


Molecular Structure, Spectral Analysis, Molecular Docking and Physicochemical Studies of 3-Bromo-2-hydroxypyridine Monomer and Dimer as Bromodomain Inhibitors

 ,
,  and
and
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
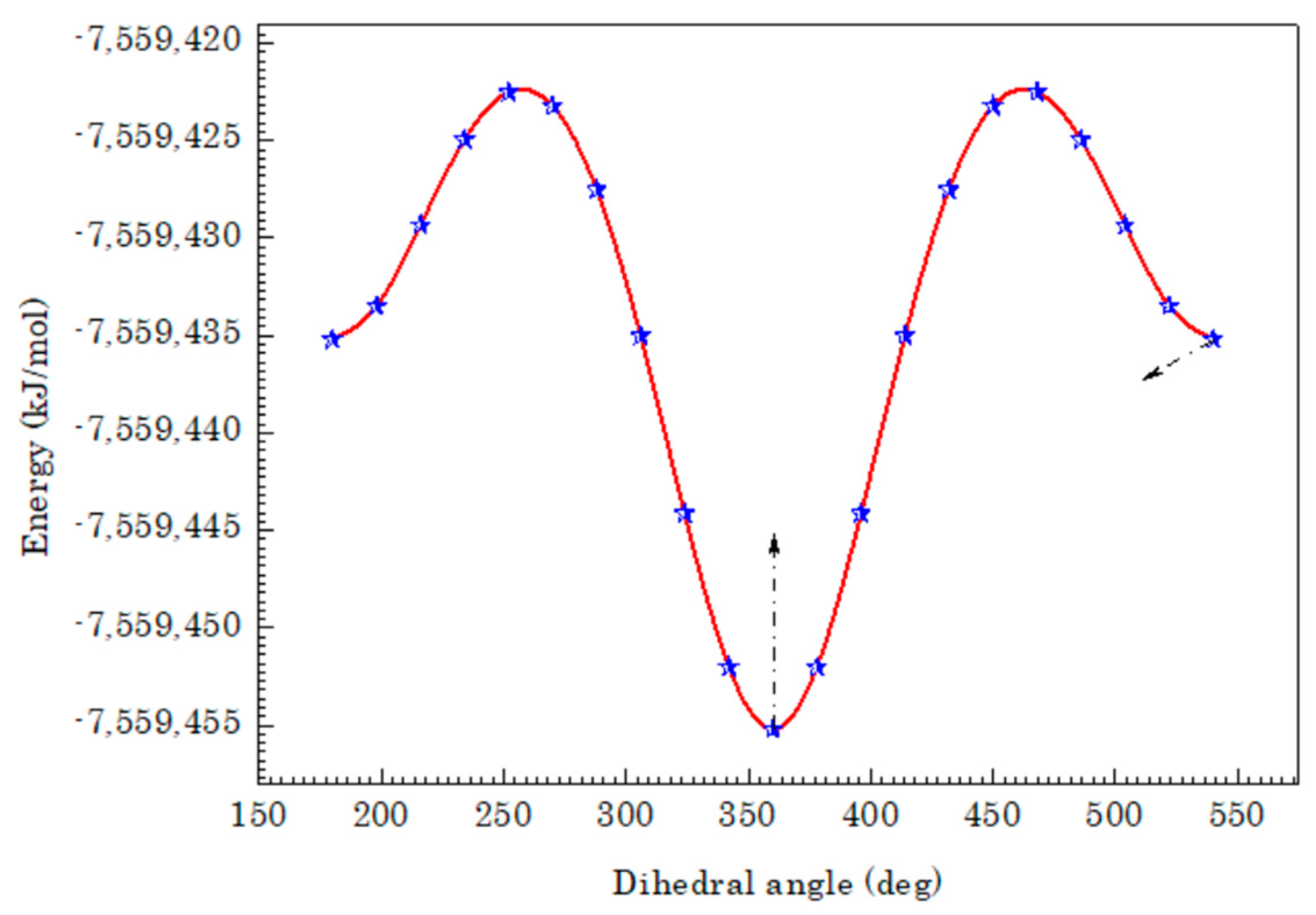
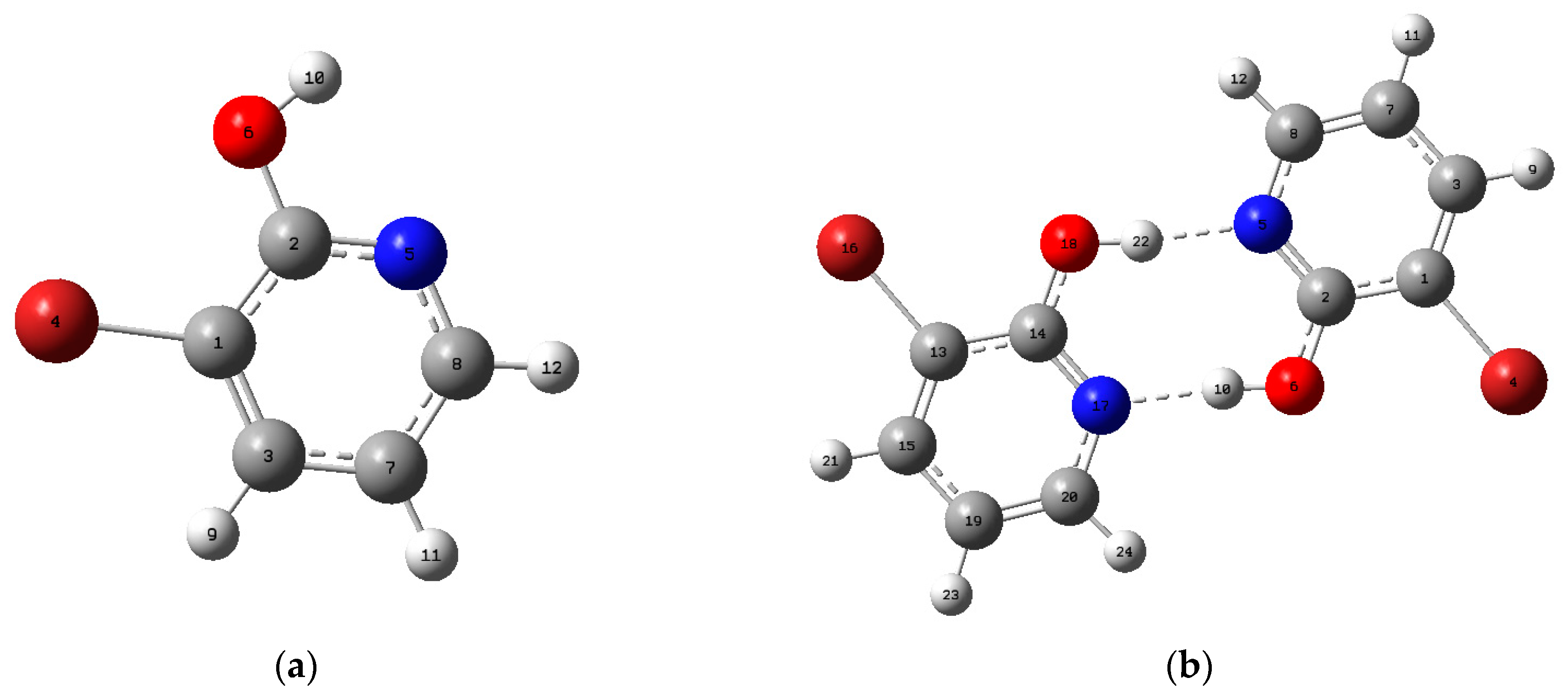
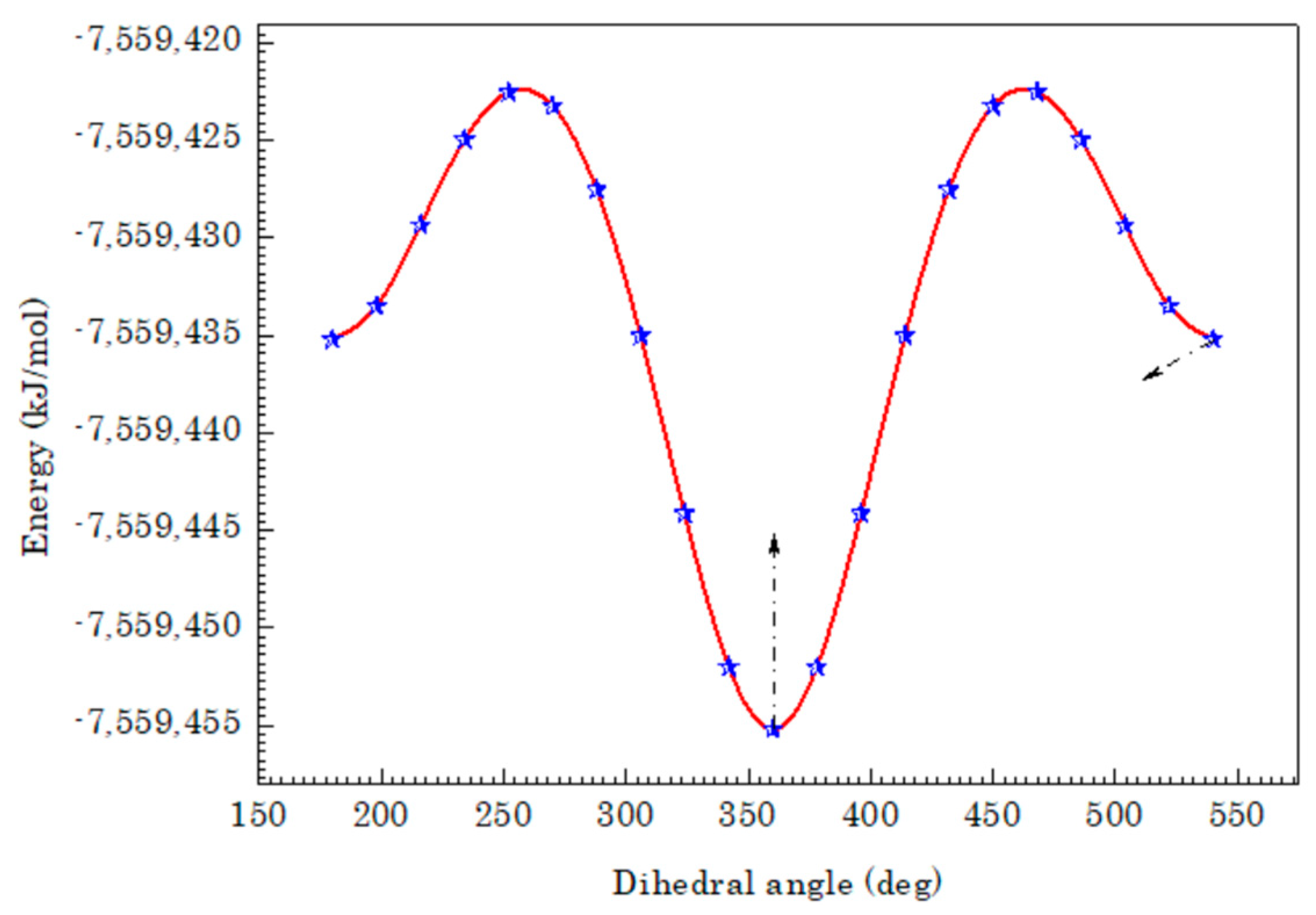
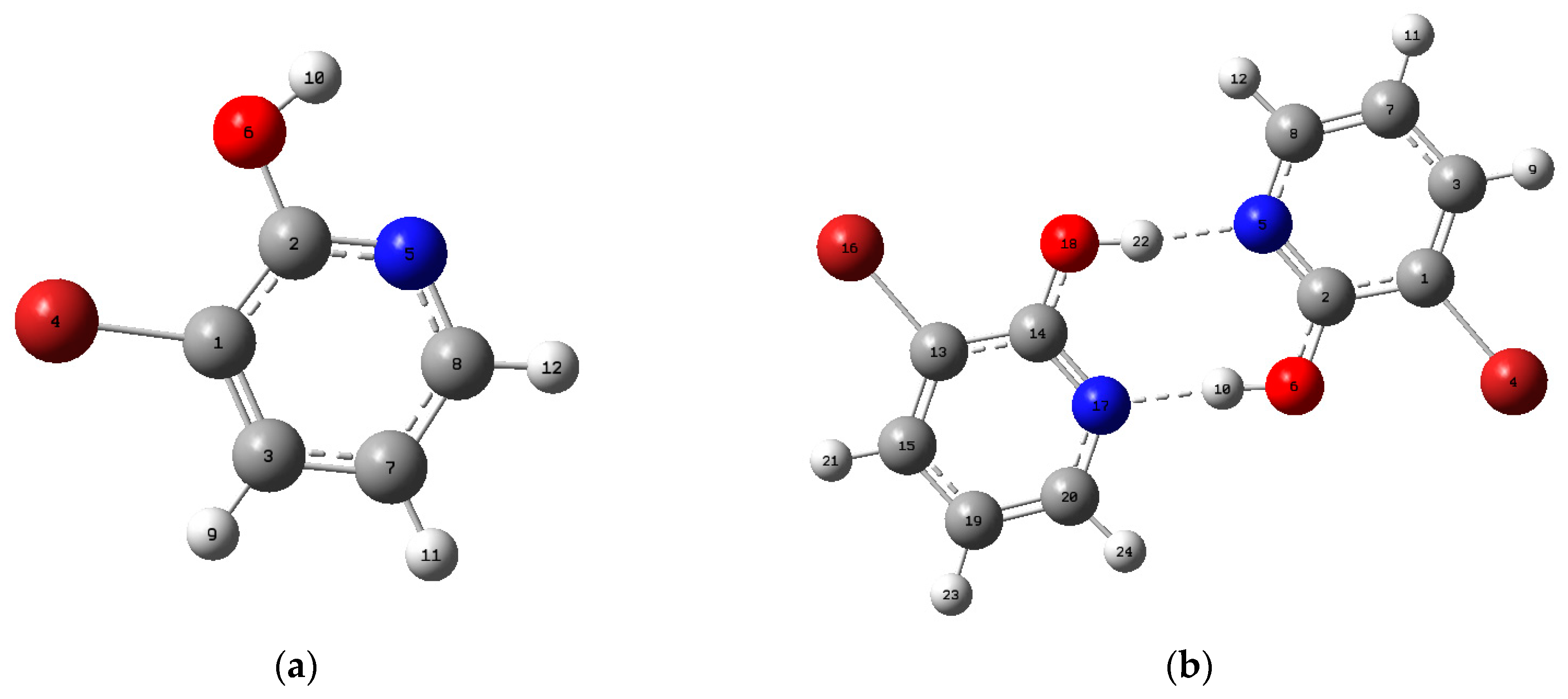
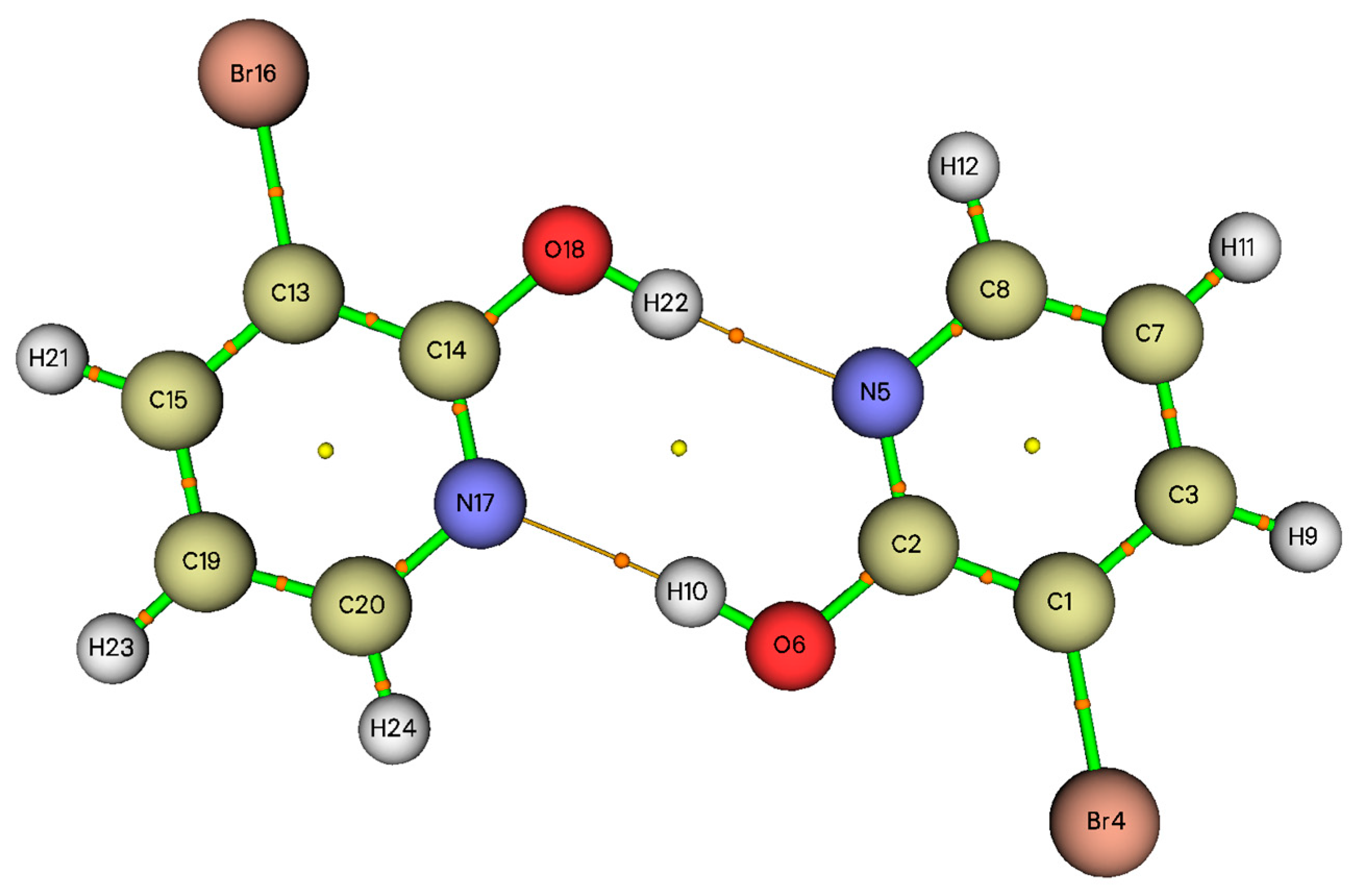
3.1. Geometric Structure
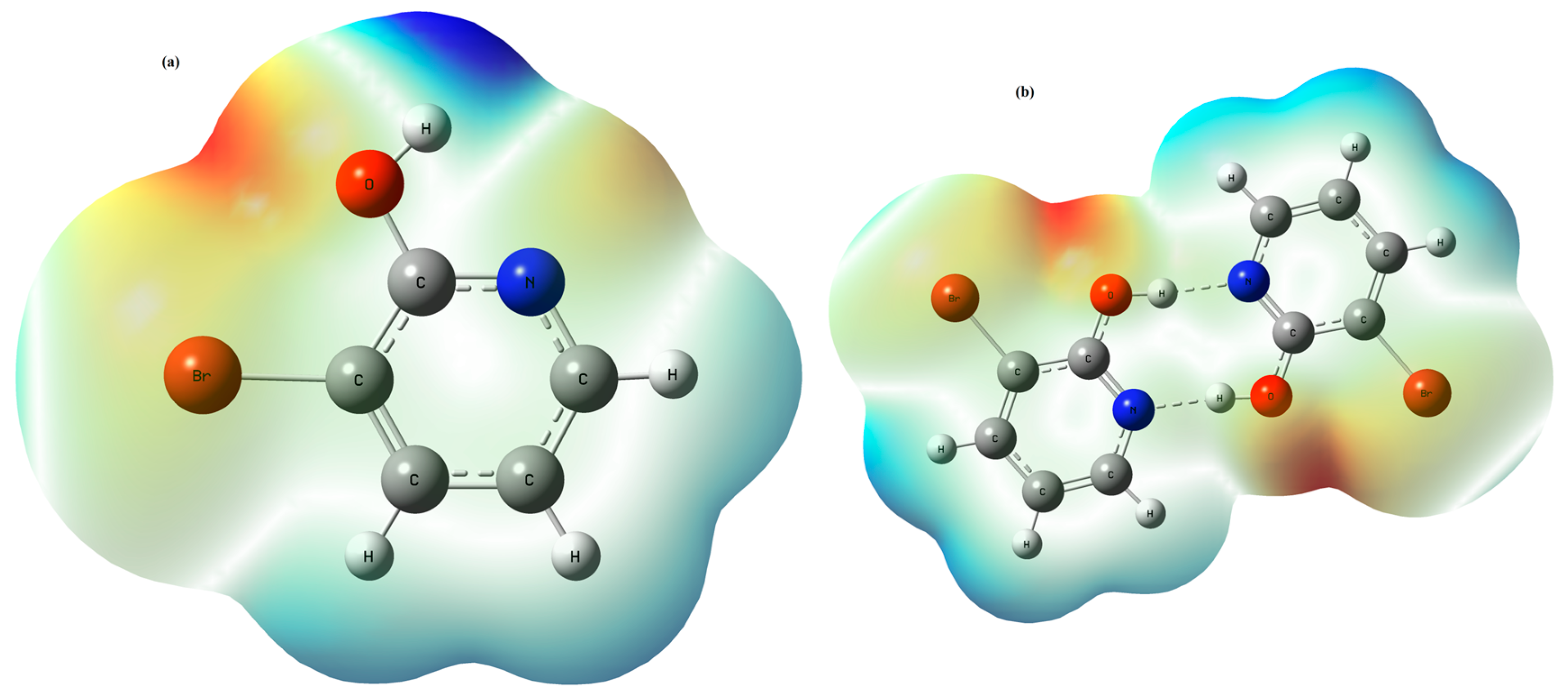
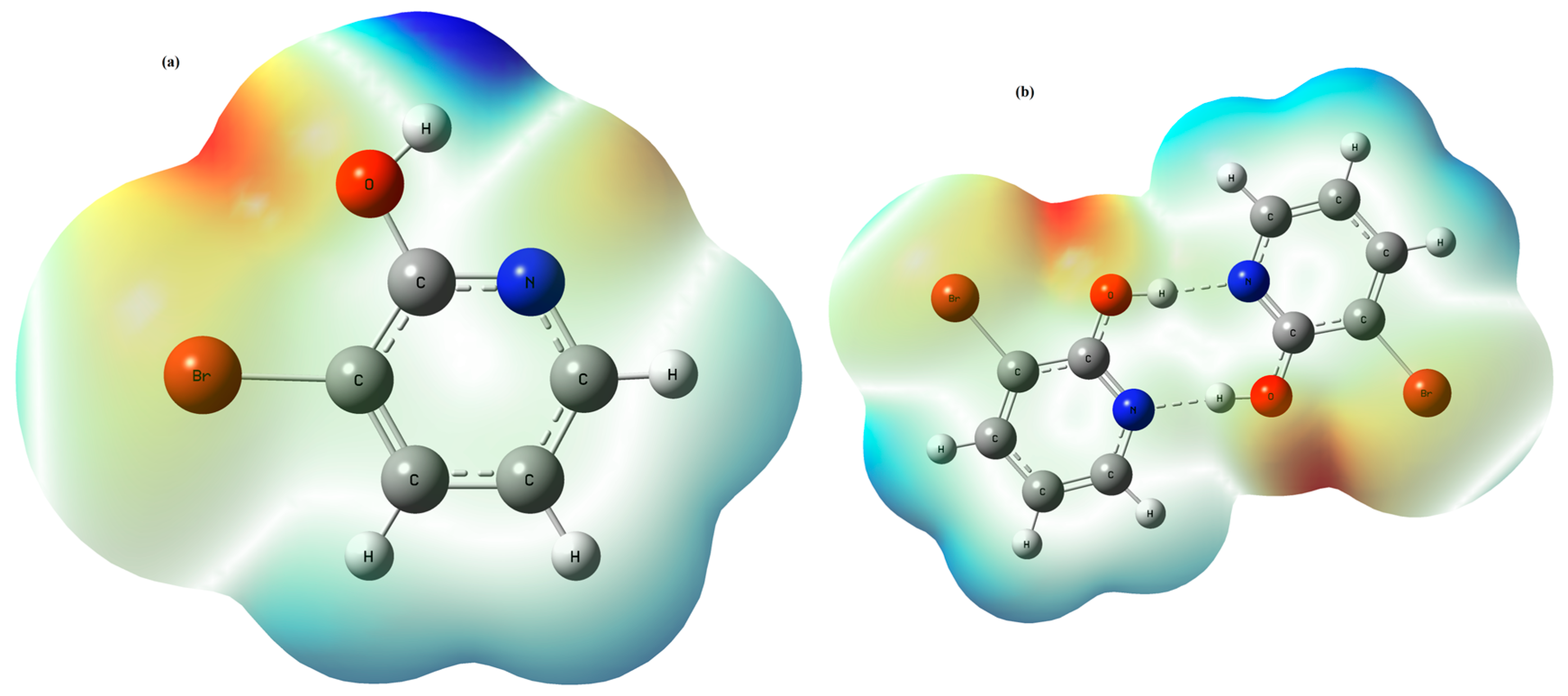
3.2. Molecular Electrostatic Potential
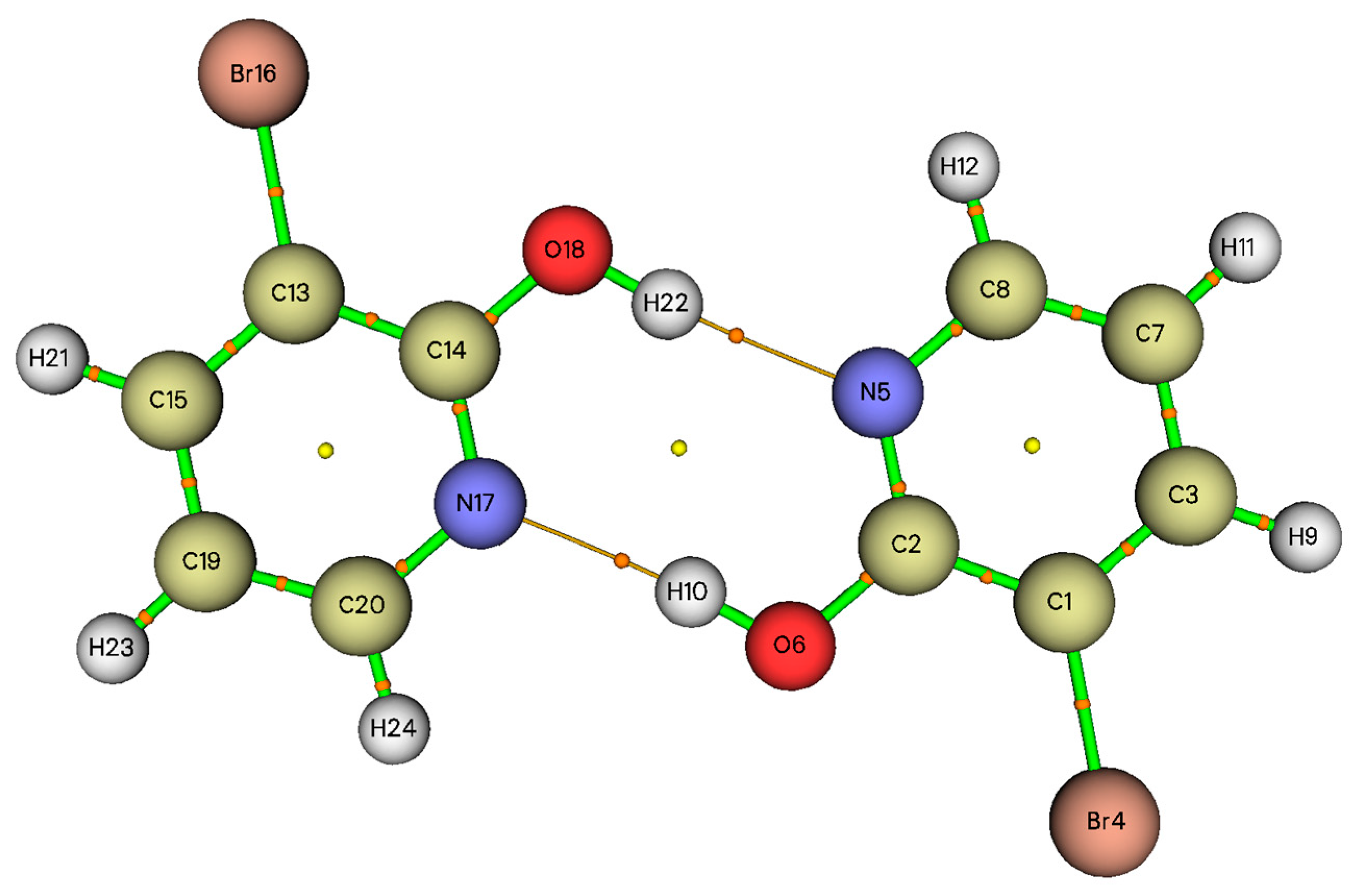
3.3. Topological Parameters
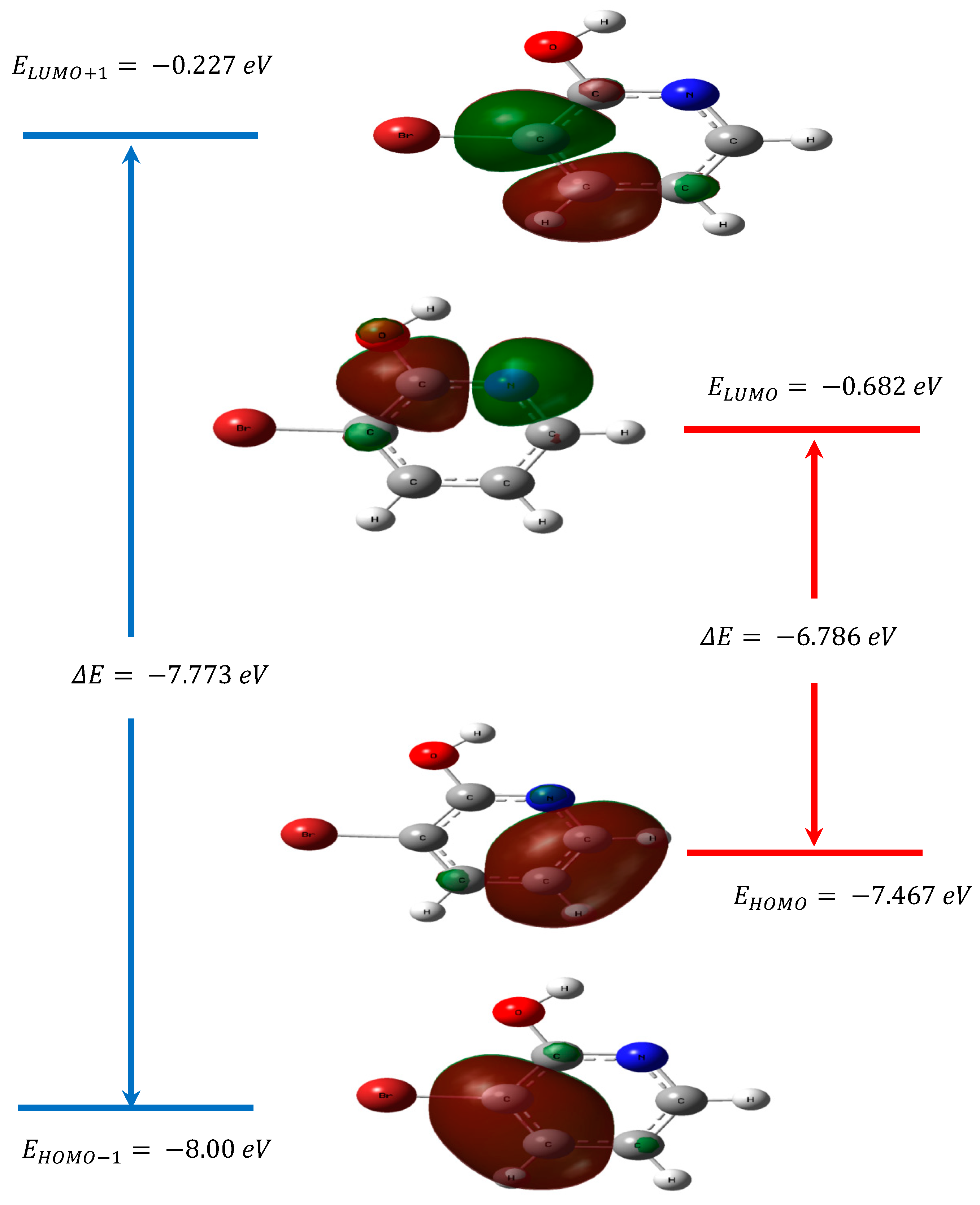
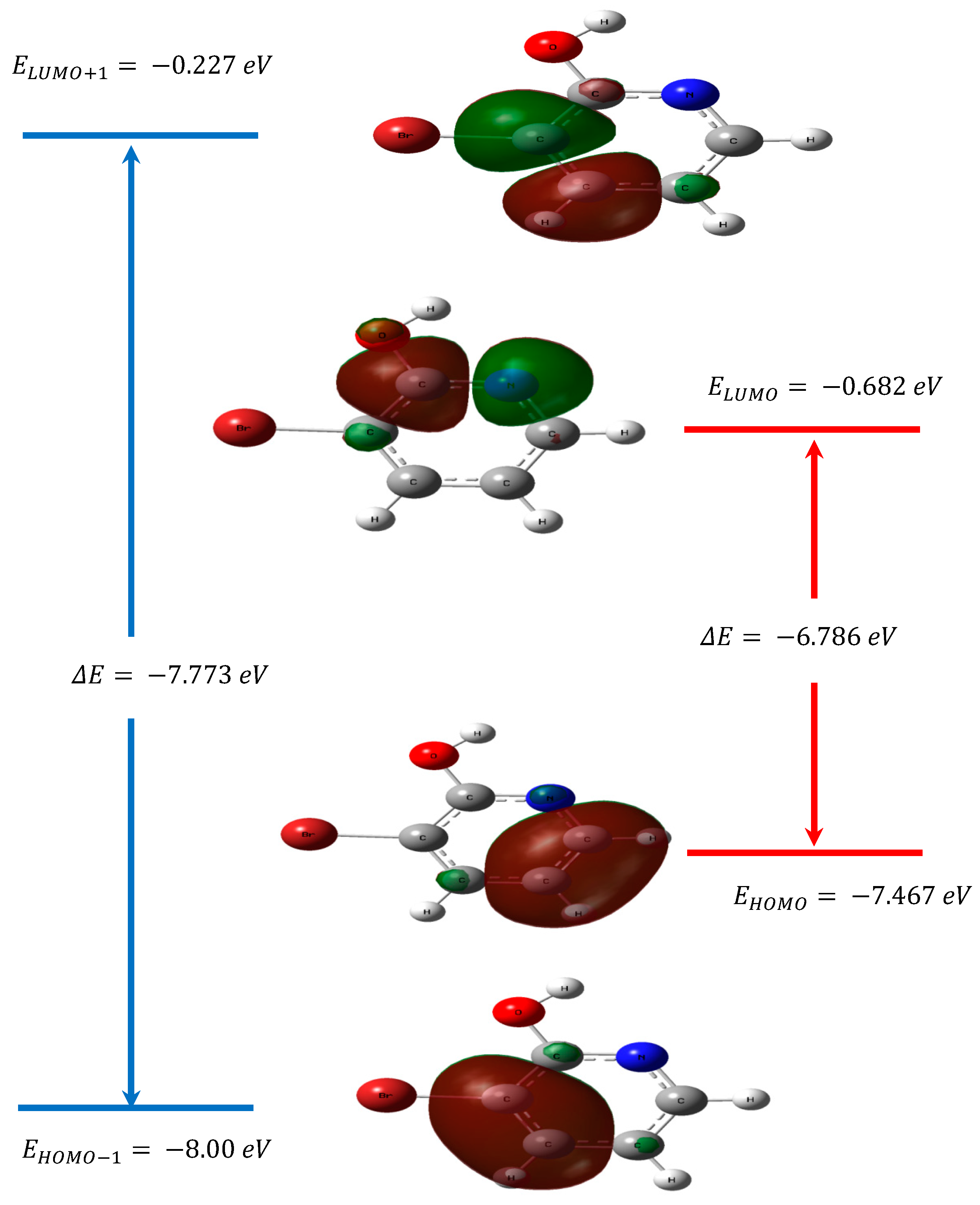
3.4. Frontier Molecular Orbitals
3.5. UV–Visible Spectral Analysis
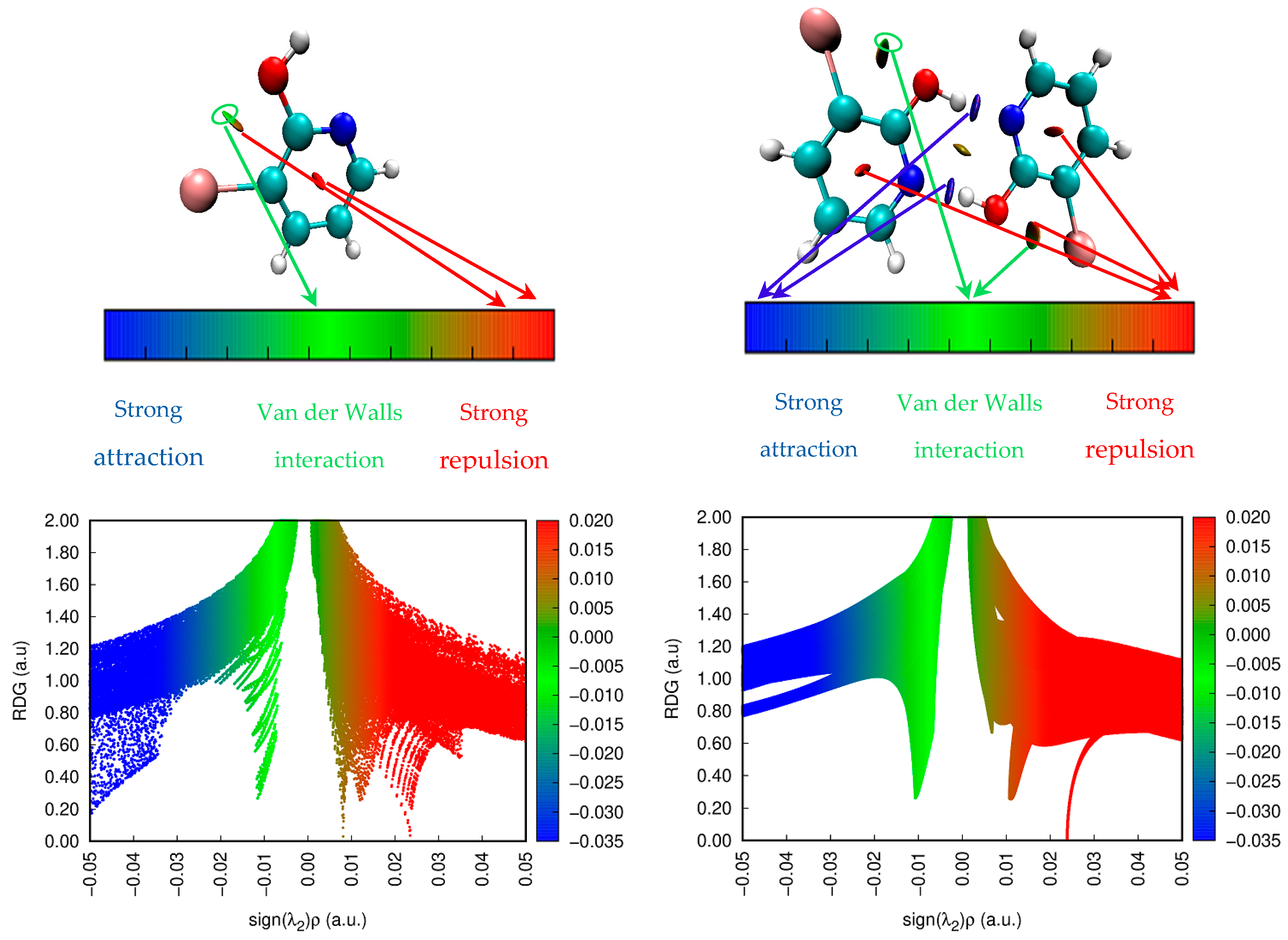
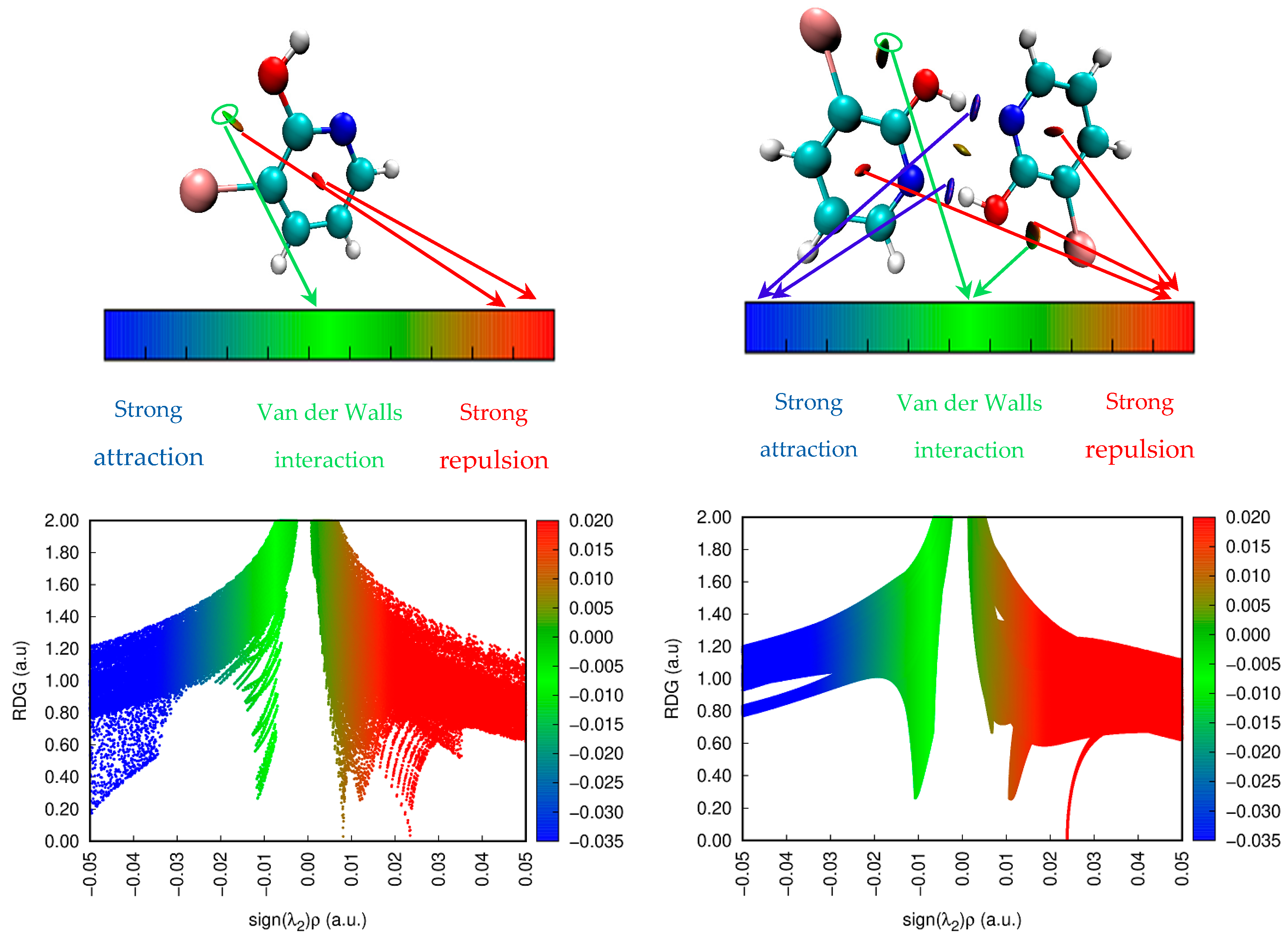
3.6. Reduced Density Gradient (RDG)
3.7. Thermodynamic Properties
3.8. ELF and LOL Analysis
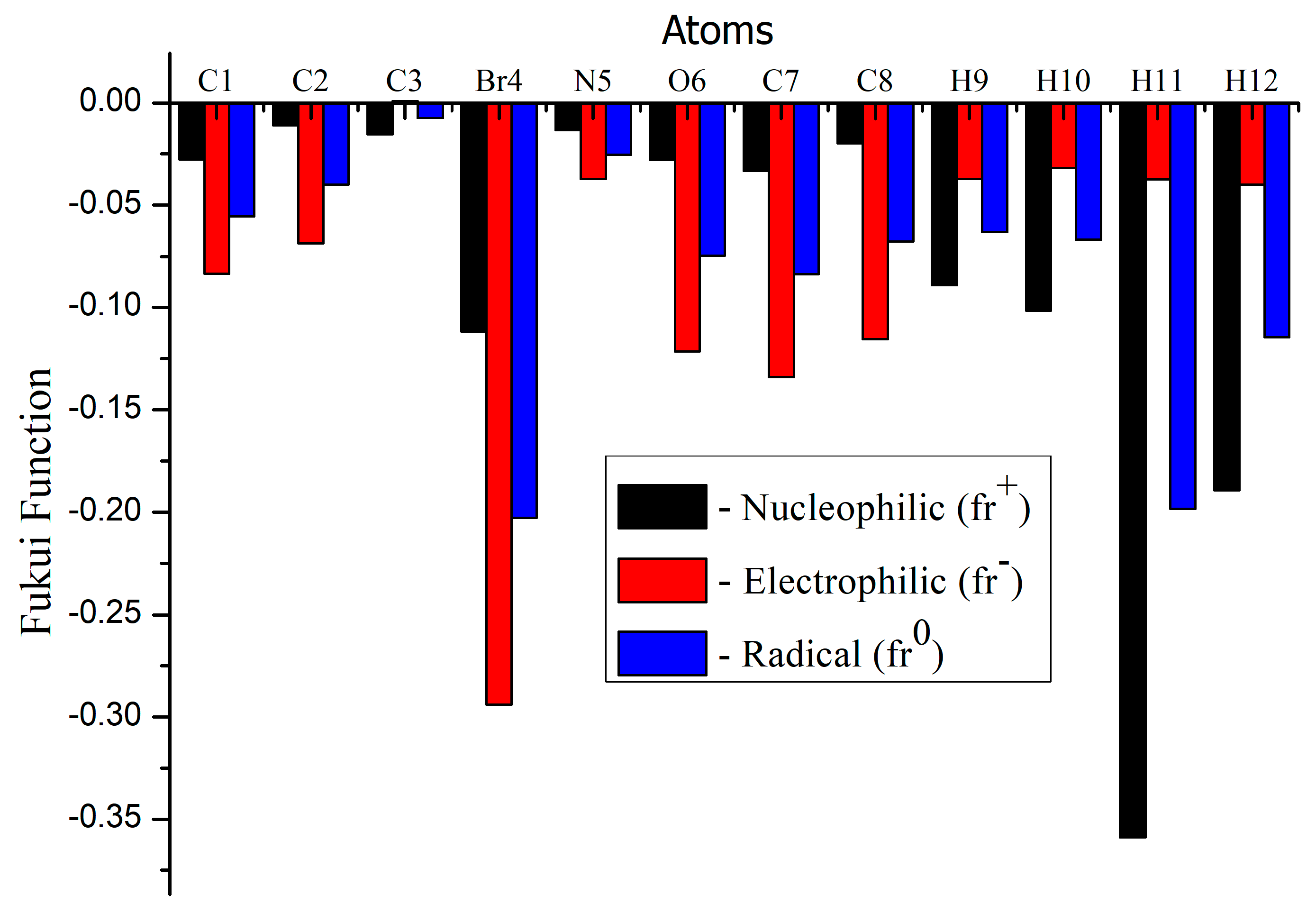
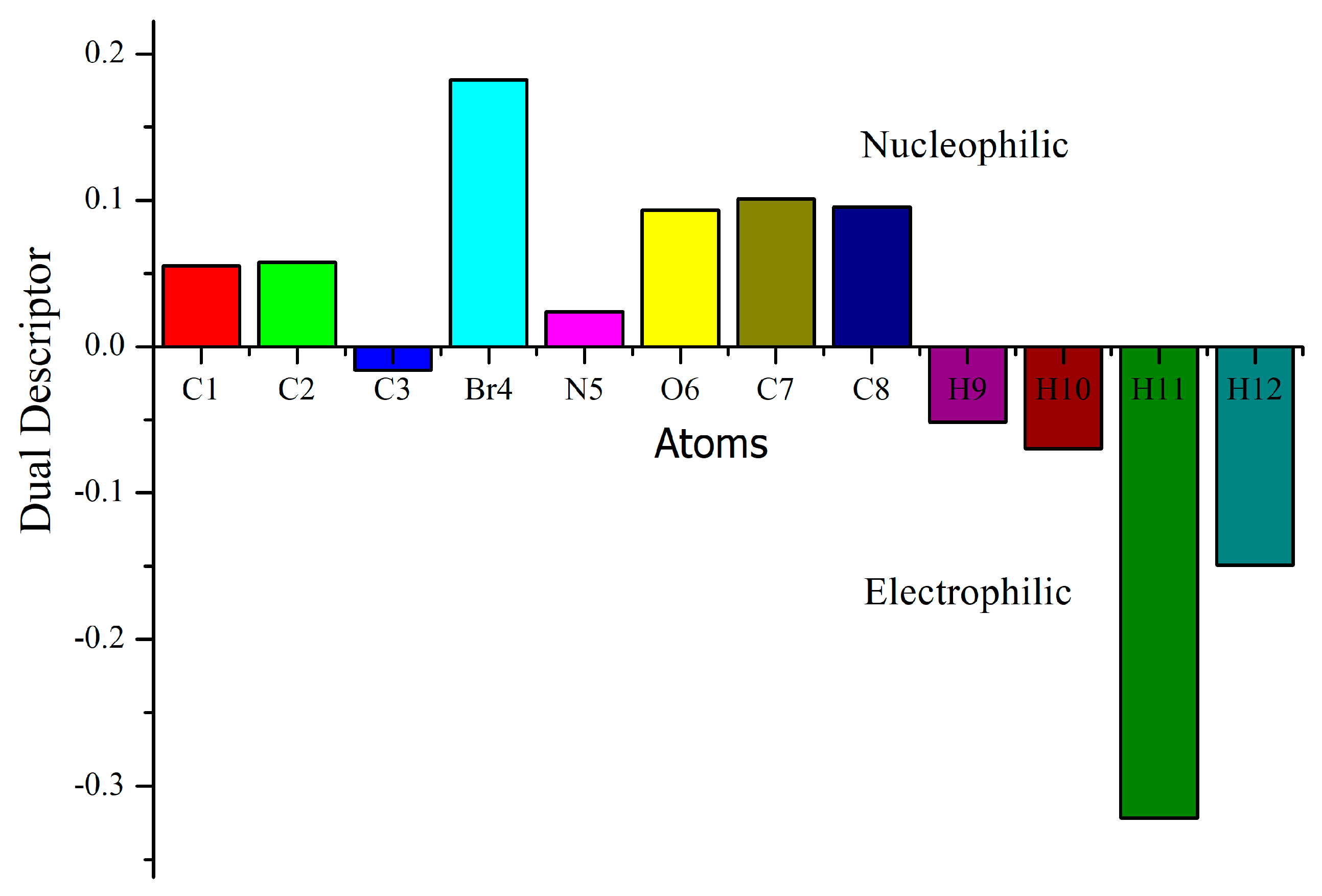
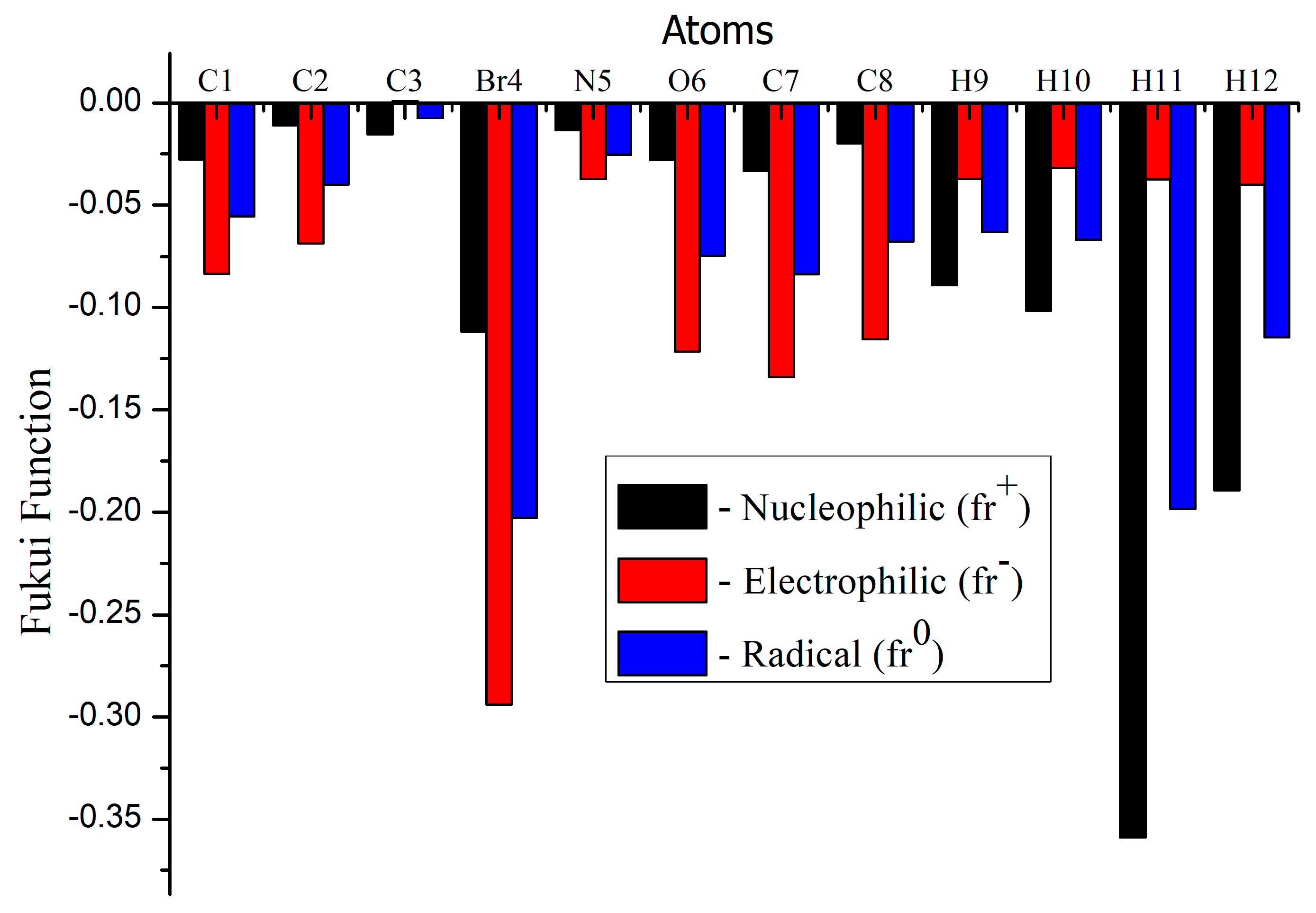
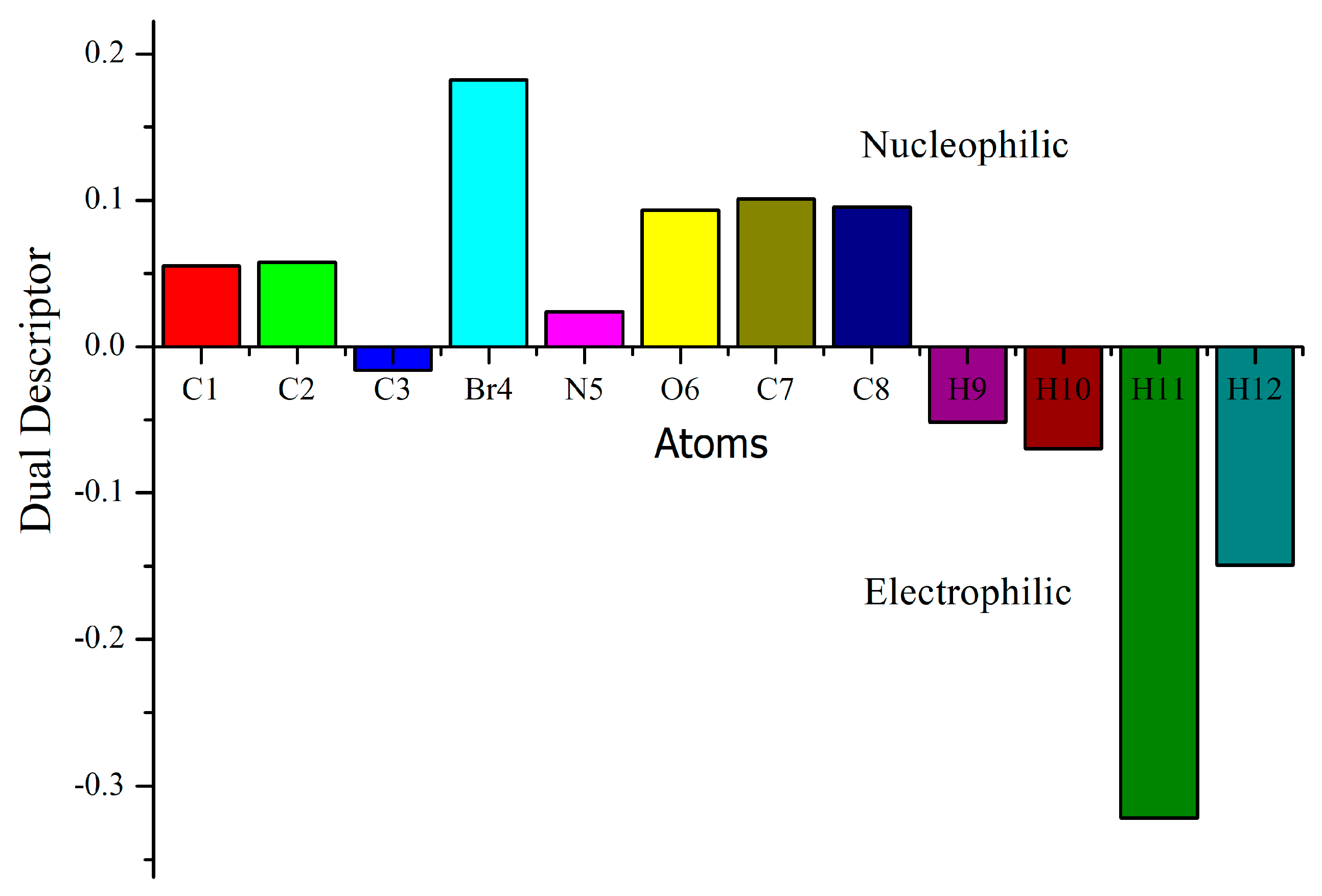
3.9. Molecular Reactivity Analysis
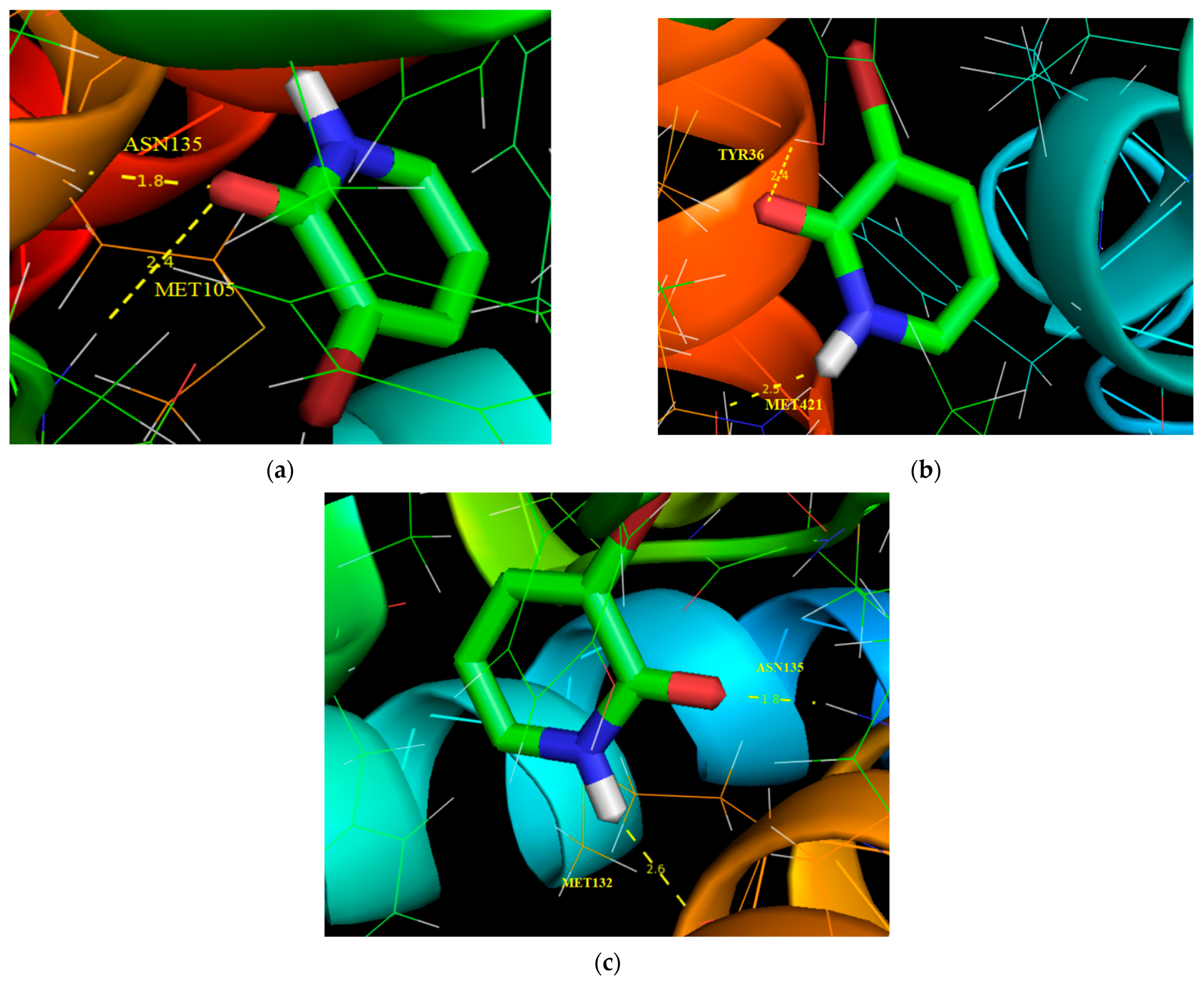
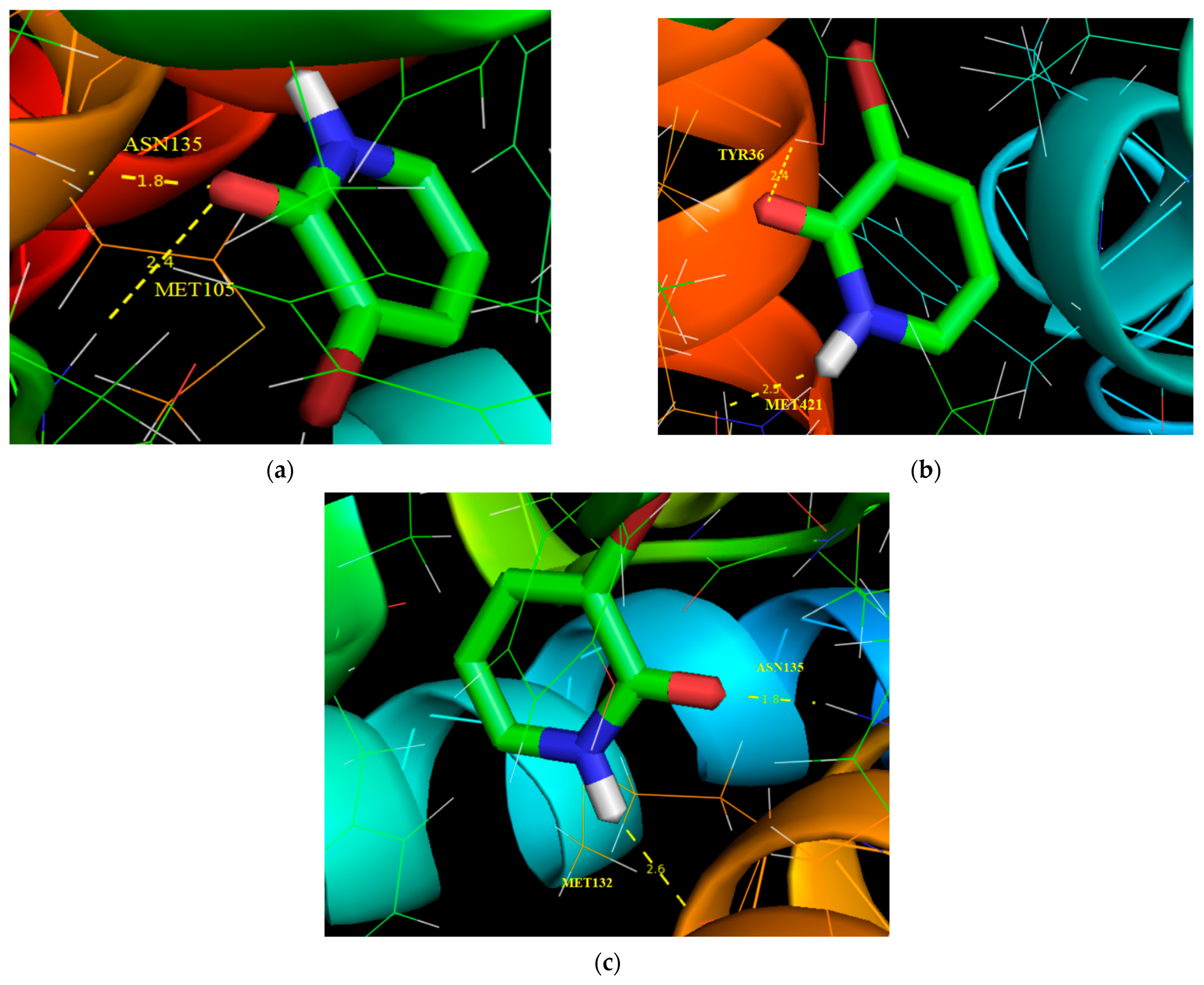
3.10. Molecular Docking Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Machura, B.; Wolff, M. A density functional study of oxorhenium(V) complexes incorporating quinoline or isoquinoline carboxylic acids: Structural, spectroscopic, and electronic properties. Struct. Chem. 2014, 25, 1607–1623. [Google Scholar] [CrossRef]
- Aziz, S.G.; Elroby, S.A.; Alyoubi, A.; Osman, O.I.; Hilal, R. Experimental and theoretical assignment of the vibrational spectra of triazoles and benzotriazoles. Identification of IR marker bands and electric response properties. J. Mol. Model. 2014, 20, 2078. [Google Scholar] [CrossRef] [PubMed]
- Michalska, J.; Boduszek, B.; Olszewski, T.K. New quinoline-2, -3, and 4-yl-(amino) methylphosphonates: Synthesis and study on the C-P bond cleavage in quinoline-2 and-4 derivatives under acidic conditions. Heteroat. Chem. 2011, 22, 617–624. [Google Scholar] [CrossRef]
- Scriven, E.F.V.; Murugan, R. Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley& Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable Potential of the α-Aminophosphonate / Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Kudzin, Z.H.; Gralak, D.K.; Andrijewski, G.; Drabowicz, J.; Luczak, J. Simultaneous analysis of biologically active aminoalkanephosphonic acids. J chromatogr. A 2003, 998, 183–199. [Google Scholar] [CrossRef]
- Powers, J.C.; Odake, S.; Oleksyszyn, J.; Hori, H.; Ueda, T.; Boduszek, B.; Kam, C. Proteases--structures, mechanism and inhibitors. Agents Actions Suppl. 1993, 42, 3–18. [Google Scholar]
- Kirk-Othmer. Encyclopedia of Chemical Technology, 4th ed.; Wiley: New York, NY, USA, 1997; Volume 20. [Google Scholar]
- Sortur, V.; Yenagi, J.; Tonannavar, J. Vibrational assignments of 2-iodopyridine. Spectrochim. Acta A 2008, 69, 604–611. [Google Scholar] [CrossRef]
- Mohan, S.; Murugan, R. Laser Raman and infrared spectra of 2,6-dichloropyridine. Ind. J. Pure Appl. Phys. 1992, 30, 283–286. [Google Scholar]
- Krishnakumar, V.; John Xavier, R. Density functional theory calculations and vibrational spectra of 3,5-dibromopyridine and 3,5-dichloro-2,4,6-trifluoropyridine. Spectrochim. Acta A 2005, 61, 253–260. [Google Scholar] [CrossRef]
- Krishnakumar, V.; Dheivamalar, S.; Xavier, R.J.; Balachandran, V. Analysis of vibrational spectra of 4-amino-2,6-dichloropyridine and 2-chloro-3,5-dinitropyridine based on density functional theory calculations. Spectrochim. Acta A 2006, 65, 147–154. [Google Scholar] [CrossRef]
- Hiremath, C.S.; Yenagi, J.; Tonannavar, J.; Sundius, T. Ab initio/DFT electronic structure calculations, spectroscopic studies and normal coordinate analysis of 2-chloro-5-bromopyridine. Spectrochim. Acta A 2010, 77, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Medhi, R.N.; Barman, R.; Medhi, K.C.; Jois, S.S. Ultraviolet absorption and vibrational spectra of 2-fluoro-5-bromopyridine. Spectrochim. Acta A 2000, 56, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Pervaiz, M.; Mishra, P.; Günther, S. Bromodomain Drug Discovery—the Past, the Present, and the Future. Chem. Rec. 2018, 18, 1808–1817. [Google Scholar] [CrossRef]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Future Sci. OA 2019, 5, FSO372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korth, H.G.; Martine, I.d.H.; Mulder, P. A DFT Study on Intramolecular Hydrogen Bonding in 2-Substituted Phenols: Conformations, Enthalpies, and Correlation with Solute Parameters. J. Phys. Chem. A 2002, 106, 8779–8789. [Google Scholar] [CrossRef]
- Chowdhry, P.K. Infrared Depletion Spectroscopy of the Hydrogen-Bonded Aniline−Diethylamine (C6H5-NH2···NHC4H10) Complex Produced in Supersonic Jet. J. Phys. Chem. A 2003, 107, 5692–5696. [Google Scholar] [CrossRef]
- Chiş, V. Molecular and vibrational structure of 2, 4-dinitrophenol: FT-IR, FT-Raman and quantum chemical calculations. Chem. Phys. 2004, 300, 1–11. [Google Scholar] [CrossRef]
- Asensio, A.; Kobko, N.; Dannenberg, J.J. Cooperative Hydrogen-Bonding in Adenine−Thymine and Guanine−Cytosine Base Pairs. Density Functional Theory and Møller−Plesset Molecular Orbital Study. J. Phys. Chem. A 2003, 107, 6441–6443. [Google Scholar] [CrossRef]
- Muthu, S.; Renuga, S. Vibrational and spectroscopic investigation on the structure of 5H-dibenzo[b,f]azipine-5-carboxamide. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 114, 1–10. [Google Scholar] [CrossRef]
- Issaoui, N.; Rekik, N.; Oujia, B.; Wójcik, M.J. Anharmonic Effects on Theoretical IR Line Shapes of Medium Strong H(D) Bonds. Int. J. Quant. Chem. 2009, 109, 483–499. [Google Scholar] [CrossRef]
- Issaoui, N.; Rekik, N.; Oujia, B.; Wójcik, M.J. Theoretical infrared line shapes of H-bonds within the strong anharmonic coupling theory and fermi resonances effects. Int. J. Quant. Chem. 2010, 110, 2583–2602. [Google Scholar] [CrossRef]
- Rekik, N.; Issaoui, N.; Ghalla, H.; Oujia, B.; Wójcik, M.J. IR spectral density of H-bonds. Both intrinsic anharmonicity of the fast mode and the H-bond bridge. Part I: Anharmonic coupling parameter and temperature effects. J. Mol. Struct. Theochem. 2007, 821, 9–21. [Google Scholar] [CrossRef]
- Dinçer, M.; Avci, D.; Şekerci, M.; Atalay, Y. Molecular structure and vibrational and chemical shift assignments of 5-(2-Hydroxyphenyl)-4-(p-tolyl)-2,4-dihydro-1,2,4-triazole-3-thione by DFT and ab initio HF calculations. J. Mol. Model. 2008, 14, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Minaev, B.F.; Baryshnikov, G.V.; Minaeva, V.A. Electronic structure and spectral properties of the triarylamine-dithienosilole dyes for efficient organic solar cells. Dye. Pigment. 2012, 92, 531–536. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 85–789. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Nielsen, A.B.; Holder, A.J. Gauss View Users Manual; Gaussian Inc.: Pittsburgh, PA, USA, 2000. [Google Scholar]
- Young, D.C. Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems (Electronic), 3rd ed.; John Wiley & Sons Inc.: New York, NY, USA, 2001. [Google Scholar]
- Frisch, H.P.; Hratchian, R.D.; Dennington, T.A., II; Keith, J.; Millam, A.B.; Nielsen, A.; Holder, J.; Hiscocks, J. Gauss View, Version 5.08; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Biegler-Köning, F.; Schönbohm, J.; Bayles, D. AIM2000—A program to Analyze and Visualize Atoms in Molecules. J. Comput. Chem. 2001, 22, 545–559. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wave function analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Jacobsen, H. Localized-orbital locator (LOL) profiles of chemical bonding. Can. J. Chem. 2008, 86, 695–702. [Google Scholar] [CrossRef]
- Yang, W.; Parr, R.G. Hardness, softness, and the fukui function in the electronic theory of metals and catalysis. Proc. Nat. Acad. Sci. USA 1985, 82, 6723–6726. [Google Scholar] [CrossRef] [Green Version]
- Spingh, G.P.; Goud, R.N.; Kumar, J.P.; Sundaresan, C.N.; Nageswara, R.G. 2-Bromo-3-hydroxy-6-methylpyridine. Acta Cryst. 2013, 69, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Hobza, P.; Zahradnik, R. Intermolecular Complexes; Elsevier: Amsterdam, The Netherlands, 1988. [Google Scholar]
- van Duijneveldt, F.B.; van Duijneveldt-van de Rijdt, J.G.C.M.; van Lenthe, J.H. State of the Art in the Counterpoise Theory. Chem. Rev. 1994, 94, 1873–1885. [Google Scholar] [CrossRef]
- Kazachenko, A.; Akman, F.; Medimagh, M.; Sagaama, A.; Issaoui, N.; Malyar, Y.N.; Vasilieva, N.; Borovkova, V.S. Theoretical and experimental study of guar gum sulfation. J. Mol. Model. 2021, 27, 5. [Google Scholar] [CrossRef]
- Medimagh, M.; Issaoui, N.; Gatfaoui, A.S.; Al-Dossary, O.; Kazachenko, A.; Marouani, H.; Wojcik, M.J. Molecular modeling and biological activity analysis of new organic-inorganic hybrid: 2-(3,4-dihydroxyphenyl) ethanaminium nitrate. J. King Saud Univ.-Sci. 2021, 33, 101616. [Google Scholar] [CrossRef]
- Jomaa, I.; Noureddine, O.; Gatfaoui, S.; Issaoui, N.; Roisnel, T.; Marouani, H. Experimental, computational, and in silico analysis of (C8H14N2)2[CdCl6] compound. J. Mol. Struct. 2020, 1213, 128186. [Google Scholar] [CrossRef]
- Kosar, B.; Albayrak, C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2011, 78, 160–167. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O hydrogen bonds on the basis of the charge density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Rozas, I.; Elguero, J.; Molins, R. About the evaluation of the local kinetic, potential and total energy densities in closed-shell interactions. Chem. Phys. Lett. 2001, 336, 457–461. [Google Scholar] [CrossRef]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in structural Chemistry and Biology; Oxford University Press Inc.: New York, NY, USA, 1999. [Google Scholar]
- Kazachenko, A.S.; Issaoui, N.; Medimagh, M.; Fetisova, O.Y.; Berezhnaya, Y.D.; Elsuf’ev, E.V.; Al-Dossary, O.M.; Wojcik, M.J.; Xiang, Z.; Bousiakou, L.G. Experimental and theoretical study of the sulfamic acid-urea deep eutectic solvent. J. Mol. Liq. 2022, 363, 119859. [Google Scholar] [CrossRef]
- Fleming, I. Frontier Orbitals and Organic Chemical Reactions; John Wiley and Sons: New York, NY, USA, 1976. [Google Scholar]
- Rauk, A. Orbital Interaction Theory of Organic Chemists, 2nd ed.; Wiley-Interscience: New York, NY, USA, 2001; p. 86. [Google Scholar]
- Fukui, K. Role of Frontier Orbitals in Chemical Reactions. Science 1982, 218, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Kazachenko, A.; Akman, F.; Medimagh, M.; Issaoui, N.; Vasilieva, N.; Malyar, Y.N.; Sudakova, I.G.; Karacharov, A.; Miroshnikova, A.; Al-Dossary, O.M. Sulfation of Diethylaminoethyl-Cellulose: QTAIM Topological Analysis and Experimental and DFT Studies of the Properties. ACS Omega 2021, 6, 22603–22615. [Google Scholar] [CrossRef]
- Gree, G. The use of quantum chemical methods in corrosion inhibitor studies. Corros. Sci. 2008, 50, 2981–2992. [Google Scholar] [CrossRef]
- Lewis, D.F.; Ioannides, C.; Parke, D.V. Interaction of a series of nitriles with the alcohol-inducible isoform of P450: Computer analysis of structure-activity relationships. Xenobiotica 1994, 24, 401–408. [Google Scholar] [CrossRef]
- Kumar, P.S.; Vasudevan, K.; Prakasam, A.; Geetha, M.P.M. Anbarasan, Quantum chemistry calculations of 3-Phenoxyphthalonitrile dye sensitizer for solar cells. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2010, 77, 45–50. [Google Scholar] [CrossRef]
- Zhang, R.; Du, B.; Sun, G.; Sun, Y. Experimental and theoretical studies on o-, m- and p-chlorobenzylideneaminoantipyrines. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2010, 75, 1115–1124. [Google Scholar] [CrossRef]
- Cotton, F.A.; Wilkinson, C.W. Advanced Inorganic Chemistry, 3rd ed.; Interscience Publisher: New York, NY, USA, 1972. [Google Scholar]
- Guillaumont, D.; Nakamura, S. Calculation of the absorption wavelength of dyes using time-dependent density-functional theory (TD-DFT). Dye. Pigment. 2000, 46, 85–92. [Google Scholar] [CrossRef]
- Zangwill, A.; Soven, P. Density-functional approach to local-field effects in finite systems: Photoabsorption in the rare gases. Phys. Rev. A 1980, 21, 1561–1572. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Gorelsky, S.I. SWizard Program Revision 4.5; University of Ottawa: Ottawa, ON, Canada, 2010; Available online: http://www.sg.chem.net/ (accessed on 8 April 2022).
- Akman, F.; Issaoui, N.; Kazachenko, A.S. Intermolecular hydrogen bond interactions in the thiourea/water complexes (Thio-(H2O)n) (n = 1, …, 5): X-ray, DFT, NBO, AIM, and RDG analyses. J. Mol. Model. 2020, 26, 161. [Google Scholar] [CrossRef] [PubMed]
- Ott, J.B.; Boerio-Goates, J. Calculations from Statistical Thermodynamics; Academic Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Gunasekaran, S.; Balaji, R.A.; Kumeresan, S.; Anand, G.; Srimivasan, S. Experimental and theoretical investigations of spectroscopic properties of N-acetyl-5-methoxytryptamine. Can. J. Anal. Sci. Spectrosc. 2008, 53, 149–162. [Google Scholar]
- Sevvanthi, S.; Muthu, S.; Raja, M.; Aayisha, S.; Janani, S. PES, molecular structure, spectroscopic (FT-IR, FT-Raman), electronic (UV-Vis, HOMO-LUMO), quantum chemical and biological (docking) studies on a potent membrane permeable inhibitor: Dibenzoxepine derivative. Heilyon 2020, 6, e04724. [Google Scholar] [CrossRef]
- Pérez-Salvia, M.; Esteller, M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics 2017, 12, 323–339. [Google Scholar] [CrossRef] [Green Version]
- Muller, S.; Filippakopoulos, P.; Knapp, S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, E29. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- RCSB PDB, Protein Data Bank. Available online: https://www.rcsb.org/ (accessed on 10 January 2023).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Monomer | Dimer | ||||
|---|---|---|---|---|---|---|
| Bond Length (Å) | HF a | DFT a | Theo. b | Exp. c | HF a | DFT a |
| C1–C2 | 1.3980 | 1.4054 | 1.385 | 1.406 | 1.404 | 1.4131 |
| C1=C3 | 1.3731 | 1.3848 | 1.386 | 1.378 | 1.3692 | 1.3815 |
| C1–Br4 | 1.8891 | 1.9053 | 1.894 | 1.867 | 1.8891 | 1.9043 |
| C2=N5 | 1.3072 | 1.3253 | 1.313 | 1.334 | 1.3135 | 1.3387 |
| C2–O6 | 1.3259 | 1.3466 | 1.338 | 1.3116 | 1.322 | |
| C3–C7 | 1.3908 | 1.3970 | 1.370 | 1.375 | 1.3941 | 1.3989 |
| C3–H9 | 1.0742 | 1.0829 | 0.930 | 1.082 | 1.0742 | 1.0829 |
| N5–C8 | 1.3252 | 1.3387 | 1.347 | 1.337 | 1.3306 | 1.3429 |
| O6–H10 | 0.9445 | 0.9679 | 0.8 | 0.9592 | 1.0073 | |
| C7=C8 | 1.3753 | 1.3870 | 1.378 | 1.38 | 1.3703 | 1.3826 |
| C7–H11 | 1.0731 | 1.0825 | 0.93 | 1.083 | 1.0728 | 1.082 |
| C8–H12 | 1.0756 | 1.0852 | 1.087 | 1.0757 | 1.085 | |
| N5–H22 | 1.9333 | 1.7163 | ||||
| H10–N17 | 1.9329 | 1.7163 | ||||
| C13–C14 | 1.404 | 1.4131 | ||||
| C13=C15 | 1.3692 | 1.3815 | ||||
| C13–Br16 | 1.8891 | 1.9043 | ||||
| C14=N17 | 1.3135 | 1.3387 | ||||
| C14–O18 | 1.3116 | 1.322 | ||||
| C15–C19 | 1.3942 | 1.3989 | ||||
| C15–H21 | 1.0742 | 1.0829 | ||||
| N17–C20 | 1.3306 | 1.3429 | ||||
| O18–H22 | 0.9592 | 1.0074 | ||||
| C19=C20 | 1.3703 | 1.3826 | ||||
| C19–H23 | 1.0728 | 1.082 | ||||
| C20–H24 | 1.0757 | 1.085 | ||||
| Bond angle (°) | ||||||
| C2–C1=C3 | 118.0385 | 118.3261 | 118.4 | 118.5 | 118.5999 | 119.3112 |
| C2–C1–Br4 | 121.1088 | 120.7992 | 120.6383 | 120.008 | ||
| C3=C1–Br4 | 120.8526 | 120.8747 | 120.7618 | 120.6808 | ||
| C2–C1–N5 | 122.6162 | 122.4936 | 124.5 | 123.8 | 121.5678 | 120.5763 |
| C1–C2–O6 | 119.5209 | 119.6722 | 119.3431 | 119.9262 | ||
| N5=C2–O6 | 117.8629 | 117.8341 | 119.0891 | 119.4975 | ||
| C1=C3–C7 | 119.4275 | 119.2043 | 118.4 | 118.4 | 119.5604 | 119.3997 |
| C1=C3–H9 | 119.7958 | 119.8381 | 120.7 | 120.8 | 119.7934 | 119.7699 |
| C7–C3–H9 | 120.7767 | 120.9576 | 120.9 | 120.8 | 120.6462 | 120.8304 |
| C2=N5–C8 | 119.0116 | 118.8784 | 118 | 116.9 | 119.4235 | 119.8116 |
| C2–O6–H10 | 108.3825 | 106.6033 | 111.2939 | 111.3094 | ||
| C3–C7=C8 | 117.6366 | 118.1407 | 119.3 | 118.5 | 117.4975 | 117.972 |
| C3–C7–H11 | 121.2475 | 120.9465 | 120.5 | 121.3459 | 121.1335 | |
| C8=C7–H11 | 121.1158 | 120.9128 | 120.1 | 121.1566 | 120.8944 | |
| N5–C8=C7 | 123.2695 | 122.9568 | 122.5 | 123.8 | 123.3508 | 122.9291 |
| N5–C8–H12 | 115.8735 | 115.906 | 116.5 | 116 | 115.7486 | 115.7388 |
| C7=C8–H12 | 120.857 | 121.1372 | 121 | 121.1 | 120.9005 | 121.3321 |
| C2=N5–H22 | 124.2571 | 123.36 | ||||
| C8–N5–H22 | 116.3193 | 116.8283 | ||||
| C14–C13=C15 | 118.5977 | 119.312 | ||||
| C14–C13–Br16 | 120.6394 | 120.0077 | ||||
| C15=C13–Br16 | 120.7629 | 120.6803 | ||||
| C13–C14–N17 | 121.5697 | 120.5753 | ||||
| C13–C14–C18 | 119.3414 | 119.9295 | ||||
| N17–C14–O18 | 119.0889 | 119.4952 | ||||
| C13=C15–C19 | 119.5615 | 119.3995 | ||||
| C13=C15–H21 | 119.7949 | 119.7704 | ||||
| C19–C15–H21 | 120.6436 | 120.8302 | ||||
| H10–N17=C14 | 124.2639 | 123.3594 | ||||
| H10–N17–C20 | 116.3128 | 116.8286 | ||||
| C14=N17–C20 | 119.4233 | 119.8121 | ||||
| C14–O18–H22 | 111.2924 | 111.3165 | ||||
| C15–C19=C20 | 117.4979 | 117.9715 | ||||
| C14–C19–H23 | 121.3433 | 121.1329 | ||||
| C20=C19–H23 | 121.1588 | 120.8956 | ||||
| N17–C20=C19 | 123.3498 | 122.9296 | ||||
| N17–C20–H24 | 115.747 | 115.7384 | ||||
| Intermolecular H−bond lengths (Å) and angles (°) | ||||||
| N5·· H22 | 1.7163 | |||||
| N17·· H10 | 1.7163 | |||||
| O18–H22 | 1.0073 | |||||
| O6–H10 | 1.0073 | |||||
| O6–H10·· N17 | 174.159 | |||||
| O18–H22·· N5 | 174.159 | |||||
| Parameter | O6–H10 | N5··· H22 | H10··· N17 | RCP1 | RCP2 | NRCP |
|---|---|---|---|---|---|---|
| ρ(r) | 0.3115 | 0.0520 | 0.0520 | 0.0234 | 0.0234 | 0.0081 |
| ρ(r) | 2.1007 | 0.0998 | 0.0998 | 0.1688 | 0.1688 | 0.0310 |
| λ1 | −1.5439 | −0.0914 | −0.0914 | −0.0194 | −0.0194 | −0.0068 |
| λ2 | −1.5196 | −0.0870 | −0.0870 | 0.0914 | 0.0914 | 0.0109 |
| λ3 | 0.9628 | 0.2783 | 0.2783 | 0.0968 | 0.0968 | 0.0269 |
| |λ1|/λ3 | 1.6035 | 0.3284 | 0.3284 | 0.2004 | 0.2004 | 0.2527 |
| [(λ1/λ2) − 1] | 0.5927 | 0.0107 | 0.0107 | −0.007 | −0.0077 | −0.0011 |
| H | 0.0675 | 0.0356 | 0.0356 | 0.0344 | 0.0344 | 0.0066 |
| G | 0.6603 | 0.0463 | 0.0463 | 0.0267 | 0.0267 | 0.0055 |
| V | 0.0160 | 0.0505 | 0.0505 | −1.2122 | −1.2122 | −1.6238 |
| G/ρ(rc) | 0.2169 | 0.6854 | 0.6854 | 1.474 | 1.474 | 0.8222 |
| Parameters | TD–DFT/B3LYP/6–311++G(d,p) | |||
|---|---|---|---|---|
| Methanol | Water | DMSO | Gas | |
| Etotal (Hartree) | −2879.157 | −2897.158 | −2897.158 | −2897.158 |
| EHOMO (eV) | −6.8787 | −6.880 | −6.880 | −6.880 |
| ELUMO (eV) | −1.4751 | −1.475 | −1.475 | −1.475 |
| ΔEHOMO–LUMO gap (eV) | 5.404 | 5.406 | 5.405 | 5.406 |
| EHOMO–1 (eV) | −7.959 | −7.961 | −7.960 | −7.961 |
| ELUMO+1 (eV) | −0.749 | −0.749 | −0.749 | −0.7488 |
| λ (nm) | E (eV) | f (a.u.) | Major Contribution (%) | |
|---|---|---|---|---|
| Gas | 260.3 | 4.76 | 0.1431 | H→L (91) |
| 240.3 | 5.15 | 0.0 | H→L + 2 (97) | |
| 234.7 | 5.28 | 0.0045 | H-2→L (94) | |
| 220.5 | 5.62 | 0.0767 | H-1→L (33), H→L + 1 (62) | |
| 209.8 | 5.90 | 0.0192 | H→L+3 (98) | |
| DMSO | 260.8 | 4.75 | 0.1506 | H→L (91) |
| 240.4 | 5.15 | 0.0 | H→L+2 (97) | |
| 234.8 | 5.28 | 0.0047 | H-2→L (94) | |
| 220.8 | 5.61 | 0.0807 | H-1→L (32), H→L + 1 (63) | |
| 209.8 | 5.90 | 0.0196 | H→L+3 (98) | |
| Water | 260.3 | 4.76 | 0.1431 | H→L (91) |
| 240.3 | 5.15 | 0.0 | H→L+2 (97) | |
| 234.7 | 5.28 | 0.0045 | H-2→L (94) | |
| 220.5 | 5.62 | 0.0767 | H-1→L (33), H→L + 1 (62) | |
| 209.8 | 5.90 | 0.0192 | H→L+3 (98) | |
| Methanol | 260.4 | 4.76 | 0.1428 | H→L (91) |
| 240.3 | 5.15 | 0.0 | H→L + 2 (97) | |
| 234.8 | 5.28 | 0.0045 | H-2→L (94) | |
| 220.6 | 5.62 | 0.0765 | H-1→L (33), H→L + 1 (62) | |
| 209.8 | 5.90 | 0.0191 | H→L+3 (98) | |
| Chloroform | 261.9 | 4.73 | 0.1541 | H→L (91) |
| 240.4 | 5.15 | 0.0 | H→L+2 (97) | |
| 236.0 | 5.25 | 0.0047 | H-1→L (93) | |
| 221.7 | 5.59 | 0.0823 | H-2→L (32), H→L + 1 (63) | |
| 210.4 | 5.89 | 0.0193 | H→L + 3 (97) |
| T (K) | S (J/mol·K) | Cp (J/mol·K) | H (kJ/mol) |
|---|---|---|---|
| 100 | 267.79 | 50.60 | 3.90 |
| 200 | 311.88 | 80.65 | 10.44 |
| 298.15 | 349.82 | 111.23 | 19.86 |
| 300 | 350.51 | 111.79 | 20.07 |
| 400 | 386.63 | 139.91 | 32.69 |
| 500 | 420.43 | 163.00 | 47.88 |
| 600 | 451.82 | 181.26 | 65.13 |
| 700 | 480.89 | 195.68 | 84.00 |
| 800 | 507.80 | 207.25 | 104.17 |
| 900 | 532.78 | 216.71 | 125.38 |
| 1000 | 556.03 | 224.57 | 147.46 |
| Atom | Natural Charges | Fukui Functions | Dual Descriptor | ||||
|---|---|---|---|---|---|---|---|
| 0, 1 (N) | N + 1 (−1, 2) | N − 1 (1, 2) | fr+ | fr− | fr0 | Δf | |
| C1 | −0.18612 | −0.21400 | −0.10282 | −0.02788 | −0.08330 | −0.05559 | 0.05542 |
| C2 | 0.50801 | 0.49680 | 0.57671 | −0.01121 | −0.06870 | −0.03996 | 0.05749 |
| C3 | −0.17061 | −0.18618 | −0.17137 | −0.01557 | 0.00076 | −0.00741 | −0.01633 |
| Br4 | 0.09704 | −0.01467 | 0.39100 | −0.11171 | −0.29396 | −0.20284 | 0.18225 |
| N5 | −0.51337 | −0.52676 | −0.47605 | −0.01339 | −0.03732 | −0.02536 | 0.02393 |
| O6 | −0.65520 | −0.68317 | −0.53381 | −0.02797 | −0.12139 | −0.07468 | 0.09342 |
| C7 | −0.26329 | −0.29652 | −0.12926 | −0.03323 | −0.13403 | −0.08363 | 0.10080 |
| C8 | 0.06479 | 0.04476 | 0.18025 | −0.02003 | −0.11546 | −0.06775 | 0.09543 |
| H9 | 0.22433 | 0.13549 | 0.26162 | −0.08884 | −0.03729 | −0.06307 | −0.05155 |
| H10 | 0.48401 | 0.38233 | 0.51591 | −0.10168 | −0.03190 | −0.06679 | −0.06978 |
| H11 | 0.21809 | −0.14101 | 0.25560 | −0.35910 | −0.03751 | −0.19831 | −0.32159 |
| H12 | 0.19233 | 0.00292 | 0.23223 | −0.18941 | −0.03990 | −0.11466 | −0.14951 |
| C1 | −0.18612 | −0.21400 | −0.10282 | −0.02788 | −0.08330 | −0.05559 | 0.05542 |
| C2 | 0.50801 | 0.49680 | 0.57671 | −0.01121 | −0.06870 | −0.03996 | 0.05749 |
| Protein (PDB ID) | Bonded Residues | Bond Distance (Å) | Estimated Inhibition Constant (μm) | Binding Energy (kcal/mol) | Reference RMSD (Å) |
|---|---|---|---|---|---|
| 5IBN | ASN135 | 1.8 | 351.98 | −4.71 | 17.207 |
| MET105 | 2.4 | ||||
| 3U5K | TYR36 | 2.4 | 454.17 | −4.56 | 79.38 |
| MET421 | 2.5 | ||||
| 6CD5 | ASN135 | 1.8 | 757.63 | −4.26 | 14.506 |
| MET132 | 2.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lefi, N.; Kazachenko, A.S.; Raja, M.; Issaoui, N.; Kazachenko, A.S. Molecular Structure, Spectral Analysis, Molecular Docking and Physicochemical Studies of 3-Bromo-2-hydroxypyridine Monomer and Dimer as Bromodomain Inhibitors. Molecules 2023, 28, 2669. https://doi.org/10.3390/molecules28062669
Lefi N, Kazachenko AS, Raja M, Issaoui N, Kazachenko AS. Molecular Structure, Spectral Analysis, Molecular Docking and Physicochemical Studies of 3-Bromo-2-hydroxypyridine Monomer and Dimer as Bromodomain Inhibitors. Molecules. 2023; 28(6):2669. https://doi.org/10.3390/molecules28062669
Chicago/Turabian StyleLefi, Nizar, Aleksandr S. Kazachenko, Murugesan Raja, Noureddine Issaoui, and Anna S. Kazachenko. 2023. "Molecular Structure, Spectral Analysis, Molecular Docking and Physicochemical Studies of 3-Bromo-2-hydroxypyridine Monomer and Dimer as Bromodomain Inhibitors" Molecules 28, no. 6: 2669. https://doi.org/10.3390/molecules28062669
APA StyleLefi, N., Kazachenko, A. S., Raja, M., Issaoui, N., & Kazachenko, A. S. (2023). Molecular Structure, Spectral Analysis, Molecular Docking and Physicochemical Studies of 3-Bromo-2-hydroxypyridine Monomer and Dimer as Bromodomain Inhibitors. Molecules, 28(6), 2669. https://doi.org/10.3390/molecules28062669