
Development of Activity Rules and Chemical Fragment Design for In Silico Discovery of AChE and BACE1 Dual Inhibitors against Alzheimer’s Disease

 ,
,  ,
,  , ,
, , 
Abstract
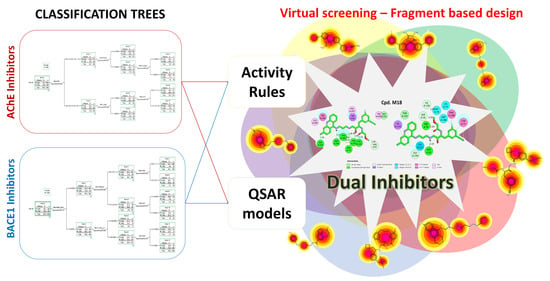
1. Introduction
2. Results and Discussion
2.1. Data Analysis and Activity Threshold Selection
2.2. Machine Learning Models
2.3. Identification of AChE and BACE1 Inhibitory Activity Rules
2.4. Reposition of Dual-Targeted Inhibitors
2.5. Design of Novel Dual-Targeted Inhibitors
2.6. Comparison with Previous Studies
3. Materials and Methods
3.1. Data Curation and Labeling
3.2. Descriptor Calculation and Training Set Selection
3.3. Development and Validation of Machine Learning Models
- -
- CART tree was constructed using the impurity Gini-based binary classification tree algorithm implemented in IBM SPSS Statistics v.22.0 [61]. For the AChE-CART model, a maximum tree depth of 9 and a minimum number of 150 cases in the parent node and 40 in the child node were configured. In the case of BACE1-CART, the model was built with the following parameters: maximum tree depth of 5 and a minimum of 100 cases in the parent node and 20 cases in the child node. For both models, the tree pruning technique was used to avoid overfitting and the prior probability in all categories was reset to the same value before running the models.
- -
- The CHAID model was created by splitting data into mutually exclusive and exhaustive subsets with the best description of the dependent variable. A major difference between the CART and CHAID algorithm is that CART produces binary splits that imply two possible outcomes, whereas CHAID can generate multiple branches of a single root/parent node [61]. The parameters of AChE-CHAID models were set as follows: maximum tree depth: 3, minimum number of cases in parent node: 60, minimum number of cases in child node: 18. For the BACE1-CHAID model, the following parameters were set as follows: maximum tree depth: 3, minimum number of cases in parent node: 100, minimum number of cases in child node: 45.
- -
- RF is a multiple classifiers system (MCS) consisting of a collection of tree-structured classifiers. Significant improvements in classification accuracy have been achieved by growing an ensemble of trees and having them vote for the most popular class [62]. In this work, the RF models were built based on variables selected from the CART and CHAID algorithms. The RF algorithm implemented in Statistica 11.0 was used. The importance of selected variables was evaluated by a normalized importance scale. As a result, 172 and 117 variables were selected for the AChE-RF and BACE1-RF models, respectively. The hyper-parameters set for the AChE-RF model included a size of 200 trees, a number of predictors of 8, a subsample proportion of 0.5, a maximum number of levels of 5, a minimum number of cases of 50, a maximum number of nodes of 100, and a minimum number of subordinate nodes of 10. The BACE1-RF model had the following parameters: 200 trees, the number of predictors is 7, the proportion of subsample is 0.45, the maximum number of levels is 5, the minimum number of cases is 50, the maximum number of nodes is 100, and the minimum number in child nodes is 10.
3.4. Rule-Based Query Selection and Virtual Design of Dual-Target Inhibitors
3.5. Docking Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Levey, A.I. Progress with Treatments for Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1762–1763. [Google Scholar] [CrossRef] [PubMed]
- Association, A. 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement 2023, 19, 1598–1695. [Google Scholar] [CrossRef]
- Monfared, A.A.T.; Byrnes, M.J.; White, L.A.; Zhang, Q. Alzheimer’s Disease: Epidemiology and Clinical Progression. Neurol. Ther. 2022, 11, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Yarns, B.C.; Holiday, K.A.; Carlson, D.M.; Cosgrove, C.K.; Melrose, R.J. Pathophysiology of Alzheimer’s Disease. Psychiatr. Clin. N. Am. 2022, 45, 663–676. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Fratila, O.; Brata, R.; Bungau, S. Exploring the Potential of Therapeutic Agents Targeted towards Mitigating the Events Associated with Amyloid-β Cascade in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7443. [Google Scholar] [CrossRef]
- Viayna, E.; Sola, I.; Bartolini, M.; De Simone, A.; Tapia-Rojas, C.; Serrano, F.G.; Sabaté, R.; Juárez-Jiménez, J.; Pérez, B.; Luque, F.J.; et al. Synthesis and Multitarget Biological Profiling of a Novel Family of Rhein Derivatives as Disease-Modifying Anti-Alzheimer Agents. J. Med. Chem. 2014, 57, 2549–2567. [Google Scholar] [CrossRef]
- Li, Q.; He, S.; Chen, Y.; Feng, F.; Qu, W.; Sun, H. Donepezil-based multi-functional cholinesterase inhibitors for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 158, 463–477. [Google Scholar] [CrossRef]
- Oset-Gasque, M.J.; Marco-Contelles, J.L. Tacrine-Natural-Product Hybrids for Alzheimer’s Disease Therapy. Curr. Med. Chem. 2020, 27, 4392–4400. [Google Scholar] [CrossRef]
- Mihai, D.P.; Nitulescu, G.M.; Ion, G.N.D.; Ciotu, C.I.; Chirita, C.; Negres, S. Computational Drug Repurposing Algorithm Targeting TRPA1 Calcium Channel as a Potential Therapeutic Solution for Multiple Sclerosis. Pharmaceutics 2019, 11, 446. [Google Scholar] [CrossRef]
- Nitulescu, G.; Nitulescu, G.M.; Zanfirescu, A.; Mihai, D.P.; Gradinaru, D. Candidates for Repurposing as Anti-Virulence Agents Based on the Structural Profile Analysis of Microbial Collagenase Inhibitors. Pharmaceutics 2021, 14, 62. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.; Dhanjal, J.K.; Goyal, S.; Tyagi, C.; Hamid, R.; Grover, A. Development of Dual Inhibitors against Alzheimer’s Disease Using Fragment-Based QSAR and Molecular Docking. BioMed. Res. Int. 2014, 2014, 979606. [Google Scholar] [CrossRef] [PubMed]
- Dhamodharan, G.; Mohan, C.G. Machine learning models for predicting the activity of AChE and BACE1 dual inhibitors for the treatment of Alzheimer’s disease. Mol. Divers. 2022, 26, 1501–1517. [Google Scholar] [CrossRef] [PubMed]
- Stern, N.; Gacs, A.; Tátrai, E.; Flachner, B.; Hajdú, I.; Dobi, K.; Bágyi, I.; Dormán, G.; Lőrincz, Z.; Cseh, S.; et al. Dual Inhibitors of AChE and BACE-1 for Reducing Aβ in Alzheimer’s Disease: From In Silico to In Vivo. Int. J. Mol. Sci. 2022, 23, 13098. [Google Scholar] [CrossRef]
- Pham-The, H.; Nam, N.-H.; Nga, D.-V.; Hai, D.T.; Dieguez-Santana, K.; Marrero-Poncee, Y.; Castillo-Garit, J.A.; Casanola-Martin, G.M.; Le-Thi-Thu, H. Learning from Multiple Classifier Systems: Perspectives for Improving Decision Making of QSAR Models in Medicinal Chemistry. Curr. Top. Med. Chem. 2017, 17, 3269–3288. [Google Scholar] [CrossRef]
- Cañizares-Carmenate, Y.; Nam, N.-H.; Díaz-Amador, R.; Thuan, N.T.; Dung, P.T.P.; Torrens, F.; Pham-The, H.; Perez-Gimenez, F.; Castillo-Garit, J.A. Ligand-based discovery of new potential acetylcholinesterase inhibitors for Alzheimer’s disease treatment. SAR QSAR Environ. Res. 2022, 33, 49–61. [Google Scholar] [CrossRef]
- Lan, T.T.; Anh, D.T.; Hai, P.-T.; Dung, D.T.M.; Huong, L.T.T.; Park, E.J.; Jeon, H.W.; Kang, J.S.; Thuan, N.T.; Han, S.-B.; et al. Design, synthesis, and bioevaluation of novel oxoindolin-2-one derivatives incorporating 1-benzyl-1H-1,2,3-triazole. Med. Chem. Res. 2020, 29, 396–408. [Google Scholar] [CrossRef]
- Le-Nhat-Thuy, G.; Thi, N.N.; Pham-The, H.; Thi, T.A.D.; Thi, H.N.; Thi, T.H.N.; Hoang, S.N.; Nguyen, T.V. Synthesis and biological evaluation of novel quinazoline-triazole hybrid compounds with potential use in Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2020, 30, 127404. [Google Scholar] [CrossRef]
- Pham-The, H.; Casañola-Martin, G.; Garrigues, T.; Bermejo, M.; Gonzalez-Alvarez, I.; Nguyen-Hai, N.; Cabrera-Pérez, M.; Le-Thi-Thu, H. Exploring different strategies for imbalanced ADME data problem: Case study on Caco-2 permeability modeling. Mol. Divers. 2016, 20, 93–109. [Google Scholar] [CrossRef]
- Perez-Castillo, Y.; Sánchez-Rodríguez, A.; Tejera, E.; Cruz-Monteagudo, M.; Borges, F.; Cordeiro, M.N.D.S.; Le-Thi-Thu, H.; Pham-The, H. A desirability-based multi objective approach for the virtual screening discovery of broad-spectrum anti-gastric cancer agents. PLoS ONE 2018, 13, e0192176. [Google Scholar] [CrossRef]
- Pham-The, H.; González-Álvarez, I.; Bermejo, M.; Garrigues, T.; Le-Thi-Thu, H.; Cabrera-Pérez, M. The Use of Rule-Based and QSPR Approaches in ADME Profiling: A Case Study on Caco-2 Permeability. Mol. Inf. 2013, 32, 459–479. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Crippen, G.M. Atomic Physicochemical Parameters for Three-Dimensional Structure-Directed Quantitative Structure-Activity Relationships I. Partition Coefficients as a Measure of Hydrophobicity. J. Comput. Chem. 1986, 7, 565–577. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; Volume 11, p. 967. [Google Scholar]
- Estrada, E. Edge Adjacency Relationships and a Novel Topological Index Related to Molecular Volume. J. Chem. Inf. Comput. Sci. 1995, 35, 31–33. [Google Scholar] [CrossRef]
- Roy, K.; Ghosh, G. Introduction of Extended Topochemical Atom (ETA) Indices in the Valence Electron Mobile (VEM) Environment as Tools for QSAR/QSPR Studies. Internet Electron. Internet Electron. J. Mol. Des. 2003, 2, 599–620. [Google Scholar]
- Ivanciuc, O.; Balaban, T.-S.; Balaban, A.T. Chemical graphs with degenerate topological indices based on information on distances. J. Math. Chem. 1993, 14, 21–33. [Google Scholar] [CrossRef]
- Labute, P. A widely applicable set of descriptors. J. Mol. Graph. Model. 2000, 18, 464–477. [Google Scholar] [CrossRef]
- Domínguez, J.L.; Villaverde, M.C.; Sussman, F. Effect of pH and ligand charge state on BACE-1 fragment docking performance. J. Comput. Aided Mol. Des. 2013, 27, 403–417. [Google Scholar] [CrossRef]
- Descamps, O.; Spilman, P.; Zhang, Q.; Libeu, C.P.; Poksay, K.; Gorostiza, O.; Campagna, J.; Jagodzinska, B.; Bredesen, D.E.; John, V. AβPP-Selective BACE Inhibitors (ASBI): Novel Class of Therapeutic Agents for Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 37, 343–355. [Google Scholar] [CrossRef]
- Pérez-Areales, F.J.; Di Pietro, O.; Espargaró, A.; Vallverdú-Queralt, A.; Galdeano, C.; Ragusa, I.M.; Viayna, E.; Guillou, C.; Clos, M.V.; Pérez, B.; et al. Shogaol–huprine hybrids: Dual antioxidant and anticholinesterase agents with β-amyloid and tau anti-aggregating properties. Bioorganic Med. Chem. 2014, 22, 5298–5307. [Google Scholar] [CrossRef]
- Galdeano, C.; Viayna, E.; Sola, I.; Formosa, X.; Camps, P.; Badia, A.; Clos, M.V.; Relat, J.; Ratia, M.; Bartolini, M.; et al. Huprine–Tacrine Heterodimers as Anti-Amyloidogenic Compounds of Potential Interest against Alzheimer’s and Prion Diseases. J. Med. Chem. 2012, 55, 661–669. [Google Scholar] [CrossRef]
- Sola, I.; Castellà, S.; Viayna, E.; Galdeano, C.; Taylor, M.C.; Gbedema, S.Y.; Pérez, B.; Clos, M.V.; Jones, D.C.; Fairlamb, A.H.; et al. Synthesis, biological profiling and mechanistic studies of 4-aminoquinoline-based heterodimeric compounds with dual trypanocidal–antiplasmodial activity. Bioorganic Med. Chem. 2015, 23, 5156–5167. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Deng, H.; Huang, S.; Yang, J.; Wang, S.; Yin, B.; Zheng, T.; Zhang, D.; Liu, J.; Gao, G.; et al. Design, synthesis and evaluation of novel 5,6,7-trimethoxyflavone–6-chlorotacrine hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2015, 25, 1541–1545. [Google Scholar] [CrossRef] [PubMed]
- Nepovimova, E.; Korabecny, J.; Dolezal, R.; Babkova, K.; Ondrejicek, A.; Jun, D.; Sepsova, V.; Horova, A.; Hrabinova, M.; Soukup, O.; et al. Tacrine–Trolox Hybrids: A Novel Class of Centrally Active, Nonhepatotoxic Multi-Target-Directed Ligands Exerting Anticholinesterase and Antioxidant Activities with Low In Vivo Toxicity. J. Med. Chem. 2015, 58, 8985–9003. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Cavalli, A.; Valgimigli, L.; Bartolini, M.; Rosini, M.; Andrisano, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Drug Design Strategy: From a Dual Binding Site Acetylcholinesterase Inhibitor to a Trifunctional Compound against Alzheimer’s Disease. J. Med. Chem. 2007, 50, 6446–6449. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, A.; Gallelli, A.; Manini, M.; Anzini, M.; Mennuni, L.; Makovec, F.; Menziani, M.C.; Alcaro, S.; Ortuso, F.; Vomero, S. Further Studies on the Interaction of the 5-Hydroxytryptamine3 (5-HT3) Receptor with Arylpiperazine Ligands. Development of a New 5-HT3 Receptor Ligand Showing Potent Acetylcholinesterase Inhibitory Properties. J. Med. Chem. 2005, 48, 3564–3575. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Andrisano, V.; Bartolini, M.; Banzi, R.; Melchiorre, C. Propidium-Based Polyamine Ligands as Potent Inhibitors of Acetylcholinesterase and Acetylcholinesterase-Induced Amyloid-β Aggregation. J. Med. Chem. 2005, 48, 24–27. [Google Scholar] [CrossRef]
- Lan, J.-S.; Xie, S.-S.; Li, S.-Y.; Pan, L.-F.; Wang, X.-B.; Kong, L.-Y. Design, synthesis and evaluation of novel tacrine-(β-carboline) hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Bioorganic Med. Chem. 2014, 22, 6089–6104. [Google Scholar] [CrossRef]
- Jiang, X.-Y.; Chen, T.-K.; Zhou, J.-T.; He, S.-Y.; Yang, H.-Y.; Chen, Y.; Qu, W.; Feng, F.; Sun, H.-P. Dual GSK-3β/AChE Inhibitors as a New Strategy for Multitargeting Anti-Alzheimer’s Disease Drug Discovery. ACS Med. Chem. Lett. 2018, 9, 171–176. [Google Scholar] [CrossRef]
- Rizzo, S.; Bisi, A.; Bartolini, M.; Mancini, F.; Belluti, F.; Gobbi, S.; Andrisano, V.; Rampa, A. Multi-target strategy to address Alzheimer’s disease: Design, synthesis and biological evaluation of new tacrine-based dimers. Eur. J. Med. Chem. 2011, 46, 4336–4343. [Google Scholar] [CrossRef]
- Di Pietro, O.; Pérez-Areales, F.J.; Juárez-Jiménez, J.; Espargaró, A.; Clos, M.V.; Pérez, B.; Lavilla, R.; Sabaté, R.; Luque, F.J.; Muñoz-Torrero, D. Tetrahydrobenzo[h](1,6)naphthyridine-6-chlorotacrine hybrids as a new family of anti-Alzheimer agents targeting β-amyloid, tau, and cholinesterase pathologies. Eur. J. Med. Chem. 2014, 84, 107–117. [Google Scholar] [CrossRef]
- Hepnarova, V.; Korabecny, J.; Matouskova, L.; Jost, P.; Muckova, L.; Hrabinova, M.; Vykoukalova, N.; Kerhartova, M.; Kucera, T.; Dolezal, R.; et al. The concept of hybrid molecules of tacrine and benzyl quinolone carboxylic acid (BQCA) as multifunctional agents for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 150, 292–306. [Google Scholar] [CrossRef]
- Hamada, Y.; Igawa, N.; Ikari, H.; Ziora, Z.; Nguyen, J.-T.; Yamani, A.; Hidaka, K.; Kimura, T.; Saito, K.; Hayashi, Y.; et al. β-Secretase inhibitors: Modification at the P4 position and improvement of inhibitory activity in cultured cells. Bioorganic Med. Chem. Lett. 2006, 16, 4354–4359. [Google Scholar] [CrossRef] [PubMed]
- Pennington, L.D.; Whittington, D.A.; Bartberger, M.D.; Jordan, S.R.; Monenschein, H.; Nguyen, T.T.; Yang, B.H.; Xue, Q.M.; Vounatsos, F.; Wahl, R.C.; et al. Hydroxyethylamine-based inhibitors of BACE1: P1–P3 macrocyclization can improve potency, selectivity, and cell activity. Bioorganic Med. Chem. Lett. 2013, 23, 4459–4464. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, A.; McGaughey, G.B.; Sheridan, R.P.; Good, A.C.; Warren, G.; Mathieu, M.; Muchmore, S.W.; Brown, S.P.; Grant, J.A.; Haigh, J.A.; et al. Molecular Shape and Medicinal Chemistry: A Perspective. J. Med. Chem. 2010, 53, 3862–3886. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.A.C.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Tosaki, A.; Kaneko, K.; Hisano, T.; Sakurai, T.; Nukina, N. Crystal structure of an active form of BACE1, an enzyme responsible for amyloid β protein production. Mol. Cell. Biol. 2008, 28, 3663–3671. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; Mallender, W.D.; Inestrosa, N.C.; Rosenberry, T.L. Thioflavin T Is a Fluorescent Probe of the Acetylcholinesterase Peripheral Site That Reveals Conformational Interactions between the Peripheral and Acylation Sites. J. Biol. Chem. 2001, 276, 23282–23287. [Google Scholar] [CrossRef]
- Zhang, P.; Xu, S.; Zhu, Z.; Xu, J. Multi-target design strategies for the improved treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 176, 228–247. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The Properties of Known Drugs. 1. Molecular Frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Obaid, R.J.; Naeem, N.; Mughal, E.U.; Al-Rooqi, M.M.; Sadiq, A.; Jassas, R.S.; Moussa, Z.; Ahmed, S.A. Inhibitory potential of nitrogen, oxygen and sulfur containing heterocyclic scaffolds against acetylcholinesterase and butyrylcholinesterase. RSC Adv. 2022, 12, 19764–19855. [Google Scholar] [CrossRef]
- Low, J.D.; Bartberger, M.D.; Chen, K.; Cheng, Y.; Fielden, M.R.; Gore, V.; Hickman, D.; Liu, Q.; Sickmier, E.A.; Vargas, H.M.; et al. Development of 2-aminooxazoline 3-azaxanthene β-amyloid cleaving enzyme (BACE) inhibitors with improved selectivity against Cathepsin D. MedChemComm 2017, 8, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Stachel, S.J.; Coburn, C.A.; Sankaranarayanan, S.; Price, E.A.; Pietrak, B.L.; Huang, Q.; Lineberger, J.; Espeseth, A.S.; Jin, L.; Ellis, J.; et al. Macrocyclic Inhibitors of β-Secretase: Functional Activity in an Animal Model. J. Med. Chem. 2006, 49, 6147–6150. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, A.; Butler, T.W.; Chapin, D.S.; Chen, Y.L.; DeMattos, S.B.; Ives, J.L.; Jones, S.B.; Liston, D.R.; Nagel, A.A. 5,7-Dihydro-3-[2-[1-(phenylmethyl)-4-piperidinyl]ethyl]-6H-pyrrolo[3,2-f]-1,2-benzisoxazol-6-one: A Potent and Centrally-Selective Inhibitor of Acetylcholinesterase. J. Med. Chem. 1995, 38, 2802–2808. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Basak, S.; Mukherjee, A.; Basu, A. Design and Study of In Silico Binding Dynamics of Certain Isoxazole Bearing Leads Against Aβ-42 and BACE-1 Loop in Protein Fibrillation. Lett. Drug Des. Discov. 2022, 19, 192–213. [Google Scholar] [CrossRef]
- Hu, H.; Chen, Z.; Xu, X.; Xu, Y. Structure-Based Survey of the Binding Modes of BACE1 Inhibitors. ACS Chem. Neurosci. 2019, 10, 880–889. [Google Scholar] [CrossRef]
- Tran, T.-S.; Le, M.-T.; Tran, T.-D.; Tran, T.-H.; Thai, K.-M. Design of Curcumin and Flavonoid Derivatives with Acetylcholinesterase and Beta-Secretase Inhibitory Activities Using in Silico Approaches. Molecules 2020, 25, 3644. [Google Scholar] [CrossRef]
- Huong, T.T.L.; Van Cuong, L.; Huong, P.T.; Thao, T.P.; Huong, L.-T.; Dung, P.T.P.; Oanh, D.T.K.; Huong, N.T.M.; Quan, H.-V.; Vu, T.K.; et al. Exploration of some indole-based hydroxamic acids as histone deacetylase inhibitors and antitumor agents. Chem. Pap. 2017, 71, 1759–1769. [Google Scholar] [CrossRef]
- Viceconti, M.; Pappalardo, F.; Rodriguez, B.; Horner, M.; Bischoff, J.; Tshinanu, F.M. In silico trials: Verification, validation and uncertainty quantification of predictive models used in the regulatory evaluation of biomedical products. Methods 2021, 185, 120–127. [Google Scholar] [CrossRef]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef]
- Krzywinski, M.; Altman, N. Classification and regression trees. Nat. Methods 2017, 14, 757–758. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Pham-The, H.; Casañola-Martin, G.; Diéguez-Santana, K.; Nguyen-Hai, N.; Ngoc, N.; Vu-Duc, L.; Le-Thi-Thu, H. Quantitative structure–activity relationship analysis and virtual screening studies for identifying HDAC2 inhibitors from known HDAC bioactive chemical libraries. SAR QSAR Environ. Res. 2017, 28, 199–220. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Sahigara, F.; Mansouri, K.; Ballabio, D.; Mauri, A.; Consonni, V.; Todeschini, R. Comparison of Different Approaches to Define the Applicability Domain of QSAR Models. Molecules 2012, 17, 4791–4810. [Google Scholar] [CrossRef] [PubMed]
- Velkoborsky, J.; Hoksza, D. Scaffold analysis of PubChem database as background for hierarchical scaffold-based visualization. J. Chemin 2016, 8, 74. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Liu, A.; Guang, H.; Zhu, L.; Du, G.; Lee, S.M.Y.; Wang, Y. 3D-QSAR analysis of a new type of acetylcholinesterase inhibitors. Sci. China C Life Sci. 2007, 50, 726–730. [Google Scholar] [CrossRef]
- Roy, K.K.; Dixit, A.; Saxena, A.K. An investigation of structurally diverse carbamates for acetylcholinesterase (AChE) inhibition using 3D-QSAR analysis. J. Mol. Graph. Model. 2008, 27, 197–208. [Google Scholar] [CrossRef]
- Chaudhaery, S.S.; Roy, K.K.; Saxena, A.K. Consensus Superiority of the Pharmacophore-Based Alignment, Over Maximum Common Substructure (MCS): 3D-QSAR Studies on Carbamates as Acetylcholinesterase Inhibitors. J. Chem. Inf. Model. 2009, 49, 1590–1601. [Google Scholar] [CrossRef]
- Wang, K.; Hu, X.; Wang, Z.; Yan, A. Classification of Acetylcholinesterase Inhibitors and Decoys by a Support Vector Machine. Comb. Chem. High Throughput Screen. 2012, 15, 492–502. [Google Scholar] [CrossRef]
- Yan, A.; Wang, K. Quantitative structure and bioactivity relationship study on human acetylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 3336–3342. [Google Scholar] [CrossRef] [PubMed]
- Deb, P.K.; Sharma, A.; Piplani, P.; Akkinepally, R.R. Molecular docking and receptor-specific 3D-QSAR studies of acetylcholinesterase inhibitors. Mol. Divers. 2012, 16, 803–823. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Liu, C.; Zhao, L.; Zhang, H. 3D-QSAR study of multi-target-directed AchE inhibitors based on autodocking. Med. Chem. Res. 2012, 21, 245–256. [Google Scholar] [CrossRef]
- Gharaghani, S.; Khayamian, T.; Ebrahimi, M. Molecular dynamics simulation study and molecular docking descriptors in structure-based QSAR on acetylcholinesterase (AChE) inhibitors. SAR QSAR Environ. Res. 2013, 24, 773–794. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-P.; Weng, X.; Ning, F.-X.; Ou, J.-B.; Hou, J.-Q.; Luo, H.-B.; Li, D.; Huang, Z.-S.; Huang, S.-L.; Gu, L.-Q. 3D-QSAR studies of azaoxoisoaporphine, oxoaporphine, and oxoisoaporphine derivatives as anti-AChE and anti-AD agents by the CoMFA method. J. Mol. Graph. Model. 2013, 41, 61–67. [Google Scholar] [CrossRef]
- Wong, K.Y.; Mercader, A.G.; Saavedra, L.M.; Honarparvar, B.; Romanelli, G.P.; Duchowicz, P.R. QSAR analysis on tacrine-related acetylcholinesterase inhibitors. J. Biomed. Sci. 2014, 21, 84. [Google Scholar] [CrossRef]
- Vats, C.; Dhanjal, J.K.; Goyal, S.; Bharadvaja, N.; Grover, A. Computational design of novel flavonoid analogues as potential AChE inhibitors: Analysis using group-based QSAR, molecular docking and molecular dynamics simulations. Struct. Chem. 2015, 26, 467–476. [Google Scholar] [CrossRef]
- Lee, S.; Barron, M.G. Development of 3D-QSAR Model for Acetylcholinesterase Inhibitors Using a Combination of Fingerprint, Molecular Docking, and Structure-Based Pharmacophore Approaches. Toxicol. Sci. 2015, 148, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Hu, J.; Wang, L.; Zhong, G.; Pan, J.; Wu, Z.; Hui, A. Combined 3D-QSAR, molecular docking, and molecular dynamics study of tacrine derivatives as potential acetylcholinesterase (AChE) inhibitors of Alzheimer’s disease. J. Mol. Model. 2015, 21, 277. [Google Scholar] [CrossRef]
- Bitam, S.; Hamadache, M.; Hanini, S. QSAR model for prediction of the therapeutic potency of N-benzylpiperidine derivatives as AChE inhibitors. SAR QSAR Environ. Res. 2017, 28, 471–489. [Google Scholar] [CrossRef]
- Daoud, I.; Melkemi, N.; Salah, T.; Ghalem, S. Combined QSAR, molecular docking and molecular dynamics study on new Acetylcholinesterase and Butyrylcholinesterase inhibitors. Comput. Biol. Chem. 2018, 74, 304–326. [Google Scholar] [CrossRef] [PubMed]
- López, A.F.F.; Martínez, O.M.M.; Hernández, H.F.C. Evaluation of Amaryllidaceae alkaloids as inhibitors of human acetylcholinesterase by QSAR analysis and molecular docking. J. Mol. Struct. 2021, 1225, 129142. [Google Scholar] [CrossRef]
- Sandhu, H.; Kumar, R.N.; Garg, P. Machine learning-based modeling to predict inhibitors of acetylcholinesterase. Mol. Divers. 2022, 26, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Hossain, T.; Islam, M.A.; Pal, R.; Saha, A. Exploring structural requirement and binding interactions of β-amyloid cleavage enzyme inhibitors using molecular modeling techniques. Med. Chem. Res. 2013, 22, 4766–4774. [Google Scholar] [CrossRef]
- Chakraborty, S.; Ramachandran, B.; Basu, S. Encompassing receptor flexibility in virtual screening using ensemble docking-based hybrid QSAR: Discovery of novel phytochemicals for BACE1 inhibition. Mol. BioSyst. 2014, 10, 2684–2692. [Google Scholar] [CrossRef]
- Gupta, K. QSAR Studies on Gallic Acid Derivatives and Molecular Docking Studies of Bace1 Enzyme – A Potent Target of Alzheimer Disease. J. Biosci. Bioeng. 2014, 1, 11–27. [Google Scholar]
- Das, S.; Majumder, T.; Sarkar, A.; Mukherjee, P.; Basu, S. Flavonoids as BACE1 inhibitors: QSAR modelling, screening and in vitro evaluation. Int. J. Biol. Macromol. 2020, 165, 1323–1330. [Google Scholar] [CrossRef]
- Ambure, P.; Roy, K. Understanding the structural requirements of cyclic sulfone hydroxyethylamines as hBACE1 inhibitors against Aβ plaques in Alzheimer's disease: A predictive QSAR approach. RSC Adv. 2016, 6, 28171–28186. [Google Scholar] [CrossRef]
- Liu, J.; Ni, J.; Wang, X.; Bi, Y. 3D-QSAR Analysis and Molecular Docking Study on Biaryl Aminothiazine BACE1 Inhibitor. China Pharm. 2018, 12, 1335–1339. [Google Scholar]
- Joseph, O.A.; Babatomiwa, K.; Niyi, A.; Olaposi, O.; Olumide, I. Molecular Docking and 3D Qsar Studies of C000000956 as a Potent Inhibitor of Bace-1. Drug Res. 2019, 69, 451–457. [Google Scholar] [CrossRef]
- Aissouq, A.E.; Toufik, H.; Lamchouri, F.; Stitou, M.; Ouammou, A. QSAR study of isonicotinamides derivatives as Alzheimr's disease inhibitors using PLS-R and ANN methods. In Proceedings of the 2019 International Conference on Intelligent Systems and Advanced Computing Sciences (ISACS), Taza, Morocco, 26–27 December 2019; pp. 1–7. [Google Scholar]
- Chetia, P.; Mazumder, M.K.; Mahanta, S.; De, B.; Dutta Choudhury, M. A novel phytochemical from Dipteris wallichii inhibits human β-secretase 1: Implications for the treatment of Alzheimer’s disease. Med. Hypotheses 2020, 143, 109839. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Ganeshpurkar, A.; Ghosh, P.; Pokle, A.V.; Kumar, D.; Singh, R.b.; Singh, S.K.; Kumar, A. Classification of beta-site amyloid precursor protein cleaving enzyme 1 inhibitors by using machine learning methods. Chem. Biol. Drug Des. 2021, 98, 1079–1097. [Google Scholar] [CrossRef] [PubMed]
- Mukerjee, N.; Das, A.; Jawarkar, R.D.; Maitra, S.; Das, P.; Castrosanto, M.A.; Paul, S.; Samad, A.; Zaki, M.E.A.; Al-Hussain, S.A.; et al. Repurposing food molecules as a potential BACE1 inhibitor for Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 878276. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Accuracy | Precision | Sensitivity | F1-Score | MCC 1 | AUC | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Training | Test | Training | Test | Training | Test | Training | Test | Training | Test | Test | |
| AChE-CART | 0.85 | 0.83 | 0.66 | 0.64 | 0.80 | 0.77 | 0.72 | 0.70 | 0.62 | 0.59 | 0.83 ± 0.024 |
| AChE-CHAID | 0.86 | 0.85 | 0.71 | 0.68 | 0.78 | 0.76 | 0.74 | 0.72 | 0.65 | 0.61 | 0.85 ± 0.024 |
| AChE-RF | 0.87 | 0.86 | 0.71 | 0.69 | 0.84 | 0.81 | 0.77 | 0.75 | 0.68 | 0.66 | 0.89 ± 0.013 |
| BACE1-CART | 0.83 | 0.82 | 0.74 | 0.72 | 0.89 | 0.87 | 0.81 | 0.79 | 0.67 | 0.64 | 0.83 ± 0.024 |
| BACE1-CHAID | 0.82 | 0.80 | 0.74 | 0.76 | 0.77 | 0.73 | 0.77 | 0.75 | 0.62 | 0.59 | 0.83 ± 0.020 |
| BACE1-RF | 0.85 | 0.83 | 0.79 | 0.75 | 0.83 | 0.85 | 0.81 | 0.80 | 0.68 | 0.66 | 0.88 ± 0.016 |
| Models | Applicability Domain (AD) | Outliers of Test Set |
|---|---|---|
| AChE-CART | Di ≤ Dk + 0.5 × Sk = 0.3311 + 0.5 × 0.3447 = 0.5035 | 121/590 |
| AChE-CHAID | Di ≤ Dk + 0.5 × Sk = 1.1011 + 0.5 × 0.7766 = 1.4894 | 130/590 |
| AChE-RF | Di ≤ Dk + 0.5 × Sk = 5.5469 + 0.5 × 3.2381 = 7.1660 | 121/590 |
| BACE1-CART | Di ≤ Dk + 0.5 × Sk = 0.3826 + 0.5 × 0.4915 = 0.6284 | 43/464 |
| BACE1-CHAID | Di ≤ Dk + 0.5 × Sk = 0.5055 + 0.5 × 0.5208 = 0.7659 | 66/464 |
| BACE1-RF | Di ≤ Dk + 0.5 × Sk = 3.9731 + 0.5 × 2.3496 = 5.1479 | 95/464 |
| Descriptors ID | Description | Descriptor Family |
|---|---|---|
| C-006 | CH2RX | Atom-centered fragments |
| H-051 | H attached to alpha C | Atom-centered fragments |
| O-058 | =O | Atom-centered fragments |
| F09[C-N] | Frequency of C-N at topological distance 9 (order 9) | 2D atom pairs |
| Yindex | Balaban Y index | Information indices |
| SpMaxA_EA(ed) | Normalized leading eigenvalue from edge adjacency mat. weighed by edge degree | Edge adjacency indices |
| SM15_EA(dm) | Spectral moment of order 15 from edge adjacency matrix weighted by dipole moment | Edge adjacency indices |
| Eta_betaP_A | Eta pi and lone pair average VEM count | ETA indices |
| Descriptors ID | Description | Descriptor Family |
|---|---|---|
| nR10 | Number of 10-membered rings | Ring descriptors |
| nCIC | Number of rings (cyclomatic number) | Ring descriptors |
| IC1 | Information content index (neighborhood symmetry of 1 order) | Information indices |
| GGI9 | Topological charge index of order 9 | 2D autocorrelations |
| SM06_EA(ri) | Spectral moment of order 6 from edge adjacency matrix weighted by resonance integral | Edge adjacency indices |
| P_VSA_e_3 | P_VSA-like on Sanderson electronegativity, bin 3 | P_VSA-like descriptors |
| Molecule ChEMBL ID | Activity Rules | QSAR Prediction | Docking Scores (kCal/mol) | AChE IC50 (nM) | BACE1 IC50 (nM) | Reference 1 |
|---|---|---|---|---|---|---|
| CHEMBL3355580 | BACE1 Rule 3 | 2/3 | −16.12 | 18.3 | - | [30] |
| CHEMBL3600552 | BACE1 Rule 1 | 2/3 | −11.78 | 3.46 | - | [31] |
| CHEMBL3600553 | BACE1 Rule 1 | 2/3 | −7.93 | 6.46 | - | [32] |
| CHEMBL3600554 | BACE1 Rule 1 | 2/3 | −12.14 | 10.1 | - | [32] |
| CHEMBL3600555 | BACE1 Rule 1 | 2/3 | −10.12 | 1.48 | - | [30] |
| CHEMBL3600556 | BACE1 Rule 1 | 2/3 | −7.24 | 3.53 | - | [30] |
| ChEMBL3403874 | BACE1 Rule 1 | 2/3 | −6.92 | 6.9 | - | [33] |
| CHEMBL3632989 | BACE1 Rule 1 | 3/3 | −12.63 | 80.0 | - | [34] |
| CHEMBL440983 | BACE1 Rule 3 | 2/3 | −10.77 | 6.65 | - | [35] |
| CHEMBL238230 | BACE1 Rule 3 | 2/3 | −8.27 | 1.83 | - | [35] |
| CHEMBL226335 | BACE1 Rule 2 | 2/3 | −6.16 | 12.0 | - | [29] |
| CHEMBL195241 | BACE1 Rule 3 | 2/3 | −11.67 | 4.1 | - | [36] |
| CHEMBL179455 | BACE1 Rule 1 | 2/3 | −10.54 | 1.55 | - | [37] |
| CHEMBL3343885 | BACE1 Rule 3 | 2/3 | −7.97 | 92.6 | - | [38] |
| CHEMBL4286601 | BACE1 Rule 3 | 3/3 | −10.72 | 6.3 | - | [39] |
| CHEMBL3403878 | BACE1 Rule 1 | 2/3 | −9.11 | 32.5 | - | [33] |
| CHEMBL3403877 | BACE1 Rule 1 | 2/3 | −7.75 | 17.3 | - | [33] |
| CHEMBL1819176 | BACE1 Rule 1 | 2/3 | −11.98 | 1.05 | [40] | |
| CHEMBL1196204 | BACE1 Rule 1 | 2/3 | −8.32 | 19.3 | - | [41] |
| CHEMBL3343882 | BACE1 Rule 3 | 2/3 | −10.96 | 98.2 | - | [41] |
| CHEMBL4278287 | BACE1 Rule 3 | 3/3 | −11.23 | 38.0 | - | [39] |
| CHEMBL3400187 | BACE1 Rule 1 | 2/3 | −9.54 | 21.6 | - | [33] |
| CHEMBL4210729 | BACE1 Rule 3 | 2/3 | −9.97 | 41.9 | - | [42] |
| CHEMBL4213591 | BACE1 Rule 3 | 2/3 | −12.31 | 51.7 | - | [42] |
| CHEMBL4293418 | BACE1 Rule 1 | 3/3 | −15.31 | 3.6 | - | [39] |
| CHEMBL4282154 | BACE1 Rule 3 | 3/3 | −19.64 | 2.1 | - | [39] |
| CHEMBL4215154 | BACE1 Rule 1 | 2/3 | −12.65 | 89.6 | - | [42] |
| CHEMBL4217346 | BACE1 Rule 3 | 2/3 | −11.96 | 94.1 | - | [42] |
| CHEMBL4290039 | BACE1 Rule 3 | 3/3 | −10.27 | 22.0 | - | [39] |
| CHEMBL4215217 | BACE1 Rule 1 | 2/3 | −17.45 | 74.5 | - | [42] |
| CHEMBL4278686 | BACE1 Rule 3 | 3/3 | −14.42 | 6.4 | - | [39] |
| CHEMBL4285581 | BACE1 Rule 1 | 3/3 | −14.23 | 23.0 | - | [39] |
| CHEMBL255838 | AChE Rule 1 | 2/3 | −17.70 | - | 5.6 | [43] |
| CHEMBL2407494 | AChE Rule 3 | 2/3 | −14.39 | - | 76.0 | [44] |
| Cpd. ID | AChE Rule | AChE QSAR Prediction | AChE Docking Scores (kCal/mol) | BACE1 Rule | AChE QSAR Prediction | BACE1 Docking Scores (kCal/mol) |
|---|---|---|---|---|---|---|
| M02 | - | 2/3 | −14.84 | Rule 1 | 2/3 | −10.75 |
| M06 | - | 2/3 | −12.31 | Rule 1 | 3/3 | −10.92 |
| M07 | Rule 4 | 2/3 | −15.54 | Rule 1 | 2/3 | −14.89 |
| M09 | - | 3/3 | −14.70 | Rule 2 | 2/3 | −11.80 |
| M13 | - | 3/3 | −18.30 | Rule 3 | 3/3 | −15.82 |
| M14 | Rule 4 | 3/3 | −19.30 | - | 2/3 | −16.38 |
| M17 | Rule 4 | 2/3 | −21.20 | - | 3/3 | −12.33 |
| M18 | Rule 4 | 3/3 | −18.71 | Rule 1 | 3/3 | −16.14 |
| M19 | Rule 4 | 3/3 | −16.90 | - | 2/3 | −15.32 |
| M27 | Rule 3 | 3/3 | −19.43 | Rule 1 | 3/3 | −16.11 |
| M30 | Rule 3 | 3/3 | −17.42 | Rule 1 | 3/3 | −14.75 |
| M96 | - | 3/3 | −18.80 | Rule 2 | 3/3 | −17.03 |
| M97 | - | 3/3 | −18.50 | Rule 2 | 3/3 | −16.52 |
| Year | Methods | Molecular Descriptors | Database | QSAR Model Performance | References |
|---|---|---|---|---|---|
| 2014 |
| 705 2D descriptors by vLifeMDS software | 20 1,4-dihydropyridine (DHP) derivatives | Best models:
| Goyal et al. [12] |
| 2020 |
| 2D molecular descriptors by MOE 2008.10 | 72 AChE and 215 BACE1 inhibitors (varied structures) | AChE models:
| Tran et al. [57] |
| 2022 |
| Tanimoto index (TI) calculated by OpenBabel using FP2 fingerprints |
- 3 AChE active molecule lists included 195–428 compounds - 4 BACE1 active molecule lists included 194–1317 compounds | 8 AChE models:
| Stern et al. [14] |
| 2022 | Regression algorithms:
| 2D descriptors: spatial, structural, thermodynamics, electro-topological and E-state indices | 57 AChE and 53 BACE1 inhibitors (varied structures) | AChE models:
| Dhamodharan and Mohan [13] |
| 2023 |
| 1100 and 1151 0-2D descriptors calculated using Dragon 6.0 | ChEMBL databases including 1975 AChE inhibitors and 1549 BACE1 inhibitors | AChE models:
| Current study |
| Active Inhibitor (Predicted) | Inactive Inhibitor (Predicted) | Total (Experimental) | |
|---|---|---|---|
| Active inhibitor (Experimental) | Tp | Fn | Tp + Fn (TPE) |
| Inactive inhibitor (Experimental) | Fp | Tn | Fp + Tn (TNE) |
| Total (Predicted) | Tp + Fp (TPP) | Fn + Tn (TNP) | TPE + TNE = TPP + TNP |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, L.-Q.; Baecker, D.; Mai Dung, D.T.; Phuong Nhung, N.; Thi Thuan, N.; Nguyen, P.L.; Phuong Dung, P.T.; Huong, T.T.L.; Rasulev, B.; Casanola-Martin, G.M.; et al. Development of Activity Rules and Chemical Fragment Design for In Silico Discovery of AChE and BACE1 Dual Inhibitors against Alzheimer’s Disease. Molecules 2023, 28, 3588. https://doi.org/10.3390/molecules28083588
Bao L-Q, Baecker D, Mai Dung DT, Phuong Nhung N, Thi Thuan N, Nguyen PL, Phuong Dung PT, Huong TTL, Rasulev B, Casanola-Martin GM, et al. Development of Activity Rules and Chemical Fragment Design for In Silico Discovery of AChE and BACE1 Dual Inhibitors against Alzheimer’s Disease. Molecules. 2023; 28(8):3588. https://doi.org/10.3390/molecules28083588
Chicago/Turabian StyleBao, Le-Quang, Daniel Baecker, Do Thi Mai Dung, Nguyen Phuong Nhung, Nguyen Thi Thuan, Phuong Linh Nguyen, Phan Thi Phuong Dung, Tran Thi Lan Huong, Bakhtiyor Rasulev, Gerardo M. Casanola-Martin, and et al. 2023. "Development of Activity Rules and Chemical Fragment Design for In Silico Discovery of AChE and BACE1 Dual Inhibitors against Alzheimer’s Disease" Molecules 28, no. 8: 3588. https://doi.org/10.3390/molecules28083588
APA StyleBao, L.-Q., Baecker, D., Mai Dung, D. T., Phuong Nhung, N., Thi Thuan, N., Nguyen, P. L., Phuong Dung, P. T., Huong, T. T. L., Rasulev, B., Casanola-Martin, G. M., Nam, N.-H., & Pham-The, H. (2023). Development of Activity Rules and Chemical Fragment Design for In Silico Discovery of AChE and BACE1 Dual Inhibitors against Alzheimer’s Disease. Molecules, 28(8), 3588. https://doi.org/10.3390/molecules28083588