
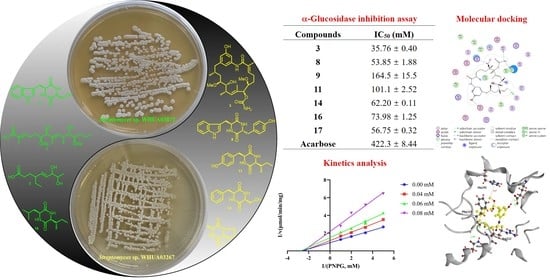
α-Glucosidase Inhibitors from Two Mangrove-Derived Actinomycetes
 ,
,  ,
, 


Abstract
:
1. Introduction
2. Results and Discussion
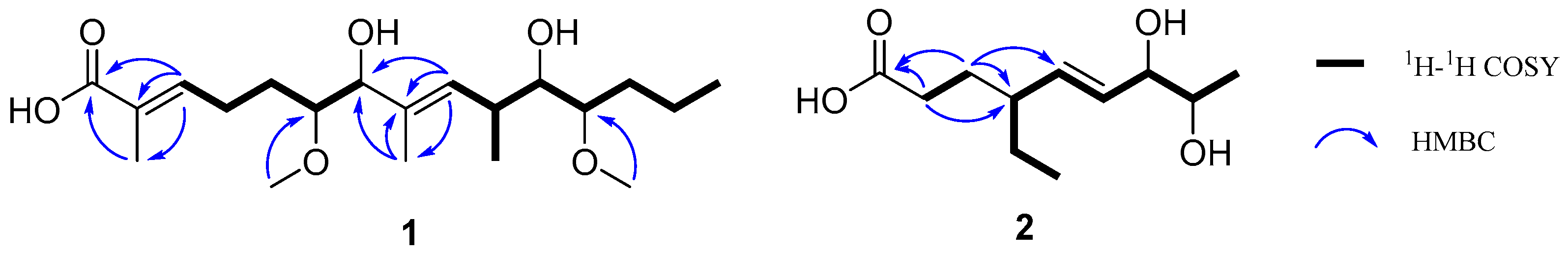
2.1. Structure Elucidation
2.2. AGS Inhibitory Activity
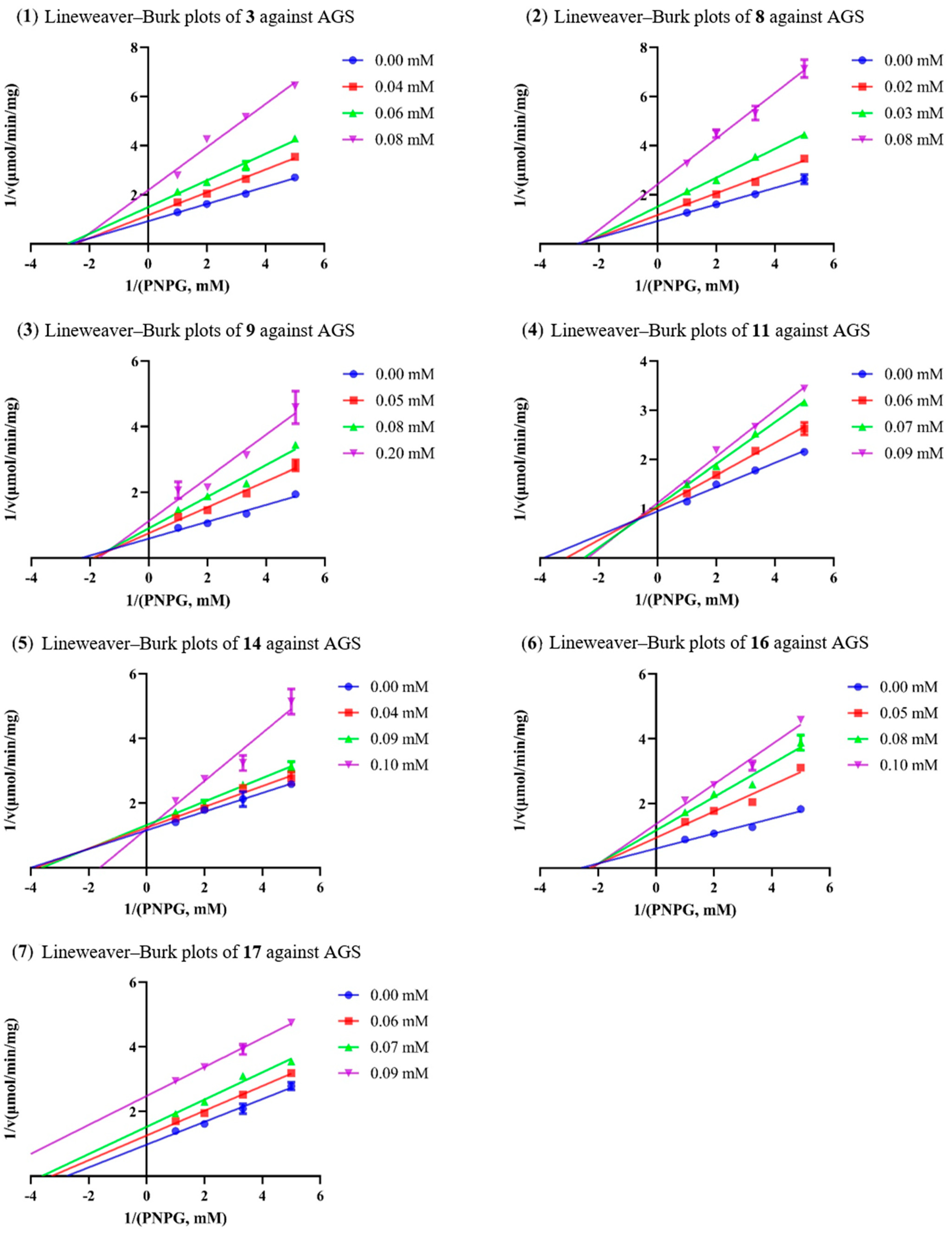
2.3. Analysis of Inhibition Kinetics
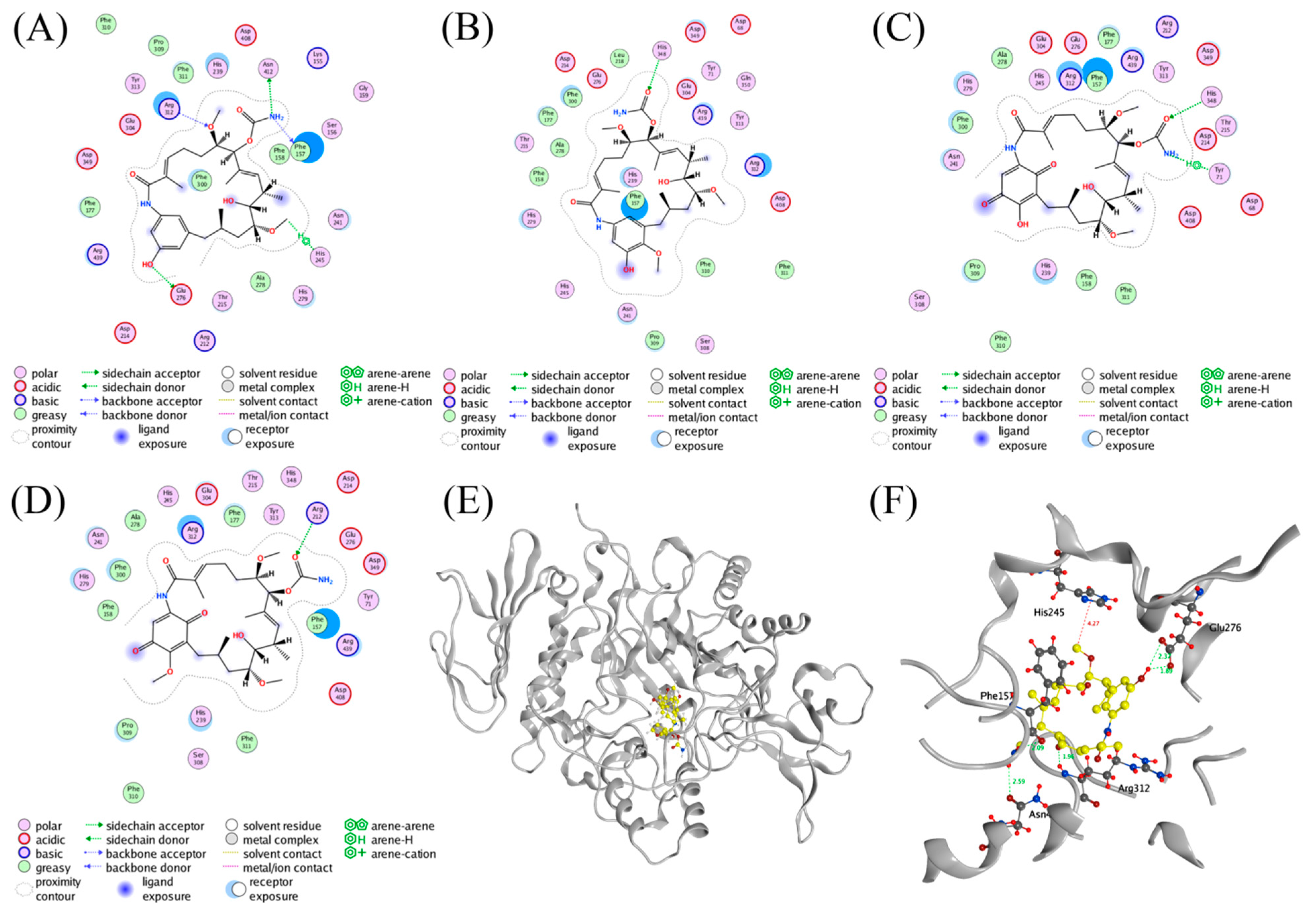
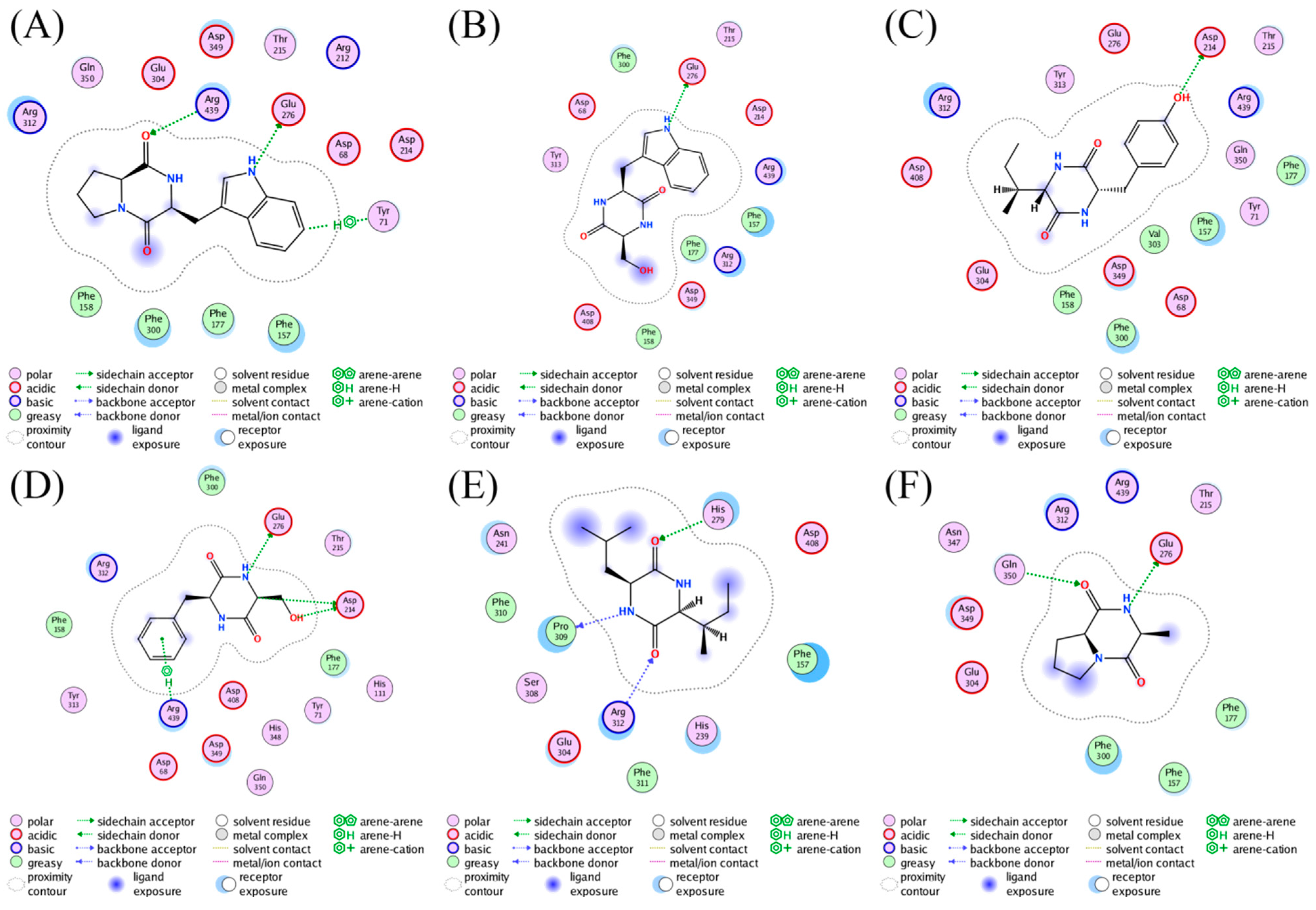
2.4. Molecular Docking Studies
2.5. In Vitro Cytotoxicity Assay
3. Materials and Methods
3.1. General Experimental Procedure
3.2. Microorganisms
3.3. Fermentation, Extraction, and Isolation
3.4. AGS Inhibition Assay
3.5. Inhibition Kinetics Analysis
3.6. Molecular Docking
3.7. Cell Culture and Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. IDF diabetes atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Santos, C.M.M.; Freitas, M.; Fernandes, E. A comprehensive review on xanthone derivatives as α-glucosidase inhibitors. Eur. J. Med. Chem. 2018, 157, 1460–1479. [Google Scholar] [CrossRef] [PubMed]
- Ahrén, B. DPP-4 inhibitors. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 517–533. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. What is diabetes. In IDF Diabetes Atlas, 6th ed.; International Diabetes Federation: Brussels, Belgium, 2013; pp. 19–27. [Google Scholar]
- Krentz, A.J.; Bailey, C.J. Oral antidiabetic agents: Current role in type 2 diabetes mellitus. Drugs 2005, 65, 385–411. [Google Scholar] [CrossRef]
- Tseng, P.S.; Ande, C.; Moremen, K.W.; Chrich, D. Influence of side chain conformation on the activity of glycosidase inhibitors. Angew. Chem. Int. Ed. 2023, 135, e202217809. [Google Scholar] [CrossRef]
- Rajasekaran, P.; Ande, C.; Vankar, Y.D. Synthesis of (5,6 & 6,6)-oxa-oxa annulated sugars as glycosidase inhibitors from 2-formyl galactal using iodocyclization as a key step. Arkivoc 2022, 2022, 5–23. [Google Scholar] [CrossRef]
- Ande, C.; Bhowmick, S.; Vankar, Y.D. Conversion of glycals into vicinal-1,2-diazides and 1,2-(or 2,1)-azidoacetates using hypervalent iodine reagents and Me3SiN3. Application in the synthesis of N-glycopeptides, pseudo-trisaccharides and an iminosugar. RSC Adv. 2017, 7, 41755–41762. [Google Scholar] [CrossRef]
- Yin, Z.H.; Zhang, W.; Feng, F.J.; Zhang, Y.; Kang, W.Y. α-Glucosidase inhibitors isolated from medicinal plants. Food Sci. Hum. Wellness 2014, 3, 136–174. [Google Scholar] [CrossRef]
- Şöhretoğlu, D.; Sari, S. Flavonoids as alpha-glucosidase inhibitors: Mechanistic approaches merged with enzyme kinetics and molecular modelling. Phytochem. Rev. 2020, 19, 1081–1092. [Google Scholar] [CrossRef]
- Wang, J.M.; Lu, S.L.; Sheng, R.L.; Fan, J.T.; Wu, W.H.; Guo, R.H. Structure-activity relationships of natural and synthetic indole-derived scaffolds as α-glucosidase inhibitors: A mini-review. Mini Rev. Med. Chem. 2020, 20, 1791–1818. [Google Scholar] [CrossRef]
- Blahova, J.; Martiniakova, M.; Babikava, M.; Kovacova, V.; Mondockova, V.; Omelka, R. Pharmaceutical drugs and natural therapeutic products for the treatment of type 2 diabetes mellitus. Pharmaceuticals 2021, 14, 806. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.; Nesi, I.; Caselli, A.; Paoli, P. Natural α-glucosidase and protein tyrosine phosphatase 1b inhibitors: A source of scaffold molecules for synthesis of new multitarget antidiabetic drugs. Molecules 2021, 26, 4818. [Google Scholar] [CrossRef]
- Dirir, A.M.; Daou, M.; Yousef, A.F.; Yousef, L.F. A review of alpha-glucosidase inhibitors from plants as potential candidates for the treatment of type-2 diabetes. Phytochem. Rev. 2021, 16, 1049–1079. [Google Scholar] [CrossRef] [PubMed]
- Ghani, U. Re-exploring promising α-glucosidase inhibitors for potential development into oral anti-diabetic drugs: Finding needle in the haystack. Eur. J. Med. Chem. 2015, 103, 133–162. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.; Nazir, M.; Saleem, M.; Al-Harrasi, A.; Elizabit; Green, I.R. Fruitful decade of fungal metabolites as anti-diabetic agents from 2010 to 2019: Emphasis on α-glucosidase inhibitors. Phytochem. Rev. 2021, 20, 145–179. [Google Scholar] [CrossRef]
- Zhou, X.F.; Chen, S.Q.; Pang, X.Y.; Cai, J.; Zhang, X.Y.; Liu, Y.H.; Zhu, Y.G.; Zhou, X.F. Natural products from mangrove sediments-derived microbes: Structural diversity, bioactivities, biosynthesis, and total synthesis. Eur. J. Med. Chem. 2022, 230, 114117. [Google Scholar] [CrossRef]
- Chen, S.H.; Cai, R.L.; Liu, Z.M.; Cui, H.; She, Z.G. Secondary metabolites from mangrove-associated fungi: Source, chemistry and bioactivities. Nat. Prod. Rep. 2022, 39, 560–595. [Google Scholar] [CrossRef]
- Ancheeva, E.; Daletos, G.; Proksch, P. Lead compounds from mangrove-associated microorganisms. Mar. Drugs 2018, 16, 319. [Google Scholar] [CrossRef]
- Xu, J. Bioactive natural products derived from mangrove-associated microbes. RSC Adv. 2015, 5, 841–892. [Google Scholar] [CrossRef]
- Ying, Y.M.; Yu, H.F.; Tong, C.P.; Shan, W.G.; Zhan, Z.J. Spiroinonotsuoxotriols A and B, two highly rearranged triterpenoids from Inonotus obliquus. Org. Lett. 2020, 22, 3377–3380. [Google Scholar] [CrossRef]
- Ma, L.F.; Yan, J.J.; Lane, H.Y.; Jin, L.C.; Qiu, F.J.; Wang, Y.J.; Xi, Z.F.; Shan, W.G.; Zhan, Z.J.; Ying, Y.M. Bioassay-guided isolation of lanostane-type triterpenoids as alpha-glucosidase inhibitors from Ganoderma hainanense. Phytochem. Lett. 2019, 29, 154–159. [Google Scholar] [CrossRef]
- Ying, Y.M.; Fang, C.A.; Yao, J.Y.; Yu, Y.; Shen, Y.; Hou, Z.N.; Wang, Z.; Zhang, W.; Shan, W.G.; Zhan, Z.J. Bergamotane sesquiterpenes with alpha-glucosidase inhibitory activity from the plant pathogenic fungus Penicillium expansum. Chem. Biodivers. 2017, 14, e1600184. [Google Scholar] [CrossRef]
- Ying, Y.M.; Zhang, L.Y.; Zhang, X.; Bai, H.B.; Liang, D.E.; Ma, L.F.; Shan, W.G.; Zhan, Z.J. Terpenoids with alpha-glucosidase inhibitory activity from the submerged culture of Inonotus obliquus. Phytochemistry 2014, 108, 171–176. [Google Scholar] [CrossRef]
- Yu, H.F.; Cheng, Y.C.; Wu, C.M.; Ran, K.; Wei, B.; Xu, Y.K.; Shan, W.G.; Ying, Y.M. Diverse diterpenoids with α-glucosidase and β-glucuronidase inhibitory activities from Euphorbia milii. Phytochemistry 2022, 196, 113106. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.M.; Yu, H.F.; Rao, G.W.; Wang, J.W.; Shan, W.G.; Zhan, Z.J. Dibenzocyclooctadiene lignans from the stems of Schisandra sphaerandra. Nat. Prod. Res. 2020, 36, 287–294. [Google Scholar] [CrossRef]
- Rao, G.W.; Yu, H.F.; Zhang, M.L.; Cheng, Y.C.; Ran, K.; Wang, J.W.; Wei, B.; Li, M.; Shan, W.G.; Zhan, Z.J.; et al. α-Glucosidase and bacterial β-glucuronidase inhibitors from the stems of Schisandra sphaerandra staph. Pharmaceuticals 2022, 15, 329. [Google Scholar] [CrossRef] [PubMed]
- Wrona, I.E.; Gozman, A.; Taldone, T.; Chiosis, G.; Panek, J.S. Synthesis of reblastatin, autolytimycin, and non-benzoquinone analogues: Potent inhibitors of heat shock protein 90. J. Org. Chem. 2010, 75, 2820–2835. [Google Scholar] [CrossRef]
- Wang, C.; Wang, L.; Fan, J.; Sun, K.; Zhu, W. Cytotoxic compounds from the deep-sea sediment-derived Streptomyces malaysiensis OUCMDZ-2167. Chin. J. Org. Chem. 2017, 37, 658–666. [Google Scholar] [CrossRef]
- Caballero, E.; Avendano, C.; Menendez, J.C. Brief total synthesis of the cell cycle inhibitor tryprostatin B and related preparation of its alanine analogue. J. Org. Chem. 2003, 68, 6944–6951. [Google Scholar] [CrossRef]
- Ivanova, V.; Laatsch, H.; Kolarova, M.; Aleksieva, K. Structure elucidation of a new natural diketopiperazine from a Microbispora aerata strain isolated from Livingston Island, Antarctica. Nat. Prod. Res. 2013, 27, 164–170. [Google Scholar] [CrossRef]
- Yan, Y.M.; Zhu, H.J.; Xiang, B.; Qi, J.J.; Wang, X.L.; Geng, F.N.; Cheng, Y.X. Chemical constituents from Periplaneta Americana and their effects on wound healing. Nat. Prod. Res. Dev. 2018, 30, 591–596. [Google Scholar] [CrossRef]
- Mehnaz, S.; Saleem, R.S.; Yameen, B.; Pianet, I.; Schnakenburg, G.; Pietraszkiewicz, H.; Valeriote, F.; Josten, M.; Sahl, H.G.; Franzblau, S.G.; et al. Lahorenoic acids A-C, ortho-dialkyl-substituted aromatic acids from the biocontrol strain Pseudomonas aurantiaca PB-St2. J. Nat. Prod. 2013, 76, 135–141. [Google Scholar] [CrossRef]
- Ding, G.Z.; Liu, Y.B.; Ma, S.G.; Yu, S.S. Metabolites of Aspergillus fumigatus. China J. Chin. Mater. Med. 2012, 37, 3083–3085. [Google Scholar]
- Stark, T.; Hofmann, T. Structures, sensory activity, and dose/response functions of 2,5-diketopiperazines in roasted cocoa nibs (Theobroma cacao). J. Agric. Food Chem. 2005, 53, 7222–7231. [Google Scholar] [CrossRef] [PubMed]
- Furtado, N.; Pupo, M.T.; Ivone, C.; Campo, V.L.; Duarte, M.; Bastos, J.K. Diketopiperazines produced by an Aspergillus fumigatus Brazilian strain. J. Braz. Chem. Soc. 2005, 16, 1448–1453. [Google Scholar] [CrossRef]
- Isaka, M.; Palasarn, S.; Rachtawee, P.; Vimuttipong, S.; Kongsaeree, P. Unique diketopiperazine dimers from the insect pathogenic fungus Verticillium hemipterigenum BCC 1449. Org. Lett. 2005, 7, 2257–2260. [Google Scholar] [CrossRef] [PubMed]
- Laville, R.; Nguyen, T.B.; Moriou, C.; Petek, S.; Debitus, C.; Al-Mourabit, A. Marine natural occurring 2,5-diketopiperazines: Isolation, synthesis and optical properties. Heterocycles 2015, 90, 1351–1366. [Google Scholar] [CrossRef]
- Selvakumar, S.; Sivasankaran, D.; Singh, V.K. Enantioselective Henry reaction catalyzed by C2-symmetric chiral diamine–copper (II) complex. Org. Biomol. Chem. 2009, 7, 3156–3162. [Google Scholar] [CrossRef]
- Campbell, J.; Lin, Q.; Geske, G.D.; Blackwell, H.E. New and unexpected insights into the modulation of LuxR-type quorum sensing by cyclic dipeptides. ACS Chem. Biol. 2009, 4, 1051–1059. [Google Scholar] [CrossRef]
- Zheng, D.; Han, L.; Jiang, Y.; Cao, Y.R.; Liu, J.; Chen, X.; Li, Y.Q.; Huang, X.S. Structure elucidation of four prenylindole derivatives from Streptomyces sp. isolated from Ailuropoda melanoleuca feces. Magn. Reson. Chem. 2013, 51, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Lee, D.; Park, M.; Lee, J.C.; Park, H.J.; Kang, K.S.; Kim, C.E.; Beemelmanns, C.; Kim, K.H. Absolute configuration and corrected NMR assignment of 17-hydroxycyclooctatin, a fused 5-8-5 tricyclic diterpene. J. Nat. Prod. 2020, 83, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.S.; Li, S.R.; Wang, Y.Y.; Hao, H.L.; Shen, Y.M.; Lu, C.H. 16,17-Dihydroxycyclooctatin, a new diterpene from Streptomyces sp. LZ35. Drug. Discov. Ther. 2013, 7, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.Y.S.; Graziani, E.; Waters, B.; Pan, W.; Li, X.; McDermott, J.; Meurer, G.; Saxena, G.; Andersen, R.J.; Davies, J. Novel natural products from soil DNA libraries in a streptomycete host. Org. Lett. 2000, 2, 2401–2404. [Google Scholar] [CrossRef]
- Matsubara, K.; Sakuda, S.; Kondo, T.; Tanaka, M.; Nishimura, T.; Suzuki, A. Morphological changes in insect BM-N4 cells induced by nocardamine. Biosci. Biotechnol. Biochem. 1998, 62, 2049–2051. [Google Scholar] [CrossRef]
- Tao, M.H.; Chen, Y.C.; Wei, X.Y.; Tan, J.W.; Zhang, W.M. Chemical constituents of the endophytic fungus Phomopsis sp. A240 isolated from Taxus chinensis var. mairei. Helv. Chim. Acta 2014, 97, 426–430. [Google Scholar] [CrossRef]
- Conti, R.; Chagas, F.O.; Caraballo-Rodriguez, A.M.; Melo, W.G.; do Nascimento, A.M.; Cavalcanti, B.C.; de Moraes, M.O.; Pessoa, C.; Costa-Lotufo, L.V.; Krogh, R.; et al. Endophytic actinobacteria from the Brazilian medicinal plant Lychnophora ericoides mart. and the biological potential of their secondary metabolites. Chem. Biodivers. 2016, 13, 727–736. [Google Scholar] [CrossRef]
- Wischang, D.; Hartung, J. Parameters for bromination of pyrroles in bromoperoxidase-catalyzed oxidations. Tetrahedron 2011, 67, 4048–4054. [Google Scholar] [CrossRef]
- Dietera, A.; Hamm, A.; Fiedler, H.P.; Goodfellow, M.; Muller, W.E.; Brun, R.; Beil, W.; Bringmann, G. Pyrocoll, an antibiotic, antiparasitic and antitumor compound produced by a novel alkaliphilic Streptomyces strain. J. Antibiot. 2003, 56, 639–646. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Oya, A.; Kudo, T.; Hirota, A. Surugapyrone A from Streptomyces coelicoflavus strain USF-6280 as a new DPPH radical-scavenger. J. Antibiot. 2010, 63, 365–369. [Google Scholar] [CrossRef]
- Kim, C.G.; Kirschning, A.; Bergon, P.; Zhou, P.; Su, E.; Sauerbrei, B.; Ning, S.; Ahn, Y.; Breuer, M.; Leistner, E.; et al. Biosynthesis of 3-amino-5-hydroxybenzoic acid, the precursor of mC7N units in ansamycin antibiotics. J. Am. Chem. Soc. 1996, 118, 7486–7491. [Google Scholar] [CrossRef]
- Yin, M.; Lu, T.; Zhao, L.X.; Chen, Y.H.; Huang, S.X.; Lohman, J.R.; Xu, L.H.; Jiang, C.L.; Shen, B. The missing C-17 O-methyltransferase in geldanamycin biosynthesis. Org. Lett. 2011, 13, 3726–3729. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.F.; Ramos, A.; Liras, P. Regulation of geldanamycin biosynthesis by cluster-situated transcription factors and the master regulator PhoP. Antibiotics 2019, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Skrzypczak, N.; Przybylski, P. Structural diversity and biological relevance of benzenoid and atypical ansamycins and their congeners. Nat. Prod. Rep. 2022, 39, 1678–1704. [Google Scholar] [CrossRef]
- Mishra, A.K.; Choi, J.; Choi, S.J.; Baek, K.H. Cyclodipeptides: An overview of their biosynthesis and biological activity. Molecules 2017, 22, 1796. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.M.; Liang, X.A.; Kong, Y.; Jia, B. Structural diversity and biological activities of indole diketopiperazine alkaloids from fungi. J. Argic. Food Chem. 2016, 64, 6659–6671. [Google Scholar] [CrossRef]
- Jhong, C.H.; Riyaphan, J.; Lin, S.H.; Chia, Y.C.; Weng, C.F. Screening alpha-glucosidase and alpha-amylase inhibitors from natural compounds by molecular docking in silico. Biofactors 2015, 41, 242–251. [Google Scholar] [CrossRef]
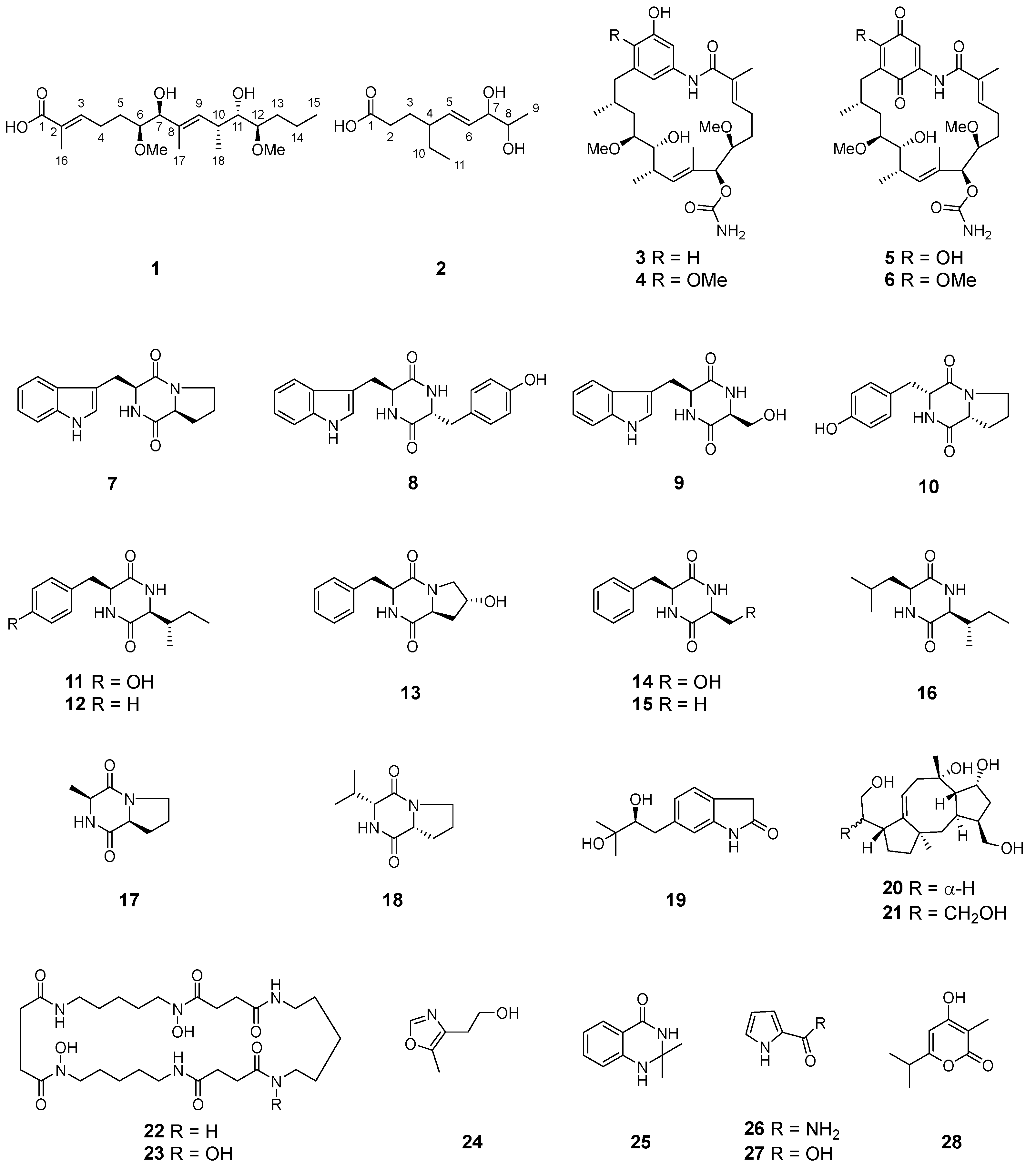

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC, Type | δH (Multi, J in Hz) | δC, Type | δH (Multi, J in Hz) | |
| 1 | 171.3, C | - | 178.5, C | |
| 2 | 129.6, C | - | 33.4, CH2 | 2.25 (m) |
| 3 | 142.9, CH | 6.77 (t, 7.2) | 31.2, CH2 | 1.76 (m) 1.50 (m) |
| 4 | 25.4, CH2 | 2.32 (m) | 45.4, CH | 1.94 (m) |
| 5 | 30.9, CH2 | 1.53 (m) 1.45 (m) | 137.8, CH | 5.46 (dd, 15.5, 8.0) |
| 6 | 83.5, CH | 3.29 (td, 7.8, 1.8) | 131.8, CH | 5.48 (dd, 15.5, 6.3) |
| 7 | 81.0, CH | 3.93 (d, 7.2) | 78.3, CH | 3.81 (dd, 6.3, 6.3) |
| 8 | 135.4, C | - | 71.8, CH | 3.60 (m) |
| 9 | 132.5, CH | 5.33 (d, 10.2) | 19.0, CH3 | 1.12 (d, 6.3) |
| 10 | 36.0, CH | 2.52 (m) | 29.1, CH2 | 1.50 (m) 1.30 (m) |
| 11 | 75.5, CH | 3.55 (dd, 9.0, 3.6) | 12.1, CH3 | 0.90 (t, 7.2) |
| 12 | 84.0, CH | 3.08 (dt, 9.0, 3.0) | - | - |
| 13 | 31.6, CH2 | 1.52 (m) 1.36 (m) | - | - |
| 14 | 20.3, CH2 | 1.55 (m) 1.28 (m) | - | - |
| 15 | 14.8, CH3 | 0.93 (t, 7.2) | - | - |
| 16 | 12.6, CH3 | 1.84 (s) | - | - |
| 17 | 12.2, CH3 | 1.66 (s) | - | - |
| 18 | 17.5, CH3 | 1.07 (d, 6.6) | - | - |
| 6-OMe | 59.1, CH3 | 3.49 (s) | - | - |
| 12-OMe | 57.3, CH3 | 3.34 (s) | - | - |
| Compounds | IC50 (μM) a | Compounds | IC50 (μM) a |
|---|---|---|---|
| 1 | NA b | 15 | NA b |
| 2 | NA b | 16 | 73.98 ± 1.25 |
| 3 | 35.76 ± 0.40 | 17 | 56.75 ± 0.32 |
| 4 | NA b | 18 | NA b |
| 5 | NA b | 19 | NA b |
| 6 | NA b | 20 | NA b |
| 7 | NA b | 21 | NA b |
| 8 | 53.85 ± 1.88 | 22 | NA b |
| 9 | 164.5 ± 15.5 | 23 | NA b |
| 10 | NA b | 24 | NA b |
| 11 | 101.1 ± 2.52 | 25 | NA b |
| 12 | NA b | 26 | NA b |
| 13 | NA b | 27 | NA b |
| 14 | 62.20 ± 0.11 | 28 | NA b |
| Acarbose | 422.3 ± 8.44 |
| Compounds | Concentration (mM) | Km (mM) | Vmax (μM/min) | Ki (μM) | Ki’ (μM) |
|---|---|---|---|---|---|
| 3 | 0.00 | 0.38 ± 0.02 | 1.08 ± 0.03 | 49.18 | 53.10 |
| 0.04 | 0.40 ± 0.03 | 0.86 ± 0.03 | |||
| 0.06 | 0.36 ± 0.04 | 0.67 ± 0.04 | |||
| 0.08 | 0.40 ± 0.05 | 0.46 ± 0.03 | |||
| 8 | 0.00 | 0.36 ± 0.04 | 1.08 ± 0.07 | 43.89 | 46.28 |
| 0.02 | 0.38 ± 0.04 | 0.85 ± 0.05 | |||
| 0.03 | 0.39 ± 0.03 | 0.66 ± 0.03 | |||
| 0.08 | 0.38 ± 0.05 | 0.41 ± 0.03 | |||
| 9 | 0.00 | 0.44 ± 0.07 | 1.70 ± 0.16 | 148.94 | 238.78 |
| 0.05 | 0.53 ± 0.11 | 1.33 ± 0.17 | |||
| 0.08 | 0.53 ± 0.11 | 1.11 ± 0.14 | |||
| 0.20 | 0.59 ± 0.22 | 0.89 ± 0.22 | |||
| 11 | 0.00 | 0.26 ± 0.02 | 1.05 ± 0.04 | 95.52 | 531.53 |
| 0.06 | 0.32 ± 0.03 | 0.98 ± 0.05 | |||
| 0.07 | 0.40 ± 0.02 | 0.94 ± 0.02 | |||
| 0.09 | 0.42 ± 0.04 | 0.90 ± 0.05 | |||
| 14 | 0.00 | 0.25 ± 0.04 | 0.86 ± 0.06 | NC a | - |
| 0.04 | 0.26 ± 0.04 | 0.81 ± 0.06 | |||
| 0.09 | 0.28 ± 0.03 | 0.76 ± 0.04 | |||
| 0.10 | 0.62 ± 0.21 | 0.83 ± 0.18 | |||
| 16 | 0.00 | 0.38 ± 0.06 | 1.66 ± 0.15 | 59.75 | 78.89 |
| 0.05 | 0.43 ± 0.09 | 1.07 ± 0.13 | |||
| 0.08 | 0.44 ± 0.10 | 0.86 ± 0.11 | |||
| 0.10 | 0.45 ± 0.07 | 0.74 ± 0.07 | |||
| 17 | 0.00 | 0.36 ± 0.05 | 1.03 ± 0.08 | NC a | NC a |
| 0.06 | 0.30 ± 0.02 | 0.79 ± 0.02 | |||
| 0.07 | 0.28 ± 0.03 | 0.66 ± 0.03 | |||
| 0.09 | 0.25 ± 0.01 | 0.47 ± 0.01 |
| Compounds | IC50 (μM) |
|---|---|
| 3 | >100 |
| 8 | >100 |
| 9 | >100 |
| 11 | >100 |
| 14 | >100 |
| 16 | >100 |
| 17 | >100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, X.; Zhang, M.; Qiu, Y.; Liu, X.; Wang, C.; Chen, J.; Zhang, H.; Wei, B.; Yu, Y.; Ying, Y.; et al. α-Glucosidase Inhibitors from Two Mangrove-Derived Actinomycetes. Molecules 2023, 28, 3822. https://doi.org/10.3390/molecules28093822
Lu X, Zhang M, Qiu Y, Liu X, Wang C, Chen J, Zhang H, Wei B, Yu Y, Ying Y, et al. α-Glucosidase Inhibitors from Two Mangrove-Derived Actinomycetes. Molecules. 2023; 28(9):3822. https://doi.org/10.3390/molecules28093822
Chicago/Turabian StyleLu, Xuejun, Manlai Zhang, Yixian Qiu, Xiuxiu Liu, Cancan Wang, Jianwei Chen, Huawei Zhang, Bin Wei, Yanlei Yu, Youmin Ying, and et al. 2023. "α-Glucosidase Inhibitors from Two Mangrove-Derived Actinomycetes" Molecules 28, no. 9: 3822. https://doi.org/10.3390/molecules28093822
APA StyleLu, X., Zhang, M., Qiu, Y., Liu, X., Wang, C., Chen, J., Zhang, H., Wei, B., Yu, Y., Ying, Y., Hong, K., & Wang, H. (2023). α-Glucosidase Inhibitors from Two Mangrove-Derived Actinomycetes. Molecules, 28(9), 3822. https://doi.org/10.3390/molecules28093822