
A Self-Immolative Linker for the pH-Responsive Release of Amides


Abstract
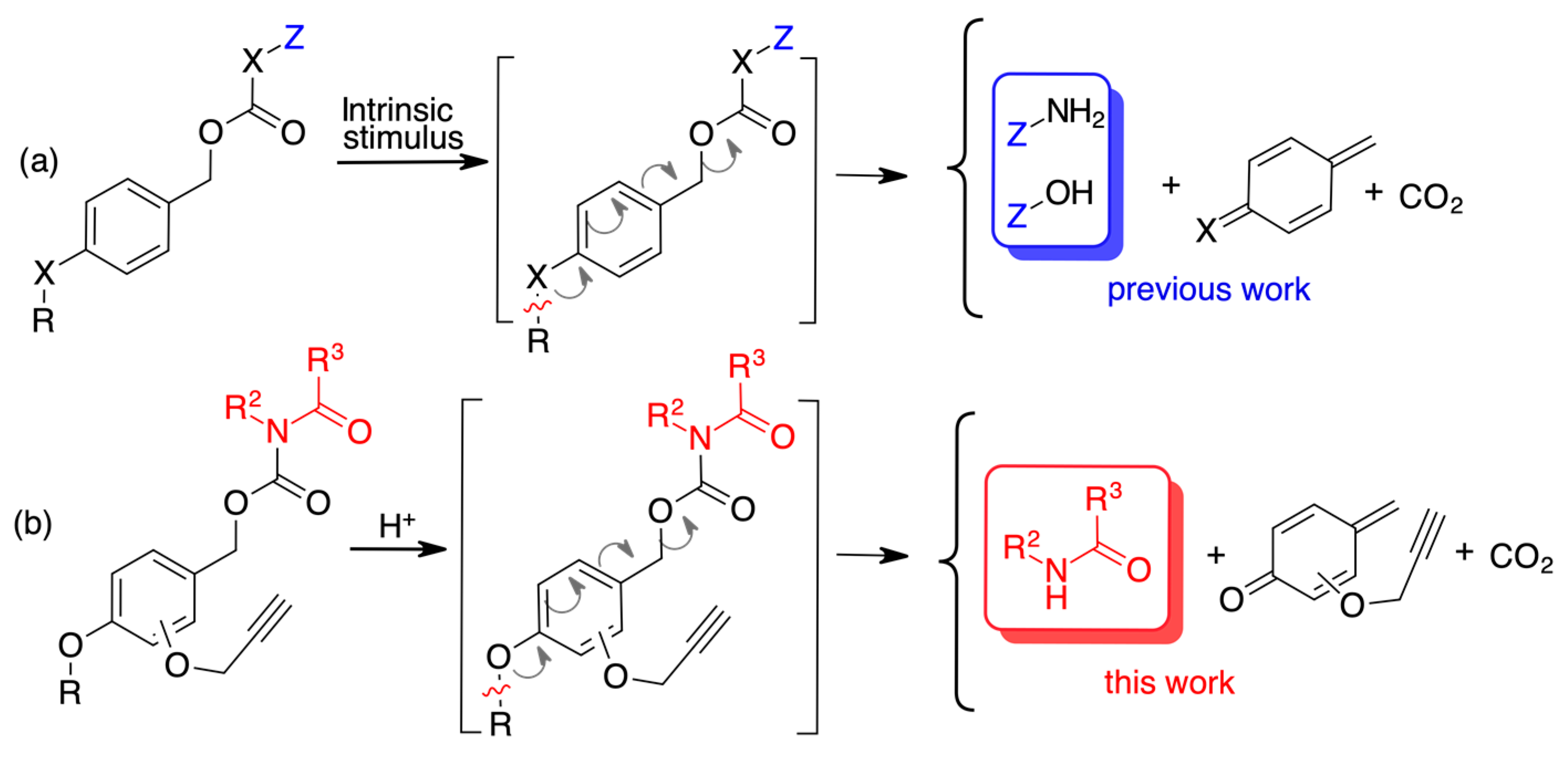
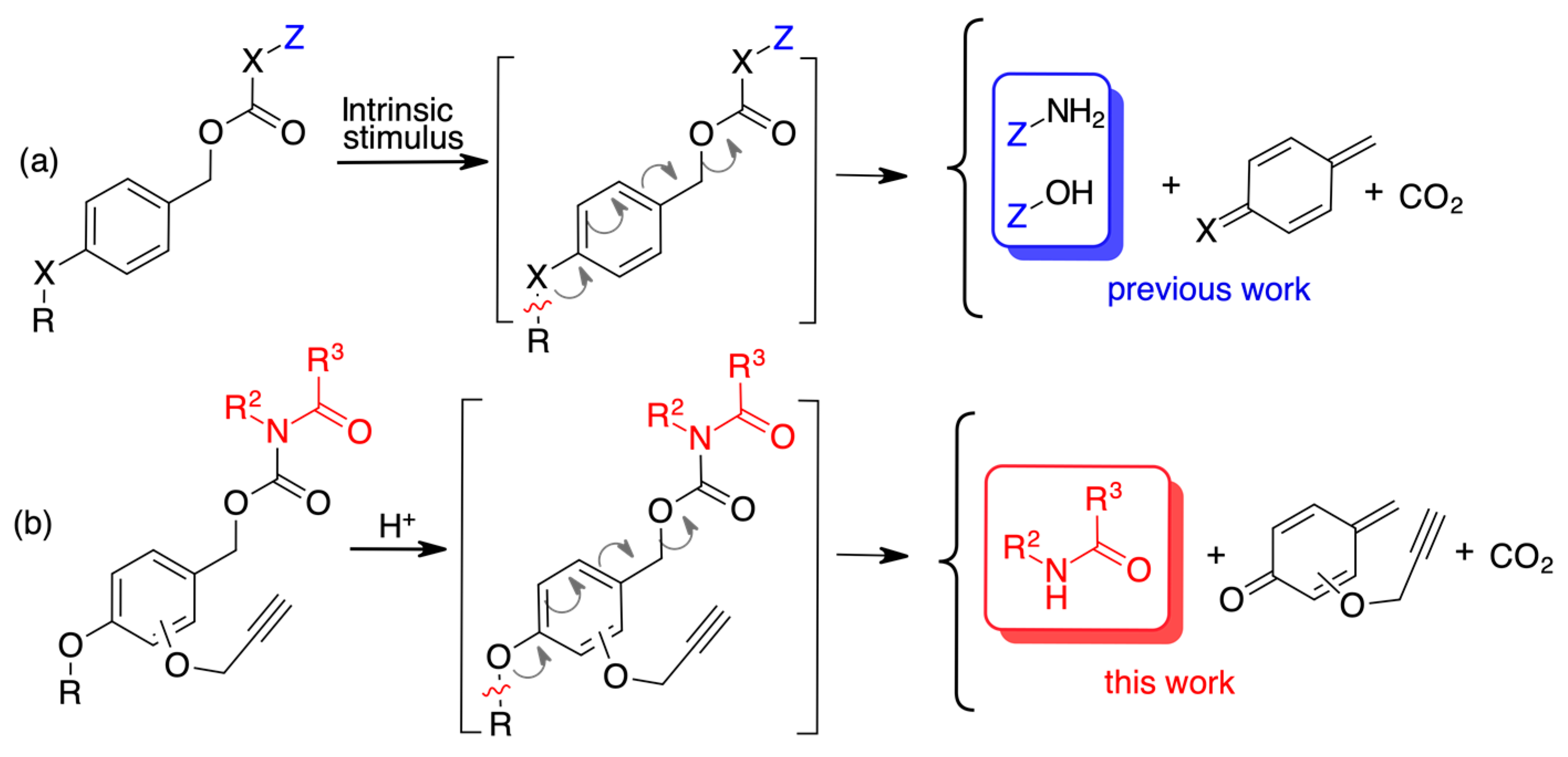
:1. Introduction
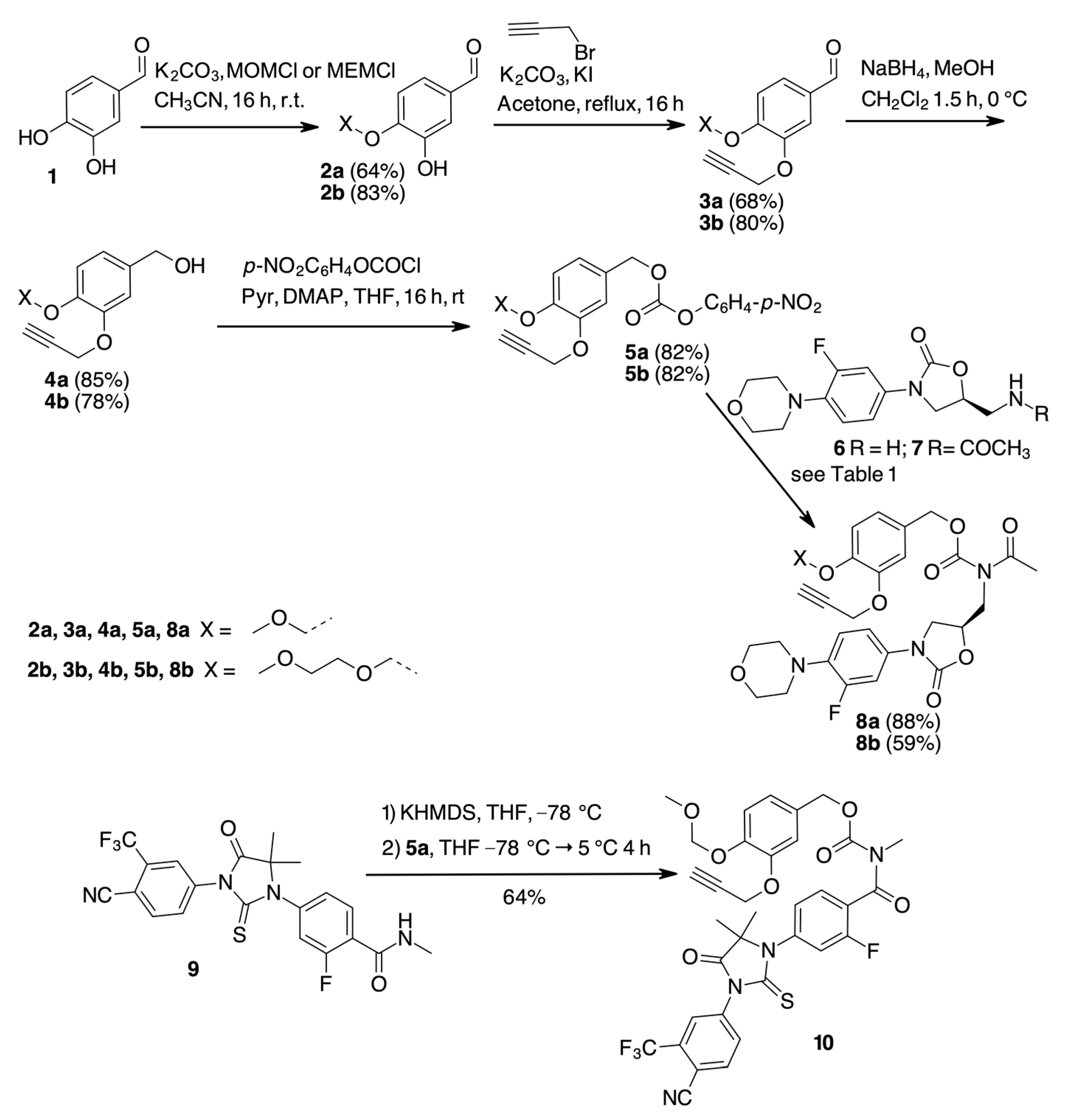
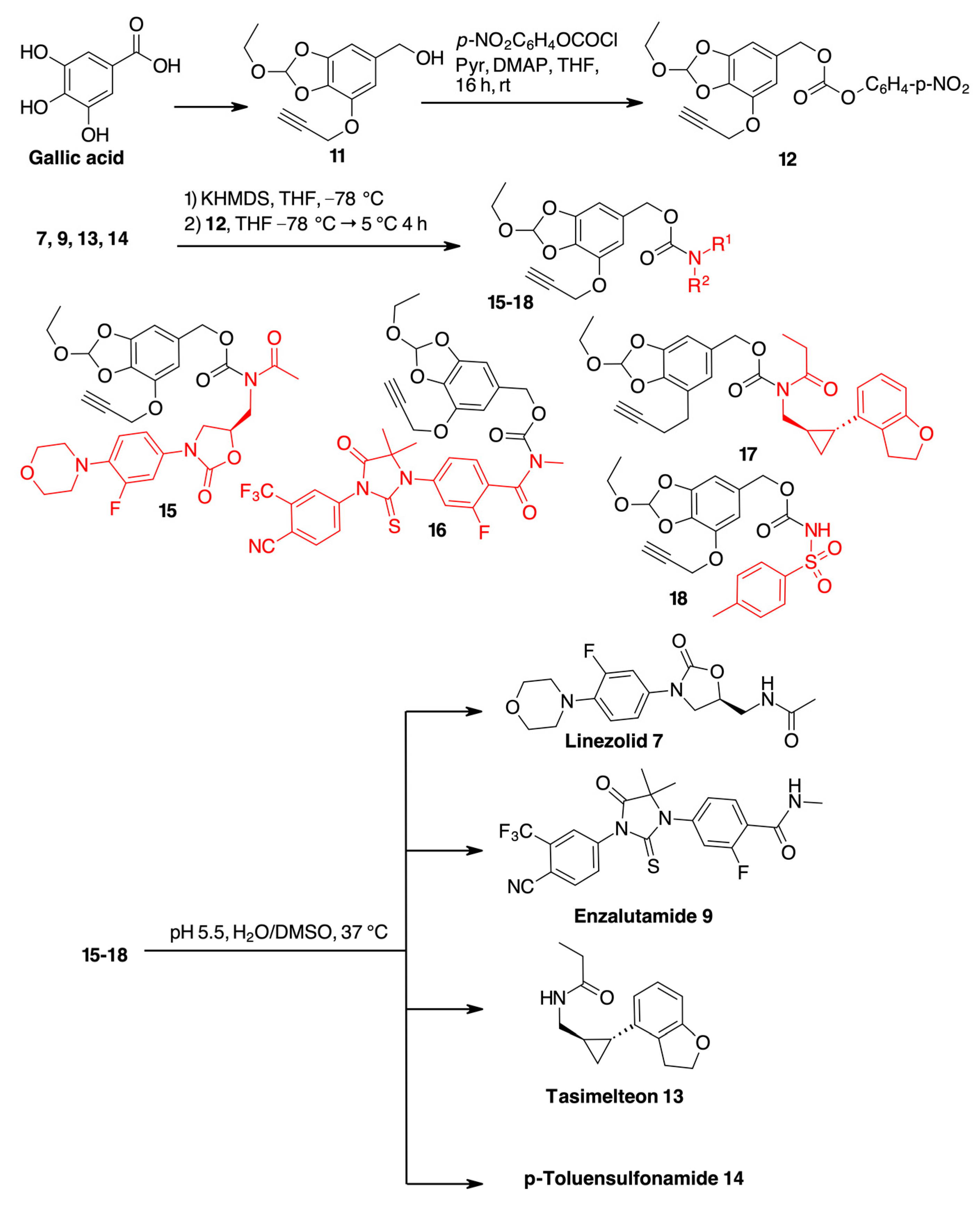
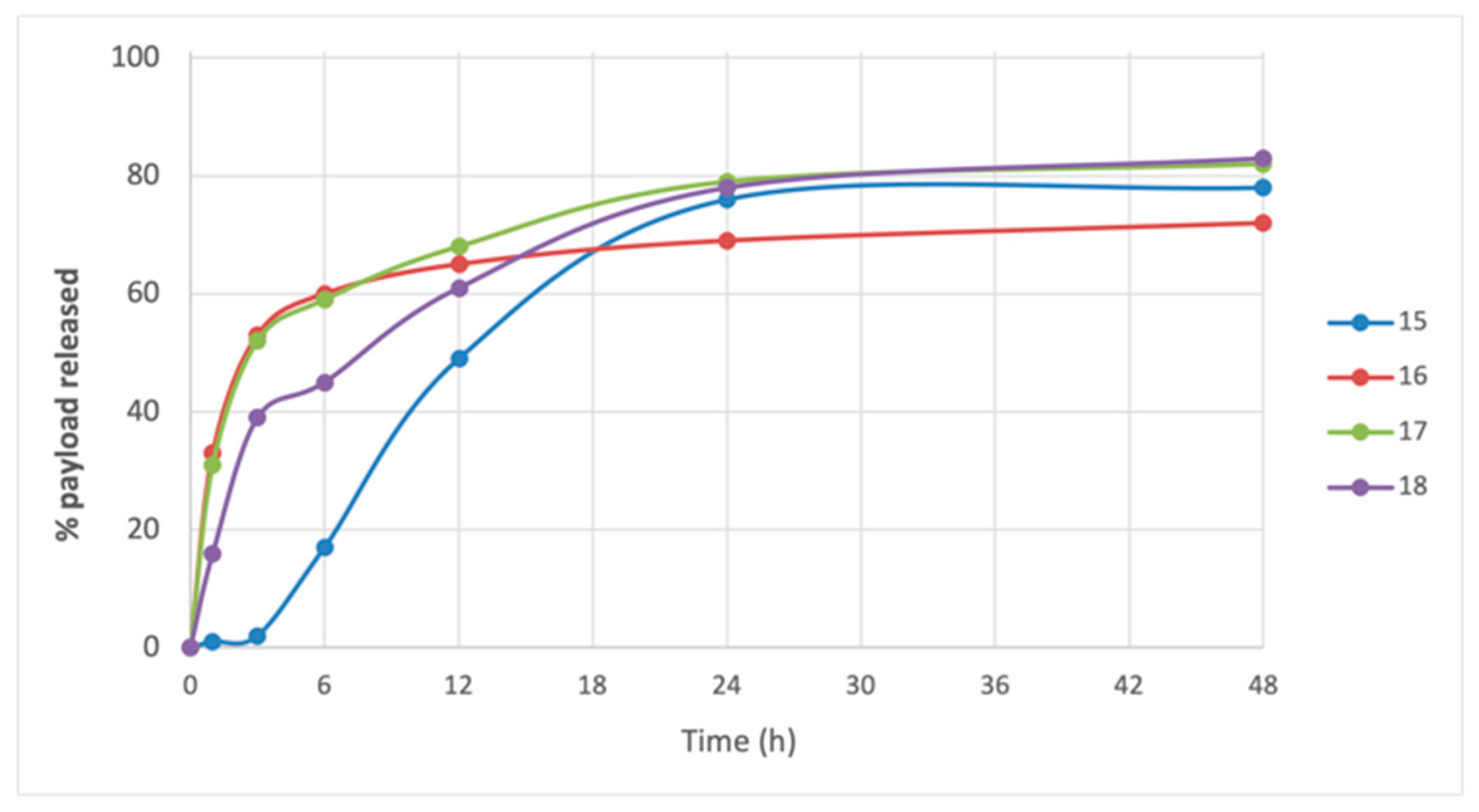
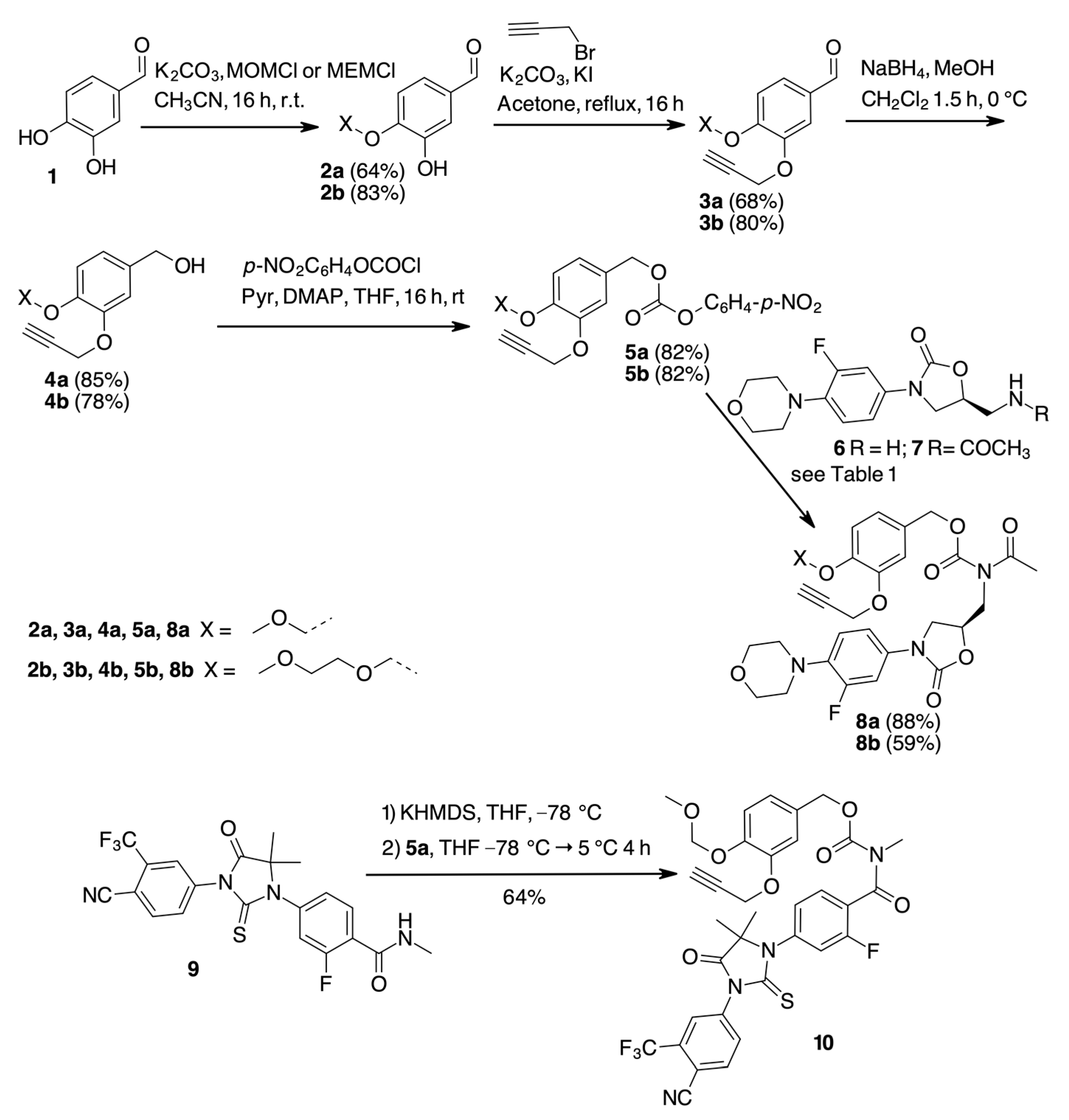
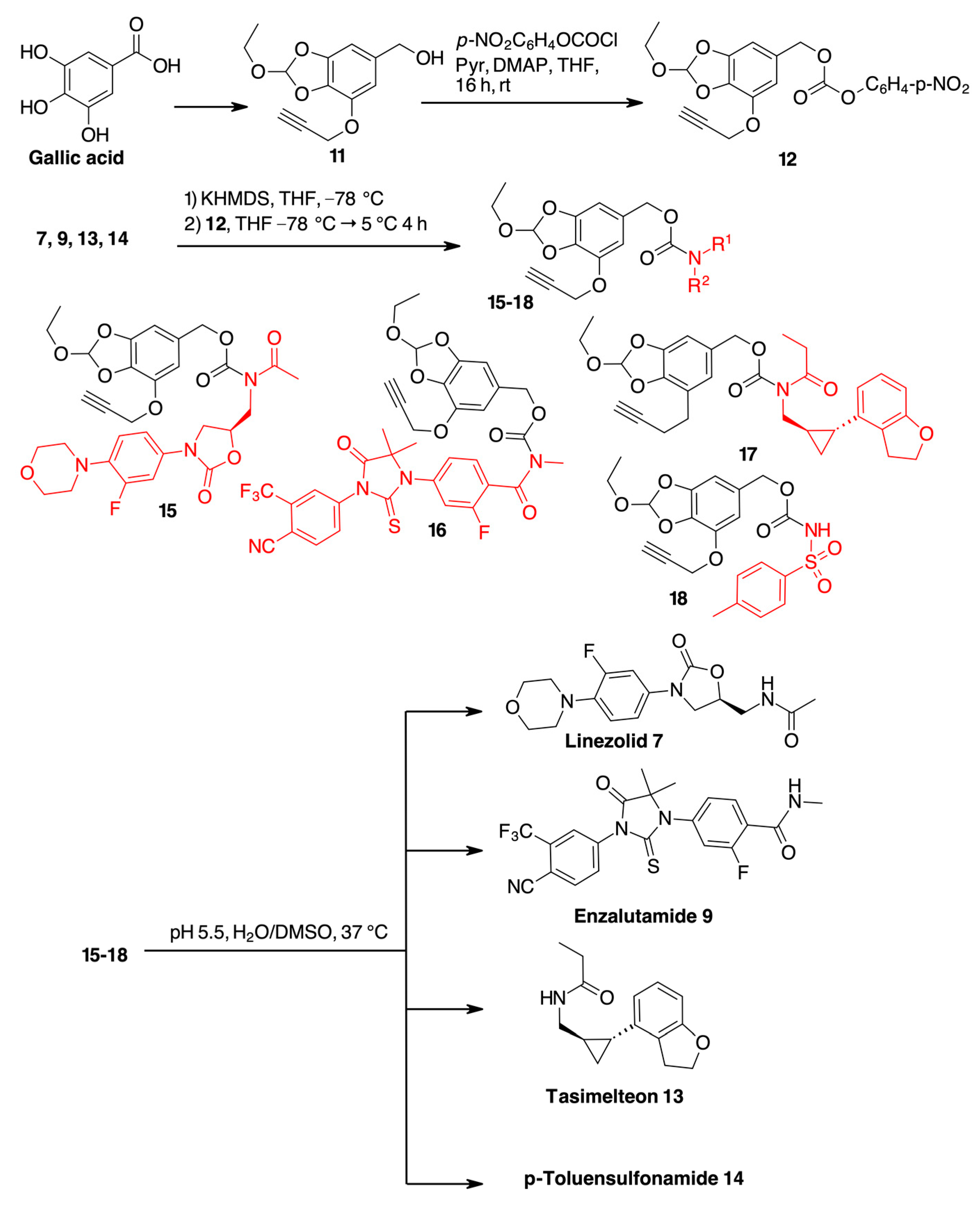
2. Results
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biju, V. Chemical Modifications and Bioconjugate Reactions of Nanomaterials for Sensing, Imaging, Drug Delivery and Therapy. Chem. Soc. Rev. 2014, 43, 744–764. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Rigol, S. The Role of Organic Synthesis in the Emergence and Development of Antibody–Drug Conjugates as Targeted Cancer Therapies. Angew. Chem. Int. Ed. 2019, 58, 11206–11241. [Google Scholar] [CrossRef] [PubMed]
- Sheyi, R.; de la Torre, B.G.; Albericio, F. Linkers: An Assurance for Controlled Delivery of Antibody-Drug Conjugate. Pharmaceutics 2022, 14, 396. [Google Scholar] [CrossRef]
- Liu, D.; Yang, F.; Xiong, F.; Gu, N. The Smart Drug Delivery System and Its Clinical Potential. Theranostics 2016, 6, 1306–1323. [Google Scholar] [CrossRef] [PubMed]
- Seidi, F.; Jenjob, R.; Crespy, D. Designing Smart Polymer Conjugates for Controlled Release of Payloads. Chem. Rev. 2018, 118, 3965–4036. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Pan, Z.; Choudhury, M.R.; Yuan, Z.; Anifowose, A.; Yu, B.; Wang, W.; Wang, B. Making Smart Drugs Smarter: The Importance of Linker Chemistry in Targeted Drug Delivery. Med. Res. Rev. 2020, 40, 2682–2713. [Google Scholar] [CrossRef] [PubMed]
- Devnarain, N.; Osman, N.; Fasiku, V.O.; Makhathini, S.; Salih, M.; Ibrahim, U.H.; Govender, T. Intrinsic Stimuli-Responsive Nanocarriers for Smart Drug Delivery of Antibacterial Agents—An in-Depth Review of the Last Two Decades. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2021, 13, e1664. [Google Scholar] [CrossRef] [PubMed]
- Alouane, A.; Labruère, R.; Le Saux, T.; Schmidt, F.; Jullien, L. Self-Immolative Spacers: Kinetic Aspects, Structure-Property Relationships, and Applications. Angew. Chem. Int. Ed. 2015, 54, 7492–7509. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable Linkers in Antibody-Drug Conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Dugal-Tessier, J.; Jain, N. Chapter 4: Non-Cleavable Linkers: Permanently Linked, for Better or for Worse. RSC Drug Discov. Ser. 2022, 2022, 136–172. [Google Scholar] [CrossRef]
- Xue, Y.; Bai, H.; Peng, B.; Fang, B.; Baell, J.; Li, L.; Huang, W.; Voelcker, N.H. Stimulus-Cleavable Chemistry in the Field of Controlled Drug Delivery. Chem. Soc. Rev. 2021, 50, 4872–4931. [Google Scholar] [CrossRef] [PubMed]
- Pillow, T. Ortho-Quinone Drugs Go Pro. Nature Chem. 2022, 14, 719–720. [Google Scholar] [CrossRef] [PubMed]
- Amides|DrugBank Online. Available online: https://go.drugbank.com/categories/DBCAT000494 (accessed on 15 July 2022).
- Bourbon, P.; Peng, Q.; Ferraudi, G.; Stauffacher, C.; Wiest, O.; Helquist, P. Development of Carbamate-Tethered Coumarins as Phototriggers for Caged Nicotinamide. Bioorg. Med. Chem. Lett. 2013, 23, 6321–6324. [Google Scholar] [CrossRef] [PubMed]
- Kahns, A.H.; Bundgaard, H. N-Acyl Derivatives as Prodrug Forms for Amides: Chemical Stability and Enzymatic Hydrolysis of Various N-Acyl and N-Alkoxycarbonyl Amide Derivatives. Int. J. Pharm. 1991, 71, 31–43. [Google Scholar] [CrossRef]
- Rannoux, C.; Roussi, F.; Martin, M.T.; Guéritte, F. Elaboration of Vinblastine Hybrids Using a Reactive in Situ Generated N-Carboxyanhydride. Org. Biomol. Chem. 2011, 9, 4873–4881. [Google Scholar] [CrossRef] [PubMed]
- Salahi, F.; Purohit, V.; Ferraudi, G.; Stauffacher, C.; Wiest, O.; Helquist, P. PHP-Tethered N-Acyl Carbamate: A Photocage for Nicotinamide. Org. Lett. 2018, 20, 2547–2550. [Google Scholar] [CrossRef]
- Thulam, V.K.; Kotte, S.C.B.; Sanjay Kumar, H.; Murali, P.M.; Mukkanti, K.; Mainker, P.S. A Novel and Efficient Method for N-Acylation of Sulfonamides and Carbamates: Their Biological Evaluation towards Anti Malassezia Activity. J. Pharm. Res. 2013, 7, 195–199. [Google Scholar] [CrossRef]
- Cui, S.; Qiao, J.; Xiong, M.P. Antibacterial and Biofilm-Eradicating Activities of PH-Responsive Vesicles against Pseudomonas Aeruginosa. Mol. Pharm. 2022, 19, 2406–2417. [Google Scholar] [CrossRef]
- Lunn, A.M.; Unnikrishnan, M.; Perrier, S. Dual PH-Responsive Macrophage-Targeted Isoniazid Glycoparticles for Intracellular Tuberculosis Therapy. Biomacromolecules 2021, 22, 3756–3768. [Google Scholar] [CrossRef]
- Gontsarik, M.; ben Mansour, A.; Hong, L.; Guizar-Sicairos, M.; Salentinig, S. PH-Responsive Aminolipid Nanocarriers for Antimicrobial Peptide Delivery. J. Colloid. Interface Sci. 2021, 603, 398–407. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Q.; Li, J.; Shang, L.; Li, J.; Chou, S.; Lyu, Y.; Shan, A. PH-Responsive Antimicrobial Peptide with Selective Killing Activity for Bacterial Abscess Therapy. J. Med. Chem. 2022, 65, 5355–5373. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.T.; Choi, S.K. Mechanisms of Drug Release in Nanotherapeutic Delivery Systems. Chem. Rev. 2015, 115, 3388–3432. [Google Scholar] [CrossRef] [PubMed]
- Sagi, A.; Weinstain, R.; Karton, N.; Shabat, D. Self-Immolative Polymers. J. Am. Chem. Soc. 2008, 130, 5434–5435. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Brindisi, M. Organic Carbamates in Drug Design and Medicinal Chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef] [Green Version]
- Abet, V.; Filace, F.; Recio, J.; Alvarez-Builla, J.; Burgos, C. Prodrug Approach: An Overview of Recent Cases. Eur. J. Med. Chem. 2017, 127, 810–827. [Google Scholar] [CrossRef]
- Hashemian, S.M.R.; Farhadi, T.; Ganjparvar, M. Linezolid: A Review of Its Properties, Function, and Use in Critical Care. Drug Des. Devel. Ther. 2018, 12, 1759–1767. [Google Scholar] [CrossRef]
- Davis, I.D.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Chi, K.N.; Chowdhury, S.; Coskinas, X.; Frydenberg, M.; Hague, W.E.; Horvath, L.G.; et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 121–131. [Google Scholar] [CrossRef]
- Catalani, E.; Buonanno, F.; Lupidi, G.; Bongiorni, S.; Belardi, R.; Zecchini, S.; Giovarelli, M.; Coazzoli, M.; de Palma, C.; Perrotta, C.; et al. The Natural Compound Climacostol as a Prodrug Strategy Based on PH Activation for Efficient Delivery of Cytotoxic Small Agents. Front. Chem. 2019, 7, 463. [Google Scholar] [CrossRef]
- McMahon, H.T.; Boucrot, E. Molecular Mechanism and Physiological Functions of Clathrin-Mediated Endocytosis. Nat. Rev. Mol. Cell. Biol. 2011, 12, 517–533. [Google Scholar] [CrossRef]
- Migliorini, F.; Cini, E.; Dreassi, E.; Finetti, F.; Ievoli, G.; Macrì, G.; Petricci, E.; Rango, E.; Trabalzini, L.; Taddei, M. A PH-Responsive Crosslinker Platform for Antibody-Drug Conjugate (ADC) Targeting Delivery. Chem. Commun. 2022, 58, 10532–10535. [Google Scholar] [CrossRef]
- Nishimon, S.; Nishimon, M.; Nishino, S. Tasimelteon for Treating Non-24-h Sleep-Wake Rhythm Disorder. Expert Opin. Pharmacother. 2019, 20, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Olalla, A.; Würdemann, M.A.; Wanner, M.J.; Ingemann, S.; van Maarseveen, J.H.; Hiemstra, H. Organocatalytic Enantioselective Pictet-Spengler Approach to Biologically Relevant 1-Benzyl-1,2,3,4-Tetrahydroisoquinoline Alkaloids. J. Org. Chem. 2015, 80, 5125–5132. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Conditions | 8, Yield [a] |
|---|---|---|
| 1 | 5a, 7, DMAP (10%), Pyr (2 eq) DMF, rt, 24 h. | -- |
| 2 | 5a, 7, DMAP (2 eq), Pyr (1 mL), DMF, rt, 24 h. | 10% |
| 3 | 5a, 7, MTBD (2 eq), DMF/THF, rt, 24 h. | 15% |
| 4 | 5a, 7, NaH (2 eq), THF, 0 °C → rt, 24 h. | -- |
| 5 | 5a, 6, DIPEA, THF rt, 24 h then Ac2O, DMAP (10%), Et3N (4 eq), DCM, rt, 24 h. [b] | 21% |
| 6 | 7, KHMDS, THF, −78 °C 1 h, then 5a, THF, −78 °C → −5 °C, 4 h. | 88% |
| Compound | H2O [a], t1/2 [b] | pH 7.4, [a] t1/2 [b] | Plasma, [a] t1/2[b] |
|---|---|---|---|
| 15 | 99%, >48 h | 94%, >48 h | 95%, >24 h |
| 16 | 99%, >48 h | 95%, >48 h | 89%, >24 h |
| 17 | 99%, >48 h | 95%, >48 h | 89%, >24 h |
| 18 | 99%, >48 h | 92%, >48 h | 84%, >24 h |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrini, A.; Ievoli, G.; Migliorini, F.; Taddei, M.; Siciliano, S. A Self-Immolative Linker for the pH-Responsive Release of Amides. Molecules 2023, 28, 2445. https://doi.org/10.3390/molecules28062445
Petrini A, Ievoli G, Migliorini F, Taddei M, Siciliano S. A Self-Immolative Linker for the pH-Responsive Release of Amides. Molecules. 2023; 28(6):2445. https://doi.org/10.3390/molecules28062445
Chicago/Turabian StylePetrini, Agnese, Giovanni Ievoli, Francesca Migliorini, Maurizio Taddei, and Sofia Siciliano. 2023. "A Self-Immolative Linker for the pH-Responsive Release of Amides" Molecules 28, no. 6: 2445. https://doi.org/10.3390/molecules28062445
APA StylePetrini, A., Ievoli, G., Migliorini, F., Taddei, M., & Siciliano, S. (2023). A Self-Immolative Linker for the pH-Responsive Release of Amides. Molecules, 28(6), 2445. https://doi.org/10.3390/molecules28062445