


An Enantiospecific Synthesis of 5-epi-α-Bulnesene


Abstract
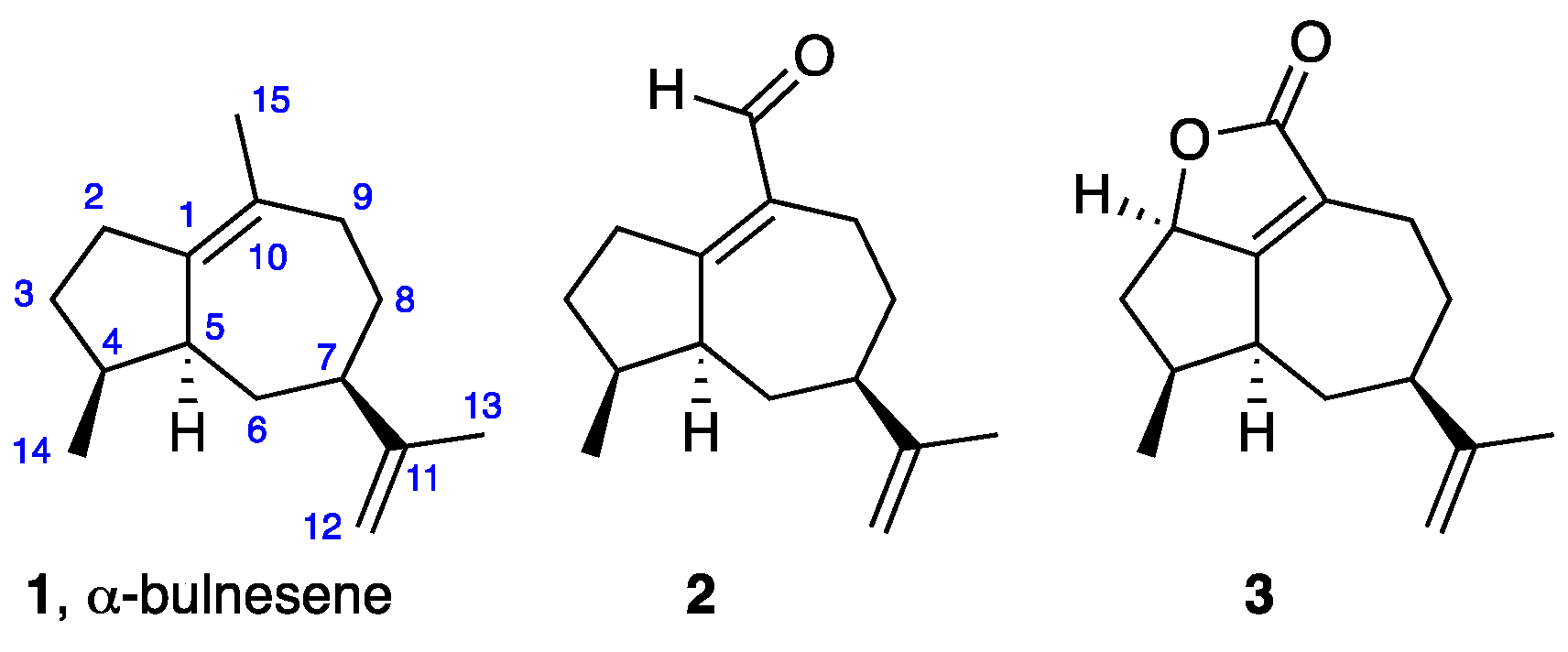
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. (2RS,5R)-2-Methyl-5-(prop-1-en-2-yl)cycloheptan-1-one (7)
3.3. (5R)-2-Methyl-5-(prop-1-en-2-yl)cyclohept-1-en-1-yl trifluoromethanesulfonate (9)
3.4. (5R)-1-(But-3-yn-1-yl)-2-methyl-5-(prop-1-en-2-yl)cyclohept-1-ene (10)
3.5. (5R)-1-(3-Iodobut-3-en-1-yl)-2-methyl-5-(prop-1-en-2-yl)cyclohept-1-ene (3)
3.6. (6R)-3-Methyl-6-(prop-1-en-2-yl)cyclohept-2-en-1-one (13)
3.7. (6R)-9-Methyl-6-(prop-1-en-2-yl)-1-vinylspiro[2.6]non-8-en-4-one (15)
3.8. (2RS,6R)-2-(But-3-en-1-yl)-3-methyl-6-(prop-1-en-2-yl)cyclohept-3-en-1-one (18)
3.9. (6R)-2-(But-3-en-1-yl)-3-methyl-6-(prop-1-en-2-yl)cyclohept-2-en-1-one (19)
3.10. (1RS,6R)-2-(But-3-en-1-yl)-3-methyl-6-(prop-1-en-2-yl)cyclohept-2-en-1-ol (12)
3.11. (3S,3aR,5R,8aR)-3,8-Dimethyl-5-(prop-1-en-2-yl)-2,3,4,5,6,8a-hexahydroazulen-3a(1H)-ol (24); (3S,3aR,5R,8aS)-3,8-dimethyl-5-(prop-1-en-2-yl)-2,3,4,5,6,8a-hexahydroazulen-3a(1H)-ol (25)
3.12. (3S,3aR,5R)-3,8-Dimethyl-5-(prop-1-en-2-yl)-1,2,3,3a,4,5,6,7-octahydroazulene [5-epi-α-bulnesene] (26)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Mohamed, R. (Ed.) Agarwood—Science Behind the Fragrance; Springer: Singapore, 2016. [Google Scholar]
- Thompson, I.D.; Lim, T.; Turjaman, M. Expensive, Exploited and Endangered—A Review of the Agarwood-Producing Genera Aquilaria and Gyrinops: CITES Considerations, Trade Patterns, Conservation, and Management. In Proceedings of the Nineteenth Meeting of the Conference of the Parties, Panama City, Panama, 14–25 November 2022. [Google Scholar]
- Kynam: The King of Agarwood. Available online: https://interactive.aljazeera.com/aje/2016/oud-agarwood-scent-from-heaven/scent-from-heaven-the-king-of-agarwood.html (accessed on 17 April 2023).
- CITES. Checklist of CITES Species. Available online: checklist.cites.org (accessed on 17 April 2023).
- Antonopoulou, M.; Compton, J.; Perry, L.S.; Al-Mubarak, R. The Trade and Use of Agarwood (Oudh) in the United Arab Emirates; TRAFFIC Southeast Asia: Petaling Jaya, Malaysia, 2010. [Google Scholar]
- Wyn, L.T.; Anak, N.A. Wood for the Trees: A Review of the Agarwood (Gaharu) Trade in Malaysia; TRAFFIC Southeast Asia: Petaling Jaya, Malaysia, 2010. [Google Scholar]
- Alternative Investment Report: Forestry—2014. Available online: https://intelligent-partnership.com/AiR/reports/Forestry-Report-2014/files/assets/common/downloads/publication.pdf (accessed on 7 April 2023).
- Shivanand, P.; Arbie, N.F.; Krishnamoorthy, S.; Ahmad, N. Agarwood—The Fragrant Molecules of a Wounded Tree. Molecules 2022, 27, 3386. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hussain, M.; Jiang, Z.; Wang, Z.; Gao, J.; Ye, F.; Mao, R.; Li, H. Aquilaria Species (Thymelaeaceae) Distribution, Volatile and Non-Volatile Phytochemicals, Pharmacological Uses, Agarwood Grading System, and Induction Methods. Molecules 2021, 27, 7708. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Isa, N.M.; Ismail, I.; Zainal, Z. Agarwood Induction: Current Developments and Future Perspectives. Front. Plant Sci. 2019, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Azren, P.D.; Lee, S.Y.; Emang, D.; Mohamed, R. History and Perspectives of Induction Technology for Agarwood Production from Cultivated Aquilaria in Asia: A Review. J. For. Res. 2019, 30, 1–11. [Google Scholar] [CrossRef]
- Chhipa, H.; Chowdhary, K.; Kaushik, N. Artificial Production of Agarwood Oil in Aquilaria sp. by Fungi: A Review. Phytochem. Rev. 2017, 16, 835–860. [Google Scholar] [CrossRef]
- Naziz, P.S.; Das, R.; Sen, S. The Scent of Stress: Evidence from the Unique Fragrance of Agarwood. Front. Plant Sci. 2019, 10, 840. [Google Scholar] [CrossRef]
- Li, W.; Chen, H.-Q.; Wang, H.; Mei, W.-L.; Dai, H.-F. Natural Products in Agarwood and Aquilaria Plants: Chemistry, Biological Activities and Biosynthesis. Nat. Prod. Rep. 2021, 38, 528–565. [Google Scholar] [CrossRef]
- Gao, M.; Han, X.; Sun, Y.; Chen, H.; Yang, Y.; Liu, Y.; Meng, H.; Gao, Z.; Xu, Y.; Zhang, Z.; et al. Overview of Sesquiterpenes and Chromones of Agarwood Originating from Four Main Species of the Genus Aquilaria. RSC Adv. 2019, 9, 4113–4130. [Google Scholar] [CrossRef]
- Kristanti, A.N.; Tanjung, M.; Amina, N.S. Secondary Metabolites of Aquilaria, a Thymelaeaceae Genus. Mini-Rev. Org. Chem. 2018, 15, 36–55. [Google Scholar] [CrossRef]
- Naef, R. The Volatile and Semi-volatile Constituents of Agarwood, the Infected Heartwood of Aquilaria Species: A Review. Flavour Fragr. J. 2011, 26, 73–89. [Google Scholar] [CrossRef]
- Niebler, J. Incense Materials. In Springer Handbook of Odor; Buettner, A., Ed.; Springer: Cham, Switzerland, 2017; pp. 63–86. [Google Scholar]
- Ishihara, M.; Tsuneya, T.; Uneyama, K. Components of the Volatile Concentrate of Agarwood. J. Essent. Oil Res. 1993, 5, 283–289. [Google Scholar] [CrossRef]
- Zviely, M.; Boix-Camps, A. Sesquiterpenoides—The Holy Fragrance Ingredients. Isr. Chem. Eng. 2015, 1, 27–31. [Google Scholar]
- Syntrivanis, L.-D.; Wong, L.L.; Robertson, J. Hydroxylation of Eleuthoside Synthetic Intermediates by P450BM3 (CYP102A1). Eur. J. Org. Chem. 2018, 2018, 6369–6378. [Google Scholar] [CrossRef]
- Harwood, L.A.; Wong, L.L.; Robertson, J. Enzymatic Kinetic Resolution by Addition of Oxygen. Angew. Chem. Int. Ed. 2021, 60, 4434–4447. [Google Scholar] [CrossRef]
- Rakotonirainy, O.; Gaydou, E.M.; Faure, R.; Bombarda, I. Sesquiterpenes from Patchouli (Pogostemon cablin) Essential Oil. Assignment of the Proton and Carbon-13 NMR Spectra. J. Essent. Oil Res. 1997, 9, 321–327. [Google Scholar] [CrossRef]
- Ishihara, M.; Tsuneya, T.; Uneyama, K. Guaiane Sesquiterpenes from Agarwood. Phytochemistry 1991, 30, 3343–3347. [Google Scholar] [CrossRef]
- Piers, E.; Cheng, K.F. Stereoselective Synthesis of α-Bulnesene. Chem. Commun. 1969, 52, 562–563. [Google Scholar] [CrossRef]
- Piers, E.; Cheng, K.F. Stereoselective Synthesis of α-Bulnesene, 4-epi-α-Bulnesene, and 5-epi-α-Bulnesene. Can. J. Chem. 1970, 48, 2234–2245. [Google Scholar] [CrossRef]
- Heathcock, C.H.; Ratcliffe, R. Stereoselective Total Synthesis of the Guaiazulenic Sesquiterpenoids α-Bulnesene and Bulnesol. J. Am. Chem. Soc. 1971, 93, 1746–1757. [Google Scholar] [CrossRef]
- Mehta, G.; Singh, B.P. Terpenes and Related Systems. XIII. Regiospecific Fragmentation of Patchoulol. Short Synthesis of α-Bulnesene. Tetrahedron Lett. 1975, 50, 4495–4498. [Google Scholar] [CrossRef]
- Robertson, J.; Peplow, M.A.; Pillai, J. The Synthesis of (±)-Heliotridane and (6S,7S)-Dihydroxyheliotridane via Sequential Hydrogen Atom Abstraction and Cyclisation. Tetrahedron Lett. 1996, 37, 5825–5828. [Google Scholar] [CrossRef]
- Lathbury, D.C.; Parsons, P.J.; Pinto, I. A Route to the Pyrrolizidine Ring System using a Novel Radical Cyclisation. Chem. Commun. 1988, 81–82. [Google Scholar] [CrossRef]
- Borthwick, A.D.; Caddick, S.; Parsons, P.J. 1,5 Allylic Abstraction, Cyclisation: A New Route to Five Membered Carbocycles. Tetrahedron Lett. 1990, 31, 6911–6914. [Google Scholar] [CrossRef]
- Borthwick, A.D.; Caddick, S.; Parsons, P.J. Approaches to Bicyclic Ring Systems via 1,5 Allylic Abstraction Cyclisation. Tetrahedron 1992, 48, 10655–10666. [Google Scholar] [CrossRef]
- Curran, D.P.; Kim, D.; Liu, H.T.; Shen, W. Translocation of Radical Sites by Intramolecular 1,5-Hydrogen Atom Transfer. J. Am. Chem. Soc. 1988, 110, 5900–5902. [Google Scholar] [CrossRef]
- Curran, D.P.; Shen, W. Radical Translocation Reactions of Vinyl Radicals: Substituent Effects on 1,5-Hydrogen-Transfer Reactions. J. Am. Chem. Soc. 1993, 115, 6051–6059. [Google Scholar] [CrossRef]
- Beaufils, F.; Dénès, F.; Becattini, B.; Renaud, P.; Schenk, K. Thiophenol-Mediated 1,5-Hydrogen Atom Abstraction: Easy Access to Mono- and Bicyclic Compounds. Adv. Synth. Catal. 2005, 347, 1587–1594. [Google Scholar] [CrossRef]
- Liu, H.J.; Majumdar, S.P. On the Regioselectivity of Boron Trifluoride Catalyzed Ring Expansion of Cycloalkanones with Ethyl Diazoacetate. Synth. Commun. 1975, 5, 125–130. [Google Scholar] [CrossRef]
- Krafft, M.E.; Holton, R.A. Preparation of a New, Highly Reactive Form of Iron(0) and its Use in Deprotonation of Carbonyl Compounds. J. Org. Chem. 1984, 49, 3669–3670. [Google Scholar] [CrossRef]
- Comins, D.L.; Dehghani, A. Pyridine-Derived Triflating Reagents: An Improved Preparation of Vinyl Triflates from Metallo Enolates. Tetrahedron Lett. 1992, 33, 6299–6302. [Google Scholar] [CrossRef]
- Fürstner, A.; De Souza, D.; Parra-Rapado, L.; Jensen, J.T. Catalysis-Based Total Synthesis of Latrunculin B. Angew. Chem. Int. Ed. 2003, 42, 5358–5360. [Google Scholar] [CrossRef]
- Scheiper, B.; Bonnekessel, M.; Krause, H.; Fürstner, A. Selective Iron-Catalyzed Cross-Coupling Reactions of Grignard Reagents with Enol Triflates, Acid Chlorides, and Dichloroarenes. J. Org. Chem. 2004, 69, 3943–3949. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Hoveyda, A.H. α-Selective Ni-Catalyzed Hydroalumination of Aryl- and Alkyl-Substituted Terminal Alkynes: Practical Syntheses of Internal Vinyl Aluminums, Halides, or Boronates. J. Am. Chem. Soc. 2010, 132, 10961–10963. [Google Scholar] [CrossRef]
- Alves, L.D.C.; Desiderá, A.L.; de Oliveira, K.T.; Newton, S.; Ley, S.V.; Brocksom, T.J. A Practical Deca-gram Scale Ring Expansion of (R)-(−)-Carvone to (R)-(+)-3-Methyl-6-isopropenyl-cyclohept-3-enone-1. Org. Biomol. Chem. 2015, 13, 7633–7642. [Google Scholar] [CrossRef]
- Meng, Z.; Fürstner, A. Total Syntheses of Scabrolide A and Nominal Scabrolide B. J. Am. Chem. Soc. 2022, 144, 1528–1533. [Google Scholar] [CrossRef]
- Cossy, J.; BouzBouz, S.; Mouza, C. A Short Total Synthesis of (±)-Isoafricanol and Two of its Isomers. Synlett 1998, 1998, 621–622. [Google Scholar] [CrossRef]
- Hughes, G.; Lautens, M.; Wu, J. The Use of 1,4-Dichlorobut-2-ene as a Synthetic Equivalent of 4-Bromo-but-1-ene. Synlett 2000, 2000, 835–837. [Google Scholar]
- Chahboun, R.; Botubol-Ares, J.M.; Durán-Peña, M.J.; Jiménez, F.; Alvarez-Manzaneda, R.; Alvarez-Manzaneda, E. Deconjugative α-Alkylation of Cyclohexenecarboxaldehydes: An Access to Diverse Terpenoids. J. Org. Chem. 2021, 86, 8742–8754. [Google Scholar] [CrossRef]
- Corey, E.J.; Pyne, S.G. Conversion of Ketones Having δ,ε-π-Functions to Cyclopentanols by Zinc-Trimethylchlorosilane. Tetrahedron Lett. 1983, 24, 2821–2824. [Google Scholar] [CrossRef]
- Belotti, D.; Cossy, J.; Pete, J.P.; Portella, C. Photoreductive Cyclization of δ,ε-Unsaturated Ketones. Tetrahedron Lett. 1985, 26, 4591–4594. [Google Scholar] [CrossRef]
- Belotti, D.; Cossy, J.; Pete, J.P.; Portella, C. Synthesis of Bicyclic Cyclopentanols by Photoreductive Cyclization of δ,ε-Unsaturated Ketones. J. Org. Chem. 1986, 51, 4196–4200. [Google Scholar] [CrossRef]
- Molander, G.A.; Kenny, C. Stereocontrolled Intramolecular Ketone-Olefin Reductive Coupling Reactions Promoted by Samarium Diiodide. Tetrahedron Lett. 1987, 28, 4367–4370. [Google Scholar] [CrossRef]
- Jasperse, C.P.; Curran, D.P. Sequential Radical Cyclization Approach to Propellane Triquinanes. Total Synthesis of (±)-Modhephene. J. Am. Chem. Soc. 1990, 112, 5601–5609. [Google Scholar] [CrossRef]
- Molander, G.A.; McKie, J.A. Samarium(II) Iodide-Induced Reductive Cyclization of Unactivated Olefinic Ketones. Sequential Radical Cyclization/Intermolecular Nucleophilic Addition and Substitution Reactions. J. Org. Chem. 1992, 57, 3132–3139. [Google Scholar] [CrossRef]
- Sugimura, T.; Futagawa, T.; Tai, A. Synthetic Approach for (+)-Africanol. Chem. Lett. 1990, 19, 2295–2298. [Google Scholar] [CrossRef]
- Marques, F.A.; Ferreira, J.T.B.; Piers, E. Synthesis of (±)-Africanol. J. Braz. Chem. Soc. 2000, 11, 502–511. [Google Scholar] [CrossRef]
- Kursanov, D.N.; Parnes, Z.N.; Bassova, G.I.; Loim, N.M.; Zdanovich, V.I. Ionic Hydrogenation of the Ethylene Bond and the Double Bond of the Carbonyl Group. Tetrahedron 1967, 23, 2235–2242. [Google Scholar] [CrossRef]
- Carey, F.A.; Tremper, H.S. Carbonium Ion-Silane Hydride Transfer Reactions. I. Scope and Stereochemistry. J. Am. Chem. Soc. 1968, 90, 2578–2583. [Google Scholar] [CrossRef]
- Carey, F.A.; Tremper, H.S. Carbonium Ion-Silane Hydride Transfer Reactions. V. tert-Alkyl Cations. J. Org. Chem. 1971, 36, 758–761. [Google Scholar] [CrossRef]
- Adlington, M.G.; Orfanopoulos, M.; Fry, J.L. A Convenient One-Step Synthesis of Hydrocarbons from Alcohols through use of the Organosilane-Boron Trifluoride Reducing System. Tetrahedron Lett. 1976, 17, 2955–2958. [Google Scholar] [CrossRef]
- Doyle, M.P.; West, C.T.; Donnelly, S.J.; McOsker, C.C. Silane Reductions in Acidic Media VIII. Boron Trifluoride Catalyzed Organosilane Reductions. Selectivity and Mechanism. J. Organomet. Chem. 1976, 117, 129–140. [Google Scholar] [CrossRef]
- Aulenta, F.; Berndt, M.; Brüdgam, I.; Hartl, H.; Sörgel, S.; Reißig, H.-U. A New, Efficient and Stereoselective Synthesis of Tricyclic and Tetracyclic Compounds by Samarium Diiodide Induced Cyclisations of Naphthyl-Substituted Arylketones—An Easy Access to Steroid-Like Skeletons. Chem. Eur. J. 2007, 13, 6047–6062. [Google Scholar] [CrossRef]
- Srikrishna, A.; Pardeshi, V.H. Enantiospecific Total Synthesis of Aciphyllene. Tetrahedron 2010, 66, 8160–8168. [Google Scholar] [CrossRef]
- Tesso, H.; König, W.A.; Kubeczka, K.-H.; Bartnik, M.; Glowniak, K. Secondary Metabolites of Peucedanum tauricum Fruits. Phytochemistry 2005, 66, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Brocksom, T.J.; Brocksom, U.; Pergentino de Sousa, D.; Frederico, D. Enantiopure Cycloheptenones from (R)-(−)-Carvone: Intermediates for Perhydroazulene Terpenoids. Tetrahedron Asymmetry 2005, 16, 3628–3632. [Google Scholar] [CrossRef]
- Hewlett, D.F.; Whitby, R.J. Intramolecular Cocyclisation of Carbonyl Groups and Unactivated Alkenes or Alkynes induced by a Low-valent Titanium Complex. Chem. Commun. 1990, 1684–1686. [Google Scholar] [CrossRef]
- Kablaoui, N.M.; Buchwald, S.L. Reductive Cyclization of Enones by a Titanium Catalyst. J. Am. Chem. Soc. 1995, 117, 6785–6786. [Google Scholar] [CrossRef]
- Quan, L.G.; Cha, J.K. Reductive Cyclization of Enones by Titanium(IV) Aryloxide and a Grignard Reagent. Tetrahedron Lett. 2001, 42, 8567–8569. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proton | Correlated with |
|---|---|
| H2-2 | H2-3, H3-15 |
| H2-3 | H2-2, H-4 |
| H-4 | H2-3, H-5, H3-14 |
| H-5 | H-4, H2-6, H3-15 |
| H2-6 | H-5, H-7 |
| H-7 | H2-6, H2-8, H3-15 |
| H2-8 | H-7, H2-9 1 |
| H2-9 | overlapped 1 |
| H2-12 | H-7, H3-13 |
| H3-13 | H2-12 |
| H3-14 | H-4 |
| H3-15 | H2-2, H-5 |
| Carbon | Expected 2JC–H | Expected 3JC–H |
|---|---|---|
| C-1 | H2-2, H-5 | H2-3, (H-4), 2 H2-6, H2-9, H-15 |
| C-2 | H2-3 | (H-4), 2 (H-5) 3 |
| C-3 | H2-2, H-4 | (H-5), 3 H3-14 |
| C-4 | H2-3, H-5, H3-14 | H2-2, H2-6 |
| C-5 | H-4, H2-6 | H2-2, H2-3, H-7, H3-14 |
| C-6 | (H-5), 3 H-7 | H-4, H2-8 |
| C-7 | H2-6, H2-8 | (H-5), 3 H2-9, H2-12, H3-13 |
| C-8 | H-7, H2-9 | H2-6 |
| C-9 | H2-8 | H-7, H3-15 |
| C-10 | H2-9, H3-15 | H2-2, H-5, H2-8 |
| C-11 | H-7, H2-12, H3-13 | H2-6, H2-8 |
| C-12 | — | H-7, H3-13 |
| C-13 | — | H-7, H2-12 |
| C-14 | H-4 | H2-3, (H-5) 3 |
| C-15 | — | H2-9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zong, J.; Robertson, J. An Enantiospecific Synthesis of 5-epi-α-Bulnesene. Molecules 2023, 28, 3900. https://doi.org/10.3390/molecules28093900
Zong J, Robertson J. An Enantiospecific Synthesis of 5-epi-α-Bulnesene. Molecules. 2023; 28(9):3900. https://doi.org/10.3390/molecules28093900
Chicago/Turabian StyleZong, Jiarui, and Jeremy Robertson. 2023. "An Enantiospecific Synthesis of 5-epi-α-Bulnesene" Molecules 28, no. 9: 3900. https://doi.org/10.3390/molecules28093900
APA StyleZong, J., & Robertson, J. (2023). An Enantiospecific Synthesis of 5-epi-α-Bulnesene. Molecules, 28(9), 3900. https://doi.org/10.3390/molecules28093900