a k-turn binding protein [91,92]. Flanking the 5′ side of the bulge is a canonical (C) helix, containing Watson–Crick base pairs, and a 3′ non-canonical (NC) helix that begins with 2-3 G-A base pairs (Figure 3B) [91,92].
The interactions between k-turns and the L7Ae family of proteins are similar to those of DNA-binding proteins [92,93]. The alpha helix enters the major groove, made possible due to the kinked shape [92,93]. There, it interacts both nonspecifically with the RNA backbone and specifically via hydrogen bonding with the guanine bases in the NC helix (Figure 3B) [91,92,93]. A hydrophobic loop in the L7Ae protein also interacts with the unpaired bases in the kink itself [91,92,93]. Together, these interactions have a binding affinity of around 10 picomolar [93]. The k-turn motif, in conjunction with the bacterial L7Ae family protein YbxF, has been used to facilitate the crystallization of a T-box riboswitch stem I domain in complex with its cognate tRNA [94]. The k-turn RNA–protein complex facilitated crystal contacts and provided phase information (see below) [94]. The k-turn motif is a useful tool to co-crystallize RNA because it can easily be added to peripheral areas of RNA (Figure 3B), and the abundance of proteins that bind the motif provides many options for co-crystallization and is known to affect the packing of the crystals [95].
4.3. Antibody Fragment Module
Antibody fragments have been used as crystallization modules or chaperones for many proteins that have proven difficult to crystallize under traditional methods [96,97,98,99,100]. In the last 10 years, antibody fragments (Fabs) have also been developed as a crystallization module for RNA. Fabs provide a large surface area for promoting crystal contacts, primarily through their beta-rich secondary structures, which can also serve as molecular replacement search models [96]. An advantage of using this module for crystallization is that the RNA does not need to be engineered, and structures of the RNA–protein complexes can be determined from the natively folded RNA. One hurdle in developing Fab crystallization modules is that RNA does not trigger the production of antibodies when introduced into an animal system [101]. Thus, a synthetic method must be applied. To produce Fabs that bind to RNA with high affinity, Piccirilli and Koldobskaya developed an M13 phage display platform to present Fab fragment libraries fused to coat proteins [102,103]. Through multiple rounds of selection, Fabs that bind the target RNA can be selected and enriched [101,104]. Hydroxyl radical protection assays performed in the presence and absence of the Fab can identify the epitope recognized on the RNA and determine whether binding disturbs the global fold [101]. This approach has been used in the crystallization of the group I intron P4-P6 domain [104] and an in vitro evolved class I RNA ligase ribozyme (Figure 3C) [90,101].
5. Solving the “Phase Problem” for RNA Crystals
Formation of electron density maps of crystallized macromolecules requires the amplitude and phase of each diffracted wave [105]. X-ray diffraction datasets collected from a crystal use predetermined X-ray energies, and the intensities of diffracted waves—or “diffraction spots”—are used to determine amplitude [105]. However, this information is essentially useless without a means to determine the phase of each wave. Phase information is necessary to offset the scattered waves when they are added together during reconstruction of the electron density map; consequently, they are critical for building structures from diffraction data [106]. Unfortunately, unlike amplitudes that can be directly measured as intensities on an X-ray detector, information regarding the phase is lost and cannot be directly observed without specific additional experimental considerations [105,106]. This inherent block between crystal diffraction data and a solved structure is referred to as the “phase problem” in crystallography. Several strategies for solving the phase problem have been developed, such as molecular replacement, various methods of isomorphous replacement, and anomalous diffraction [105,106,107]. In this section, we will discuss molecular replacement models for RNA and RNA–protein complexes, as well as isomorphous replacement with multiple different heavy metals used to support anomalous diffraction methods.
5.1. Molecular Replacement Methods
Molecular replacement (MR) is one commonly used method for solving the phase problem, especially for protein crystallography [105,106,107]. This method applies the phases of a structurally similar model to the experimental diffraction data of the target crystal in order to obtain preliminary electron density maps [105,106,107]. Although MR is applicable for estimating phases for any type of macromolecule, it is often better suited for proteins or nucleic acid–protein complexes, as nucleic acid structures make up only 1.8% of the total number of structures in the PDB [108]. This dearth of solved nucleic acid structures can make finding a suitable model for molecular replacement of an all-nucleic acid target quite difficult. MR search models should have high structural similarity to the target molecule [107]. If possible, the input of weak experimental phases determined by anomalous scattering into the search for a model will enhance the chances of success [109]. For a comprehensive review on finding MR models for RNA, see [108].
If no structures are available, a search model can also be designed by homology modeling of the target molecule, or by de novo structure predictions [110,111]. Computational structural biology powered by artificial intelligence (AI) has been revolutionary, providing powerful tools to model macromolecular structures and predict their functions. Alphafold2 is an AI system that uses deep learning algorithms to predict protein structures with astonishing accuracy and is a promising prospect for MR phasing [112]. RNA-based prediction algorithms are also being developed [113,114]: the Rosetta framework FARFAR2 (fragment assembly of RNA with full-atom refinement) uses small RNA fragments that are mended together to create predictions, and it generally performs well in recovering known native-like structures of RNA [114]. Though these advanced computational methods can be limited in the size of the macromolecules that they can predict, their potential applications in phasing will enable a robust and potentially automated pipeline to solve the phase problem in crystallography.
Molecular replacement can be a particularly convenient method for phasing RNA–protein complex crystals, especially when the protein in the complex has already been solved. For example, protein crystallization modules such as U1A [41], L7Ae family proteins [92], and Fabs [90,101] can serve as MR search models. Additionally, known RNA structures have been used, such as with a T-box leader RNA in complex with tRNA, where an existing tRNA structure was used as the search model to solve the phase of the RNA complex [115,116]. Individual homologous domains or subdomains consisting of short helical fragments can also be used [108]. The solved structures of proteins can also be used in conjunction with small RNA fragments designed to find partial MR solutions, which have been used to solve the structure of the flexizyme [105,117] and the c-di-GMP riboswitch [88].
In addition to serving as a search module in molecular replacement methods, methionine residues in protein crystallization modules can also be replaced with selenomethionine derivatives. Selenomethionine substitution of Met sites has shown to make the U1A module suitable for phase determination by multiwavelength anomalous dispersion (MAD; see below) [47,118]. This module was used in the determination of the hairpin ribozyme structure [119], where co-crystals containing selenomethionyl U1A grew readily under the same crystallization conditions as methionine-containing U1A co-crystals. This strategy has also been implemented in the co-crystallization of stem I of the T-box riboswitch that bound a selenomethionyl YbxF [94].
5.2. Isomorphous Replacement and Anomalous Scattering
Most large RNAs bind metal ions such as Mg2+ or Mn2+ that support both their structural integrity and catalytic activities [120,121]. In isomorphous replacement (IR) phasing, the native metal ions in the crystal are replaced with heavy metal ions [105,106]. This substitution results in a heavy-atom derivative crystal that shows measurable scattering intensity differences compared to the native crystal. The scattering intensity difference can then be used to determine the heavy atom positions and phases, allowing the phase of the native RNA structure to be calculated [105,106]. The IR method hinges entirely on the ability to create heavy-atom derivative crystals that are isomorphous with the natural crystal, meaning that they have the same unit cell and orientation of the molecule within the cell [105,106].
Isomorphous replacement can be performed as a single method but has often been combined with anomalous scattering [105,106]. Here, the X-ray energy is tuned to the absorption edge of the IR heavy metal, promoting excitation of inner-shell electrons [105,106]. There are two types of anomalous scattering experiments: multiwavelength anomalous diffraction (MAD), and single-wavelength anomalous diffraction (SAD). With MAD, data are collected from a single crystal at several wavelengths (usually three) to maximize absorption and anomalous diffraction. Wavelengths are chosen at the IR metal’s absorption peak, point of inflection, and at a remote point on the absorption curve of the heavy metal used for phasing [106]. SAD is measured only at the absorption edge peak and is still subject to phase ambiguity [106].
5.2.1. Isomorphous Replacement with Mg2+ Mimics
Heavy-atom derivatives are typically produced using metals that mimic Mg2+’s binding to RNA [105]. Mg2+ is crucial to the structure and folding of RNA and is frequently found coordinated to the negatively charged phosphate backbone or in the major groove at the base edge of tandem guanines [121]. The Mg2+ ion prefers octahedral geometry of coordination and can adopt a fully hydrated coordination sphere, Mg(H2O)62+, or a partially hydrated shell in which inner-sphere contacts are provided by the RNA [121]. Heavy metals that mimic the RNA binding of either the fully hydrated sphere (outer shell) or the partially hydrated shell (inner shell) Mg2+ have been used extensively for phasing RNA crystals, as they tend not to disrupt the structure [50,122,123,124]. Hexamine salts have been the predominant ions used as Mg(H2O)62+ mimics, including those of Co(III), Os(III), and Ir(III) [124]. Each of these ions adopts strict octahedral coordination geometry and exhibits nearly the same coordination distance between the ion and the amine as between Mg2+ and water [122]. Hexamine complexes tend to bind RNA almost exclusively through outer-shell contacts. This is because the NH3 group is unable to accept a hydrogen bond, unlike H2O, which means that the amine coordination shell will gravitate to negatively charged environments [122]. Amine groups within the coordination sphere of Co(NH3)6(III) also resist exchange relative to the rapid exchange observed for water in the coordination sphere of Mg(H2O)62+ [125]. Iridium and cobalt(III) hexamine salts are relatively easy to produce in the lab and have been used for phasing of RNA structures such as the 70S ribosome functional complex [126], the P4-P6 group I ribozyme domain [61] and, more recently, a group IIB intron lariat [50].
Inner-shell Mg2+ mimics typically include heavy metals in the lanthanide series, such as Yb3+, Sm3+, Ln3+, and Eu3+. Diffraction experiments on the P4-P6 group I ribozyme domain led to the observation that the unit-cell dimensions changed as a function of increasing ionic radius for lanthanides in the series from Lu3+ to Sm3+, and that the mosaic spread of the diffraction pattern increased as a function of increasing ionic radius for all lanthanides except Sm3+ [127]. Inner-sphere contacts tend to be catalytically important molecules and are more rarely found in RNA structures. Lanthanide metals have been used to successfully phase crystals of multiple RNAs such as tRNA [128,129], a hammerhead ribozyme [56], group II introns [31,50], and the Azoarcus group I ribozyme [85].
5.2.2. Engineering Heavy Metal Binding Sites
Heavy metal derivatives have historically been produced by a method affectionately referred to as “soak and pray”, where the crystal is soaked in a heavy metal atom solution with the hope that the heavy metal atoms will bind to one or more specific locations within the RNA [124]. Although this method typically results in derivatized crystals, RNA containing suitable specific sites for heavy metal binding is not predictable; thus, the process becomes highly time-consuming through rounds of trial and error [130,131,132]. To address this issue, a general “directed soaking” method has been devised by Batey and Kieft that involves engineering one or more reliable, non-structure-perturbing cation-binding sites into the RNA structure and then soaking hexamine cations into resulting RNA crystals [123,124]. Their method is based on the observation that G-U wobble pairs in A-form helices create a binding site for many cations, including hexamine complexes [127,133,134,135,136]. The identity and orientation of the base pairs that flank the wobble pairs should be taken into consideration, as this can affect cation binding [124]. This method has successfully been used with both cobalt(III) and iridium(III) hexamine, but even cesium has been shown to be effective for phasing when bound to the motif [137]. Since engineering of this site only changes a few nucleotides, it can typically be performed without perturbing the fold or function of the RNA.
5.2.3. Incorporation of Selenium into Nucleic Acids
X-ray crystallography of proteins has been greatly impacted by the utilization of selenium derivatization as a phasing module [138]. Selenium is a popular choice for phasing by anomalous diffraction and is in the same periodic family as oxygen and sulfur; thus, selenium substitution often does not cause structural perturbations [138]. Although this method typically involves substituting selenium for sulfur atoms in methionine residues, selenium has been successfully incorporated into large nucleic acids via multiple enzymatic approaches [138,139]. Small selenium-derivatized nucleic acids (60 nt. or less) can be produced easily during oligonucleotide synthesis, while larger ones can be prepared using DNA or RNA nucleotide triphosphates ((d)NTPs) incorporated by polymerase activity [138]. These Se-modified dNTP/NTPs are commercially available and can include selenium substitutions in the base, sugar, or phosphate portions. For the preparation of large selenium-derivatized RNAs, in vitro transcription using NTPαSe analogs has been used, where one of the oxygen atoms on the alpha phosphate of the NTP is replaced with selenium [138]. These analogs perform as substrates for T7 RNA polymerase just as well as natural NTPs, but certain bases may affect the activity of the resulting Se-modified RNAs. When in vitro transcriptional incorporation of NTPαSe analogs into the hammerhead ribozyme was tested, it was observed that ribozymes produced with UTPαSe and CTPαSe analogs had the same catalytic activity as the wild type [138]. However, ribozymes produced with GTPαSe had only 30% wild-type activity, and ribozymes produced with ATPαSe had very low activity [138]. This suggests that the incorporation of selenium into an RNA crystallization target may require some trial-and-error optimization. Aside from direct polymerization incorporation, Se-modified RNAs may also be produced by enzymatic ligation of two or more selenium-containing fragments. This method was used in the crystallization of a rat spliceosomal U6 snRNA stem-loop motif using T4 RNA ligase [140,141].
5.2.4. Soaking with Halogens
MAD and SAD methods combined with halogen soaking have also been used as techniques for phasing, with limited success [142]. Both bromine and iodine have been utilized, where the halides incorporate into the ordered solvent shell as anomalous scatterers. The absorption edge of bromine is achievable at all synchrotron beam lines and, although the absorption edges of iodine are not easily accessible, it does have a significant anomalous effect [142]. Soaking with this method can require high halide concentrations (0.2–1 M), and soaks should last only a few seconds because of their fast diffusion into the crystals [142].
6. Conclusions
X-ray crystallographic methods have proven to be an invaluable tool in the study of large RNAs. The purification, crystallization, and phasing strategies presented here have helped counter the inherent challenges of RNA crystallography, enabling the determination of structures of many difficult-to-crystallize RNAs. The development of new tools and techniques is continuing to improve the convenience and resolution of these methods, allowing us to understand the functions and potential applications of structured RNA in research and therapeutics. In addition, cryo-electron microscopy (cryo-EM) is gaining traction as a method of RNA structural biology. It is likely that combined approaches using X-ray crystallography, cryo-EM, and even small-angle X-ray scattering (SAXS) will become increasingly popular to fully appreciate the dynamics of structured RNA molecules and to validate structural observations.
Author Contributions
Conceptualization, C.M.S. and A.R.R.; funding acquisition, A.R.R.; visualization, R.W.J. and C.M.S.; writing—original draft preparation, R.W.J., C.M.S. and A.R.R.; writing—review and editing, R.W.J. and A.R.R. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under award number R01GM133857.
Acknowledgments
We thank the current and past members of the Robart Lab, as well as other members of the Department of Biochemistry and Molecular Medicine for their critical evaluations of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Milligan, J.F.; Groebe, D.R.; Witherell, G.W.; Uhlenbeck, O.C. Oligoribonucleotide Synthesis Using T7 RNA Polymerase and Synthetic DNA Templates. Nucleic Acids Res. 1987, 15, 8783–8798. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.; Zheng, M.; Rüdisser, S. A Simple and Efficient Method to Reduce Nontemplated Nucleotide Addition at the 3 Terminus of RNAs Transcribed by T7 RNA Polymerase. RNA 1999, 5, 1268–1272. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, L.E.; Zhou, Y.; McAllister, W.T. Termination and Slippage by Bacteriophage T7 RNA Polymerase. J. Mol. Biol. 1993, 232, 1030–1047. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, C.J.; Rathjen, P.D.; Forster, A.C.; Symons, R.H. Self-Cleavage of plus and Minus RNA Transcripts of Avocado Sunblotch Viroid. Nucleic Acids Res. 1986, 14, 3627–3640. [Google Scholar] [CrossRef] [PubMed]
- Buzayan, J.M.; Gerlach, W.L.; Bruening, G.; Keese, P.; Gould, A.R. Nucleotide Sequence of Satellite Tobacco Ringspot Virus RNA and Its Relationship to Multimeric Forms. Virology 1986, 151, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Symons, R.H. Plant Pathogenic RNAs and RNA Catalysis. Nucleic Acids Res. 1997, 25, 2683–2689. [Google Scholar] [CrossRef]
- Pata, J.D.; King, B.R.; Steitz, T.A. Assembly, Purification and Crystallization of an Active HIV-1 Reverse Transcriptase Initiation Complex. Nucleic Acids Res. 2002, 30, 4855–4863. [Google Scholar] [CrossRef][Green Version]
- Nagai, K.; Oubridge, C.; Ito, N.; Jessen, T.H.; Avis, J.; Evans, P. Crystal Structure of the U1A Spliceosomal Protein Complexed with Its Cognate RNA Hairpin. Nucleic Acids Symp. Ser. 1995, 34, 1–2. [Google Scholar]
- Ruffner, D.E.; Stormo, G.D.; Uhlenbeck, O.C. Sequence Requirements of the Hammerhead RNA Self-Cleavage Reaction. Biochemistry 1990, 29, 10695–10702. [Google Scholar] [CrossRef]
- Stage-Zimmermann, T.K.; Uhlenbeck, O.C. Hammerhead Ribozyme Kinetics. RNA 1998, 4, 875–889. [Google Scholar] [CrossRef]
- Canny, M.D.; Jucker, F.M.; Kellogg, E.; Khvorova, A.; Jayasena, S.D.; Pardi, A. Fast Cleavage Kinetics of a Natural Hammerhead Ribozyme. J. Am. Chem. Soc. 2004, 126, 10848–10849. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, S.M.; Scott, W.G. Structural Simplicity and Mechanistic Complexity in the Hammerhead Ribozyme. Prog. Mol. Biol. Transl. Sci. 2018, 159, 177–202. [Google Scholar] [CrossRef] [PubMed]
- Price, S.R.; Ito, N.; Oubridge, C.; Avis, J.M.; Nagai, K. Crystallization of RNA-Protein Complexes. I. Methods for the Large-Scale Preparation of RNA Suitable for Crystallographic Studies. J. Mol. Biol. 1995, 249, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Bassi, G.S.; Murchie, A.I.; Walter, F.; Clegg, R.M.; Lilley, D.M. Ion-Induced Folding of the Hammerhead Ribozyme: A Fluorescence Resonance Energy Transfer Study. EMBO J. 1997, 16, 7481–7489. [Google Scholar] [CrossRef]
- Hammann, C.; Lilley, D.M.J. Folding and Activity of the Hammerhead Ribozyme. Chembiochem 2002, 3, 690–700. [Google Scholar] [CrossRef]
- Perriman, R.; Delves, A.; Gerlach, W.L. Extended Target-Site Specificity for a Hammerhead Ribozyme. Gene 1992, 113, 157–163. [Google Scholar] [CrossRef]
- Koizumi, M.; Hayase, Y.; Iwai, S.; Kamiya, H.; Inoue, H.; Ohtsuka, E. Design of RNA Enzymes Distinguishing a Single Base Mutation in RNA. Nucleic Acids Res. 1989, 17, 7059–7071. [Google Scholar] [CrossRef]
- Sheldon, C.C.; Symons, R.H. Mutagenesis Analysis of a Self-Cleaving RNA. Nucleic Acids Res. 1989, 17, 5679–5685. [Google Scholar] [CrossRef]
- O’Rourke, S.M.; Estell, W.; Scott, W.G. Minimal Hammerhead Ribozymes with Uncompromised Catalytic Activity. J. Mol. Biol. 2015, 427, 2340–2347. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.G. Ribozymes. Curr. Opin. Struct. Biol. 2007, 17, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.Y.; Fedor, M.J. The GlmS Riboswitch Integrates Signals from Activating and Inhibitory Metabolites in vivo. Nat. Struct. Mol. Biol. 2011, 18, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Ferré-D’Amaré, A.R.; Scott, W.G. Small Self-Cleaving Ribozymes. Cold Spring Harb. Perspect. Biol. 2010, 2, a003574. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.J.B.; Behera, V.; Walter, N.G. Nondenaturing Purification of Co-Transcriptionally Folded RNA Avoids Common Folding Heterogeneity. PLoS ONE 2010, 5, e12953. [Google Scholar] [CrossRef] [PubMed]
- Ke, A.; Zhou, K.; Ding, F.; Cate, J.H.D.; Doudna, J.A. A Conformational Switch Controls Hepatitis Delta Virus Ribozyme Catalysis. Nature 2004, 429, 201–205. [Google Scholar] [CrossRef]
- Ke, A.; Doudna, J.A. Crystallization of RNA and RNA-Protein Complexes. Methods 2004, 34, 408–414. [Google Scholar] [CrossRef]
- Batey, R.T.; Sagar, M.B.; Doudna, J.A. Structural and Energetic Analysis of RNA Recognition by a Universally Conserved Protein from the Signal Recognition Particle. J. Mol. Biol. 2001, 307, 229–246. [Google Scholar] [CrossRef]
- Pan, J.; Woodson, S.A. Folding Intermediates of a Self-Splicing RNA: Mispairing of the Catalytic Core. J. Mol. Biol. 1998, 280, 597–609. [Google Scholar] [CrossRef]
- Kladwang, W.; Hum, J.; Das, R. Ultraviolet Shadowing of RNA Can Cause Significant Chemical Damage in Seconds. Sci. Rep. 2012, 2, 517. [Google Scholar] [CrossRef]
- Turner, D.H.; Sugimoto, N.; Freier, S.M. RNA Structure Prediction. Annu. Rev. Biophys. Biophys. Chem. 1988, 17, 167–192. [Google Scholar] [CrossRef]
- Lukavsky, P.J.; Puglisi, J.D. Large-Scale Preparation and Purification of Polyacrylamide-Free RNA Oligonucleotides. RNA 2004, 10, 889–893. [Google Scholar] [CrossRef]
- Toor, N.; Keating, K.S.; Taylor, S.D.; Pyle, A.M. Crystal Structure of a Self-Spliced Group II Intron. Science 2008, 320, 77–82. [Google Scholar] [CrossRef]
- Chan, R.T.; Robart, A.R.; Rajashankar, K.R.; Pyle, A.M.; Toor, N. Crystal Structure of a Group II Intron in the Pre-Catalytic State. Nat. Struct. Mol. Biol. 2012, 19, 555–557. [Google Scholar] [CrossRef]
- Nachtergaele, S.; He, C. The Emerging Biology of RNA Post-Transcriptional Modifications. RNA Biol. 2016, 14, 156–163. [Google Scholar] [CrossRef]
- Kudrin, P.; Meierhofer, D.; Vågbø, C.B.; Ørom, U.A.V. Nuclear RNA-Acetylation Can Be Erased by the Deacetylase SIRT7. bioRxiv 2021, arXiv:2021.04.06.438707. [Google Scholar]
- Flynn, R.A.; Pedram, K.; Malaker, S.A.; Batista, P.J.; Smith, B.A.H.; Johnson, A.G.; George, B.M.; Majzoub, K.; Villalta, P.W.; Carette, J.E.; et al. Small RNAs Are Modified with N-Glycans and Displayed on the Surface of Living Cells. Cell 2021, 184, 3109–3124.e22. [Google Scholar] [CrossRef]
- Peabody, D.S. The RNA Binding Site of Bacteriophage MS2 Coat Protein. EMBO J. 1993, 12, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-H.; Srikantan, S.; Gorospe, M. MS2-TRAP (MS2-Tagged RNA Affinity Purification): Tagging RNA to Identify Associated MiRNAs. Methods 2012, 58, 81–87. [Google Scholar] [CrossRef]
- Lim, F.; Peabody, D.S. RNA Recognition Site of PP7 Coat Protein. Nucleic Acids Res. 2002, 30, 4138–4144. [Google Scholar] [CrossRef] [PubMed]
- Fritz, S.E.; Haque, N.; Hogg, J.R. Highly Efficient in Vitro Translation of Authentic Affinity-Purified Messenger Ribonucleoprotein Complexes. RNA 2018, 24, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Youngman, E.M.; Green, R. Affinity Purification of in Vivo-Assembled Ribosomes for in Vitro Biochemical Analysis. Methods 2005, 36, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Ferré-D’Amaré, A.R. Use of the Spliceosomal Protein U1A to Facilitate Crystallization and Structure Determination of Complex RNAs. Methods 2010, 52, 159–167. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating Non-Coding RNAs in Complete Genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.L.; Garst, A.D.; Batey, R.T. Determining Structures of RNA Aptamers and Riboswitches by X-Ray Crystallography. Methods Mol. Biol. 2009, 535, 135–163. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Winker, S.; Gutell, R.R. Architecture of Ribosomal RNA: Constraints on the Sequence of “Tetra-Loops”. Proc. Natl. Acad. Sci. USA 1990, 87, 8467–8471. [Google Scholar] [CrossRef]
- Hermann, T.; Patel, D.J. Stitching Together RNA Tertiary Architectures. J. Mol. Biol. 1999, 294, 829–849. [Google Scholar] [CrossRef]
- Richardson, K.E.; Adams, M.S.; Kirkpatrick, C.C.; Gohara, D.W.; Znosko, B.M. Identification and Characterization of New RNA Tetraloop Sequence Families. Biochemistry 2019, 58, 4809–4820. [Google Scholar] [CrossRef] [PubMed]
- Ferré-D’Amaré, A.R.; Zhou, K.; Doudna, J.A. A General Module for RNA Crystallization. J. Mol. Biol. 1998, 279, 621–631. [Google Scholar] [CrossRef]
- Coonrod, L.A.; Lohman, J.R.; Berglund, J.A. Utilizing the GAAA Tetraloop/Receptor to Facilitate Crystal Packing and Determination of the Structure of a CUG RNA Helix. Biochemistry 2012, 51, 8330–8337. [Google Scholar] [CrossRef]
- Reiter, N.J.; Osterman, A.; Torres-Larios, A.; Swinger, K.K.; Pan, T.; Mondragón, A. Structure of a Bacterial Ribonuclease P Holoenzyme in Complex with tRNA. Nature 2010, 468, 784–789. [Google Scholar] [CrossRef]
- Robart, A.R.; Chan, R.T.; Peters, J.K.; Rajashankar, K.R.; Toor, N. Crystal Structure of a Eukaryotic Group II Intron Lariat. Nature 2014, 514, 193–197. [Google Scholar] [CrossRef]
- Toor, N.; Rajashankar, K.; Keating, K.S.; Pyle, A.M. Structural Basis for Exon Recognition by a Group II Intron. Nat. Struct. Mol. Biol. 2008, 15, 1221–1222. [Google Scholar] [CrossRef]
- Montange, R.K.; Batey, R.T. Structure of the S-Adenosylmethionine Riboswitch Regulatory mRNA Element. Nature 2006, 441, 1172–1175. [Google Scholar] [CrossRef]
- Grundy, F.J.; Henkin, T.M. The S Box Regulon: A New Global Transcription Termination Control System for Methionine and Cysteine Biosynthesis Genes in Gram-Positive Bacteria. Mol. Microbiol. 1998, 30, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Winkler, W.C.; Nahvi, A.; Sudarsan, N.; Barrick, J.E.; Breaker, R.R. An mRNA Structure That Controls Gene Expression by Binding S-Adenosylmethionine. Nat. Struct. Biol. 2003, 10, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Pley, H.W.; Flaherty, K.M.; McKay, D.B. Model for an RNA Tertiary Interaction from the Structure of an Intermolecular Complex between a GAAA Tetraloop and an RNA Helix. Nature 1994, 372, 111–113. [Google Scholar] [CrossRef]
- Pley, H.W.; Flaherty, K.M.; McKay, D.B. Three-Dimensional Structure of a Hammerhead Ribozyme. Nature 1994, 372, 68–74. [Google Scholar] [CrossRef]
- Costa, M.; Michel, F. Frequent Use of the Same Tertiary Motif by Self-Folding RNAs. EMBO J. 1995, 14, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Michel, F. Rules for RNA Recognition of GNRA Tetraloops Deduced by in Vitro Selection: Comparison with in Vivo Evolution. EMBO J. 1997, 16, 3289–3302. [Google Scholar] [CrossRef] [PubMed]
- Abramovitz, D.L.; Pyle, A.M. Remarkable Morphological Variability of a Common RNA Folding Motif: The GNRA Tetraloop-Receptor Interaction. J. Mol. Biol. 1997, 266, 493–506. [Google Scholar] [CrossRef]
- Murphy, F.L.; Cech, T.R. GAAA Tetraloop and Conserved Bulge Stabilize Tertiary Structure of a Group I Intron Domain. J. Mol. Biol. 1994, 236, 49–63. [Google Scholar] [CrossRef]
- Cate, J.H.; Gooding, A.R.; Podell, E.; Zhou, K.; Golden, B.L.; Kundrot, C.E.; Cech, T.R.; Doudna, J.A. Crystal Structure of a Group I Ribozyme Domain: Principles of RNA Packing. Science 1996, 273, 1678–1685. [Google Scholar] [CrossRef]
- Szewczak, A.A.; Podell, E.R.; Bevilacqua, P.C.; Cech, T.R. Thermodynamic Stability of the P4-P6 Domain RNA Tertiary Structure Measured by Temperature Gradient Gel Electrophoresis. Biochemistry 1998, 37, 11162–11170. [Google Scholar] [CrossRef]
- Baird, N.J.; Westhof, E.; Qin, H.; Pan, T.; Sosnick, T.R. Structure of a Folding Intermediate Reveals the Interplay between Core and Peripheral Elements in RNA Folding. J. Mol. Biol. 2005, 352, 712–722. [Google Scholar] [CrossRef]
- Chauhan, S.; Woodson, S.A. Tertiary Interactions Determine the Accuracy of RNA Folding. J. Am. Chem. Soc. 2008, 130, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Sosnick, T.R.; Pan, T. Modular Construction of a Tertiary RNA Structure: The Specificity Domain of the Bacillus Subtilis RNase P RNA. Biochemistry 2001, 40, 11202–11210. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakova, I.; Brenowitz, M. Perturbation of the Hierarchical Folding of a Large RNA by the Destabilization of Its Scaffold’s Tertiary Structure. J. Mol. Biol. 2005, 354, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Treiber, D.K.; Williamson, J.R. Concerted Kinetic Folding of a Multidomain Ribozyme with a Disrupted Loop-Receptor Interaction. J. Mol. Biol. 2001, 305, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Pomeranz Krummel, D.A.; Oubridge, C.; Leung, A.K.W.; Li, J.; Nagai, K. Crystal Structure of Human Spliceosomal U1 SnRNP at 5.5 Å Resolution. Nature 2009, 458, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Quigley, G.J.; Rich, A. Structural Domains of Transfer RNA Molecules. Science 1976, 194, 796–806. [Google Scholar] [CrossRef]
- Lehnert, V.; Jaeger, L.; Michel, F.; Westhof, E. New Loop-Loop Tertiary Interactions in Self-Splicing Introns of Subgroup IC and ID: A Complete 3D Model of the Tetrahymena Thermophila Ribozyme. Chem. Biol. 1996, 3, 993–1009. [Google Scholar] [CrossRef]
- Batey, R.T.; Rambo, R.P.; Doudna, J.A. Tertiary Motifs in RNA Structure and Folding. Angew. Chem. Int. Ed. Engl. 1999, 38, 2326–2343. [Google Scholar] [CrossRef]
- Gregorian, R.S.; Crothers, D.M. Determinants of RNA Hairpin Loop-Loop Complex Stability. J. Mol. Biol. 1995, 248, 968–984. [Google Scholar] [CrossRef]
- Eisinger, J. Complex Formation between Transfer RNA’S with Complementary Anticodons. Biochem. Biophys. Res. Commun. 1971, 43, 854–861. [Google Scholar] [CrossRef]
- Eisinger, J.; Gross, N. The Anticodon-Anticodon Complex. J. Mol. Biol. 1974, 88, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, H.; Söll, D.G.; Crothers, D.M. Studies of the Complex between Transfer RNAs with Complementary Anticodons. I. Origins of Enhanced Affinity between Complementary Triplets. J. Mol. Biol. 1976, 103, 499–519. [Google Scholar] [CrossRef] [PubMed]
- Labuda, D.; Grosjean, H.; Striker, G.; Pörschke, D. Codon:Anticodon and Anticodon:Anticodon Interaction: Evaluation of Equilibrium and Kinetic Parameters of Complexes Involving a G:U Wobble. Biochim. Biophys. Acta 1982, 698, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Houssier, C.; Grosjean, H. Temperature Jump Relaxation Studies on the Interactions between Transfer RNAs with Complementary Anticodons. The Effect of Modified Bases Adjacent to the Anticodon Triplet. J. Biomol. Struct. Dyn. 1985, 3, 387–408. [Google Scholar] [CrossRef]
- Romby, P.; Giegé, R.; Houssier, C.; Grosjean, H. Anticodon-Anticodon Interactions in Solution. Studies of the Self-Association of Yeast or Escherichia Coli tRNAAsp and of Their Interactions with Escherichia Coli tRNAVal. J. Mol. Biol. 1985, 184, 107–118. [Google Scholar] [CrossRef]
- Skripkin, E.; Paillart, J.C.; Marquet, R.; Ehresmann, B.; Ehresmann, C. Identification of the Primary Site of the Human Immunodeficiency Virus Type 1 RNA Dimerization in Vitro. Proc. Natl. Acad. Sci. USA 1994, 91, 4945–4949. [Google Scholar] [CrossRef]
- Paillart, J.C.; Marquet, R.; Skripkin, E.; Ehresmann, B.; Ehresmann, C. Mutational Analysis of the Bipartite Dimer Linkage Structure of Human Immunodeficiency Virus Type 1 Genomic RNA. J. Biol. Chem. 1994, 269, 27486–27493. [Google Scholar] [CrossRef]
- Brunel, C.; Marquet, R.; Romby, P.; Ehresmann, C. RNA Loop-Loop Interactions as Dynamic Functional Motifs. Biochimie 2002, 84, 925–944. [Google Scholar] [CrossRef]
- Oubridge, C.; Ito, N.; Evans, P.R.; Teo, C.H.; Nagai, K. Crystal Structure at 1.92 A Resolution of the RNA-Binding Domain of the U1A Spliceosomal Protein Complexed with an RNA Hairpin. Nature 1994, 372, 432–438. [Google Scholar] [CrossRef]
- Rupert, P.B.; Ferré-D’Amaré, A.R. Crystal Structure of a Hairpin Ribozyme-Inhibitor Complex with Implications for Catalysis. Nature 2001, 410, 780–786. [Google Scholar] [CrossRef]
- Cochrane, J.C.; Lipchock, S.V.; Strobel, S.A. Structural Investigation of the GlmS Ribozyme Bound to Its Catalytic Cofactor. Chem. Biol. 2007, 14, 97–105. [Google Scholar] [CrossRef]
- Adams, P.L.; Stahley, M.R.; Kosek, A.B.; Wang, J.; Strobel, S.A. Crystal Structure of a Self-Splicing Group I Intron with Both Exons. Nature 2004, 430, 45–50. [Google Scholar] [CrossRef]
- Ferré-D’Amaré, A.R.; Zhou, K.; Doudna, J.A. Crystal Structure of a Hepatitis Delta Virus Ribozyme. Nature 1998, 395, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Edwards, T.E.; Ferré-D’Amaré, A.R. Structural Basis for Specific, High-Affinity Tetracycline Binding by an in Vitro Evolved Aptamer and Artificial Riboswitch. Chem. Biol. 2008, 15, 1125–1137. [Google Scholar] [CrossRef]
- Kulshina, N.; Baird, N.J.; Ferré-D’Amaré, A.R. Recognition of the Bacterial Second Messenger Cyclic Diguanylate by Its Cognate Riboswitch. Nat. Struct. Mol. Biol. 2009, 16, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.D.; Lipchock, S.V.; Ames, T.D.; Wang, J.; Breaker, R.R.; Strobel, S.A. Structural Basis of Ligand Binding by a C-Di-GMP Riboswitch. Nat. Struct. Mol. Biol. 2009, 16, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Shechner, D.M.; Grant, R.A.; Bagby, S.C.; Koldobskaya, Y.; Piccirilli, J.A.; Bartel, D.P. Crystal Structure of the Catalytic Core of an RNA-Polymerase Ribozyme. Science 2009, 326, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.J.; Schmeing, T.M.; Moore, P.B.; Steitz, T.A. The Kink-Turn: A New RNA Secondary Structure Motif. EMBO J. 2001, 20, 4214–4221. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Lilley, D.M.J. The Molecular Recognition of Kink-Turn Structure by the L7Ae Class of Proteins. RNA 2013, 19, 1703–1710. [Google Scholar] [CrossRef]
- Lilley, D.M.J. The L7Ae Proteins Mediate a Widespread and Highly Functional Protein–RNA Interaction. Biochemist 2019, 41, 40–44. [Google Scholar] [CrossRef]
- Zhang, J.; Ferré-D’Amaré, A.R. Cocrystal Structure of a T-Box Riboswitch Stem I Domain in Complex with Its Cognate tRNA. Nature 2013, 500, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ferré-D’Amaré, A.R. New Molecular Engineering Approaches for Crystallographic Studies of Large RNAs. Curr. Opin. Struct. Biol. 2014, 26, 9–15. [Google Scholar] [CrossRef]
- Koide, S. Engineering of Recombinant Crystallization Chaperones. Curr. Opin. Struct. Biol. 2009, 19, 449–457. [Google Scholar] [CrossRef]
- Dutzler, R.; Campbell, E.B.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-Ray Structure of a ClC Chloride Channel at 3.0 A Reveals the Molecular Basis of Anion Selectivity. Nature 2002, 415, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Tereshko, V.; Uysal, S.; Koide, A.; Margalef, K.; Koide, S.; Kossiakoff, A.A. Toward Chaperone-Assisted Crystallography: Protein Engineering Enhancement of Crystal Packing and X-Ray Phasing Capabilities of a Camelid Single-Domain Antibody (VHH) Scaffold. Protein Sci. 2008, 17, 1175–1187. [Google Scholar] [CrossRef]
- Iwata, S.; Ostermeier, C.; Ludwig, B.; Michel, H. Structure at 2.8 A Resolution of Cytochrome c Oxidase from Paracoccus Denitrificans. Nature 1995, 376, 660–669. [Google Scholar] [CrossRef]
- Lieberman, R.L.; Culver, J.A.; Entzminger, K.C.; Pai, J.C.; Maynard, J.A. Crystallization Chaperone Strategies for Membrane Proteins. Methods 2011, 55, 293–302. [Google Scholar] [CrossRef]
- Piccirilli, J.A.; Koldobskaya, Y. Crystal Structure of an RNA Polymerase Ribozyme in Complex with an Antibody Fragment. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 2918–2928. [Google Scholar] [CrossRef] [PubMed]
- Fellouse, F.A.; Esaki, K.; Birtalan, S.; Raptis, D.; Cancasci, V.J.; Koide, A.; Jhurani, P.; Vasser, M.; Wiesmann, C.; Kossiakoff, A.A.; et al. High-Throughput Generation of Synthetic Antibodies from Highly Functional Minimalist Phage-Displayed Libraries. J. Mol. Biol. 2007, 373, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Gilbreth, R.N.; Esaki, K.; Tereshko, V.; Koide, S. High-Affinity Single-Domain Binding Proteins with a Binary-Code Interface. Proc. Natl. Acad. Sci. USA 2007, 104, 6632–6637. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.-D.; Tereshko, V.; Frederiksen, J.K.; Koide, A.; Fellouse, F.A.; Sidhu, S.S.; Koide, S.; Kossiakoff, A.A.; Piccirilli, J.A. Synthetic Antibodies for Specific Recognition and Crystallization of Structured RNA. Proc. Natl. Acad. Sci. USA 2008, 105, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, G. Crystallography Made Crystal Clear: A Guide for Users of Macromolecular Models; Elsevier Science & Technology: Burlington, VT, USA, 2006; ISBN 978-0-08-045554-9. [Google Scholar]
- Taylor, G.L. Introduction to Phasing. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 325–338. [Google Scholar] [CrossRef]
- Evans, P.; McCoy, A. An Introduction to Molecular Replacement. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 1–10. [Google Scholar] [CrossRef]
- Marcia, M.; Humphris-Narayanan, E.; Keating, K.S.; Somarowthu, S.; Rajashankar, K.; Pyle, A.M. Solving Nucleic Acid Structures by Molecular Replacement: Examples from Group II Intron Studies. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2174–2185. [Google Scholar] [CrossRef]
- Kleywegt, G.J.; Jones, T.A. Template Convolution to Enhance or Detect Structural Features in Macromolecular Electron-Density Maps. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 179–185. [Google Scholar] [CrossRef]
- Giorgetti, A.; Raimondo, D.; Miele, A.E.; Tramontano, A. Evaluating the Usefulness of Protein Structure Models for Molecular Replacement. Bioinformatics 2005, 21 (Suppl. 2), ii72–ii76. [Google Scholar] [CrossRef]
- Thompson, J.; Baker, D. Incorporation of Evolutionary Information into Rosetta Comparative Modeling. Proteins 2011, 79, 2380–2388. [Google Scholar] [CrossRef]
- McCoy, A.J.; Sammito, M.D.; Read, R.J. Implications of AlphaFold2 for Crystallographic Phasing by Molecular Replacement. Acta Crystallogr. D Struct. Biol. 2022, 78, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; McHugh, R.; Anishchenko, I.; Baker, D.; DiMaio, F. Accurate Prediction of Nucleic Acid and Protein-Nucleic Acid Complexes Using RoseTTAFoldNA. bioRxiv 2022, arXiv:2022.09.09.507333. [Google Scholar]
- Watkins, A.M.; Rangan, R.; Das, R. FARFAR2: Improved De Novo Rosetta Prediction of Complex Global RNA Folds. Structure 2020, 28, 963–976.e6. [Google Scholar] [CrossRef] [PubMed]
- Grigg, J.C.; Ke, A. Structural Determinants for Geometry and Information Decoding of TRNA by T Box Leader RNA. Structure 2013, 21, 2025–2032. [Google Scholar] [CrossRef]
- Grigg, J.C.; Price, I.R.; Ke, A. TRNA Fusion to Streamline RNA Structure Determination: Case Studies in Probing Aminoacyl-TRNA Sensing Mechanisms by the T-Box Riboswitch. Crystals 2022, 12, 694. [Google Scholar] [CrossRef]
- Xiao, H.; Murakami, H.; Suga, H.; Ferré-D’Amaré, A.R. Structural Basis of Specific tRNA Aminoacylation by a Small in Vitro Selected Ribozyme. Nature 2008, 454, 358–361. [Google Scholar] [CrossRef]
- Ferré-D’Amaré, A.R.; Doudna, J.A. Methods to Crystallize RNA. In Current Protocols in Nucleic Acid Chemistry; Wiley & Sons: New York, NY, USA, 2001; Chapter 7, Unit 7.6. [Google Scholar] [CrossRef]
- Rupert, P.B.; Ferré-D’Amaré, A.R. Crystallization of the Hairpin Ribozyme: Illustrative Protocols. Methods Mol. Biol. 2004, 252, 303–311. [Google Scholar] [CrossRef]
- Pyle, A. Metal Ions in the Structure and Function of RNA. J. Biol. Inorg. Chem. 2002, 7, 679–690. [Google Scholar] [CrossRef]
- Wedekind, J.E. Metal Ion Binding and Function in Natural and Artificial Small RNA Enzymes from a Structural Perspective. Met. Ions Life Sci. 2011, 9, 299–345. [Google Scholar]
- Jenkins, J.L.; Wedekind, J.E. The Quick and the Dead: A Guide to Fast Phasing of Small Ribozyme and Riboswitch Crystal Structures. Methods Mol. Biol. 2016, 1490, 265–280. [Google Scholar] [CrossRef]
- Batey, R.T.; Kieft, J.S. Soaking Hexammine Cations into RNA Crystals to Obtain Derivatives for Phasing Diffraction Data. Methods Mol. Biol. 2016, 1320, 219–232. [Google Scholar] [CrossRef]
- Keel, A.Y.; Rambo, R.P.; Batey, R.T.; Kieft, J.S. A General Strategy to Solve the Phase Problem in RNA Crystallography. Structure 2007, 15, 761–772. [Google Scholar] [CrossRef]
- Jou, R.; Cowan, J.A. Ribonuclease H Activation by Inert Transition-Metal Complexes. Mechanistic Probes for Metallocofactors: Insights on the Metallobiochemistry of Divalent Magnesium Ion. J. Am. Chem. Soc. 1991, 113, 6685–6686. [Google Scholar] [CrossRef]
- Cate, J.H.; Yusupov, M.M.; Yusupova, G.Z.; Earnest, T.N.; Noller, H.F. X-Ray Crystal Structures of 70S Ribosome Functional Complexes. Science 1999, 285, 2095–2104. [Google Scholar] [CrossRef]
- Cate, J.H.; Doudna, J.A. Metal-Binding Sites in the Major Groove of a Large Ribozyme Domain. Structure 1996, 4, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Suddath, F.L.; Quigley, G.J.; McPherson, A.; Sussman, J.L.; Wang, A.H.; Seeman, N.C.; Rich, A. Three-Dimensional Tertiary Structure of Yeast Phenylalanine Transfer RNA. Science 1974, 185, 435–440. [Google Scholar] [CrossRef]
- Robertus, J.D.; Ladner, J.E.; Finch, J.T.; Rhodes, D.; Brown, R.S.; Clark, B.F.; Klug, A. Structure of Yeast Phenylalanine tRNA at 3 A Resolution. Nature 1974, 250, 546–551. [Google Scholar] [CrossRef]
- Golden, B.L. Heavy Atom Derivatives of RNA. In Methods in Enzymology; RNA—Ligand Interactions, Part A; Academic Press: Cambridge, MA, USA, 2000; Volume 317, pp. 124–132. [Google Scholar]
- Golden, B.L.; Gooding, A.R.; Podell, E.R.; Cech, T.R. X-Ray Crystallography of Large RNAs: Heavy-Atom Derivatives by RNA Engineering. RNA 1996, 2, 1295–1305. [Google Scholar]
- Wedekind, J.E.; McKay, D.B. Purification, Crystallization, and X-Ray Diffraction Analysis of Small Ribozymes. Meth. Enzymol. 2000, 317, 149–168. [Google Scholar] [CrossRef]
- Masquida, B.; Westhof, E. On the Wobble GoU and Related Pairs. RNA 2000, 6, 9–15. [Google Scholar] [CrossRef]
- Varani, G.; McClain, W.H. The G·U Wobble Base Pair. EMBO Rep. 2000, 1, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Colmenarejo, G.; Tinoco, I. Structure and Thermodynamics of Metal Binding in the P5 Helix of a Group I Intron Ribozyme. J. Mol. Biol. 1999, 290, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Stefan, L.R.; Zhang, R.; Levitan, A.G.; Hendrix, D.K.; Brenner, S.E.; Holbrook, S.R. MeRNA: A Database of Metal Ion Binding Sites in RNA Structures. Nucleic Acids Res. 2006, 34, D131–D134. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.D.; Rambo, R.P.; Van Tyne, D.; Batey, R.T. Structure of the SAM-II Riboswitch Bound to S-Adenosylmethionine. Nat. Struct. Mol. Biol. 2008, 15, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Huang, Z. Selenium Derivatization of Nucleic Acids for X-Ray Crystal-Structure and Function Studies. Chem. Biodivers. 2010, 7, 753–785. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Sheng, J.; Carrasco, N.; Huang, Z. Selenium Derivatization of Nucleic Acids for Crystallography. Nucleic Acids Res. 2007, 35, 477–485. [Google Scholar] [CrossRef]
- Höbartner, C.; Rieder, R.; Kreutz, C.; Puffer, B.; Lang, K.; Polonskaia, A.; Serganov, A.; Micura, R. Syntheses of RNAs with up to 100 Nucleotides Containing Site-Specific 2′-Methylseleno Labels for Use in X-Ray Crystallography. J. Am. Chem. Soc. 2005, 127, 12035–12045. [Google Scholar] [CrossRef]
- Höbartner, C.; Micura, R. Chemical Synthesis of Selenium-Modified Oligoribonucleotides and Their Enzymatic Ligation Leading to an U6 SnRNA Stem-Loop Segment. J. Am. Chem. Soc. 2004, 126, 1141–1149. [Google Scholar] [CrossRef]
- Dauter, M.; Dauter, Z. Many Ways to Derivatize Macromolecules and Their Crystals for Phasing. Methods Mol. Biol. 2017, 1607, 349–356. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).