

Tuning of the Electrostatic Potentials on the Surface of the Sulfur Atom in Organic Molecules: Theoretical Design and Experimental Assessment


Abstract
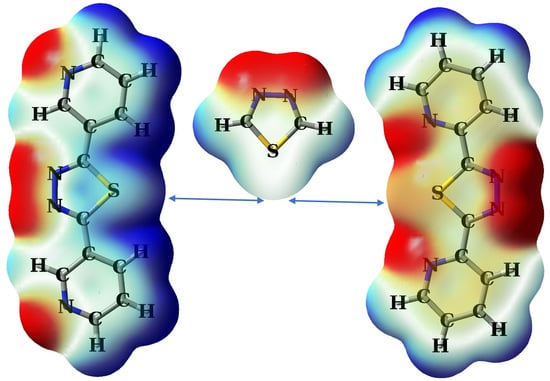
1. Introduction
2. Results
2.1. Theoretical Design
2.2. Crystallographic Assessment
3. Materials and Methods
3.1. Computational Methods
3.2. Experimental Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mustafa, M.; Winum, J.-Y. The importance of sulfur-containing motifs in drug design and discovery. Expert Opin. Drug Discov. 2022, 17, 501–512. [Google Scholar] [CrossRef]
- Tilby, M.J.; Willis, M.C. How do we address neglected sulfur pharmacophores in drug discovery? Expert Opin. Drug Discov. 2021, 16, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur containing scaffolds in drugs: Synthesis and application in medicinal chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. [Google Scholar] [CrossRef] [PubMed]
- Ilardi, E.A.; Vitaku, E.; Njardarson, J.T. Data-mining for sulfur and fluorine: An evaluation of pharmaceuticals to reveal opportunities for drug design and discovery. J. Med. Chem. 2014, 57, 2832–2842. [Google Scholar] [CrossRef]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; Pennington, L.D.; Meanwell, N.A. A survey of the role of noncovalent sulfur interactions in drug design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [CrossRef]
- Cheng, P.; Yang, Y. Narrowing the band gap: The key to high-performance organic photovoltaics. Acc. Chem. Res. 2020, 53, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Li, Y.; Zhan, X. Small molecule semiconductors for high-efficiency organic photovoltaics. Chem. Soc. Rev. 2012, 41, 4245–4272. [Google Scholar] [CrossRef]
- Huang, H.; Yang, L.; Facchetti, A.; Marks, T.J. Organic and polymeric semiconductors enhanced by noncovalent conformational locks. Chem. Rev. 2017, 117, 10291–10318. [Google Scholar] [CrossRef]
- Chand, A.; Sahoo, D.K.; Rana, A.; Jena, S.; Biswal, H.S. The prodigious hydrogen bonds with sulfur and selenium in molecular assemblies, structural biology, and functional materials. Acc. Chem. Res. 2020, 53, 1580–1592. [Google Scholar] [CrossRef]
- Burling, F.T.; Goldstein, B.M. Computational studies of nonbonded sulfur-oxygen and selenium-oxygen interactions in the thiazole and selenazole nucleosides. J. Am. Chem. Soc. 1992, 114, 2313–2320. [Google Scholar] [CrossRef]
- Wojtkowiak, K.; Michalczyk, M.; Zierkiewicz, W.; Jezierska, A.; Panek, J.J. Chalcogen bond as a factor stabilizing ligand conformation in the binding pocket of carbonic anhydrase IX receptor mimic. Int. J. Mol. Sci. 2022, 23, 13701. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Participation of S and Se in hydrogen and chalcogen bonds. CrystEngComm 2021, 23, 6821–6837. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Politzer, P. σ-Hole bonding: Molecules containing group VI atoms. J. Mol. Model. 2007, 13, 1033–1038. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Simultaneous σ-hole and hydrogen bonding by sulfur- and selenium-containing heterocycles. Int. J. Quantum Chem. 2008, 108, 2770–2781. [Google Scholar] [CrossRef]
- Scrocco, E.; Tomasi, J. Electronic molecular structure, reactivity and intermolecular forces: An euristic interpretation by means of electrostatic molecular potentials. Adv. Quantum Chem. 1978, 11, 115–193. [Google Scholar]
- Murray, J.S.; Politzer, P. The electrostatic potential: An overview. WIREs Comput. Mol. Sci. 2011, 1, 153–163. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond. Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Ji, B. Chalcogen bond: A sister noncovalent bond to halogen bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond. Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Reed, A.E.; Curitss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Houk, K.N. Through-space effects of substituents dominate molecular electrostatic potentials of substituted arenes. J. Chem. Theory Comput. 2009, 5, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond. Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. A 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Dennington II, R.D.; Keith, T.A.; Millam, J.M. GaussView, Revision 6.0.16; Semichem, Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sun, T.; Zhang, Y.; Wang, Y.B. The benzene⋯naphthalene complex: A more challenging system than the benzene dimer for newly developed computational methods. J. Chem. Phys. 2015, 143, 114312. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, Y.; Wang, Y.B. Highly accurate benchmark calculations of the interaction energies in the complexes C6H6⋯C6X6 (X = F, Cl, Br, and I). Int. J. Quantum Chem. 2017, 117, e25345. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the difference of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Eliseeva, A.A.; Ivanov, D.M.; Novikov, A.S.; Kukushkin, V.Y. Recognition of the π-hole donor ability of iodopentafluorobenzene—A conventional σ-hole donor for crystal engineering involving halogen bonding. CrystEngComm 2019, 21, 616–628. [Google Scholar] [CrossRef]
- Katkova, S.A.; Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Zolotarev, A.A.; Boyarskiy, V.P.; Kukushkin, V.Y. (Isocyano group π-hole)⋯[dz2-MII] interactions of (isocyanide)[MII] complexes, in which positively charged metal centers (d8-M=Pt, Pd) act as nucleophiles. Chem. Eur. J. 2019, 25, 8590–8598. [Google Scholar] [CrossRef]
- Mikherdov, A.S.; Novikov, A.S.; Kinzhalov, M.A.; Zolotarev, A.A.; Boyarskiy, V.P. Intra-/intermolecular bifurcated chalcogen bonding in crystal structure of thiazole/thiadiazole derived binuclear (diaminocarbene)PdII complexes. Crystals 2018, 8, 112. [Google Scholar] [CrossRef]
- Kolari, K.; Sahamies, J.; Kalenius, E.; Novikov, A.S.; Kukushkin, V.Y.; Haukka, M. Metallophilic interactions in polymeric group 11 thiols. Solid State Sci. 2016, 60, 92–98. [Google Scholar] [CrossRef]
- Nenajdenko, V.G.; Shikhaliyev, N.G.; Maharramov, A.M.; Bagirova, K.N.; Suleymanova, G.T.; Novikov, A.S.; Khrustalev, V.N.; Tskhovrebov, A.G. Halogenated diazabutadiene dyes: Synthesis, structures, supramolecular features, and theoretical Studies. Molecules 2020, 25, 5013. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian~16 Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Niu, C.-Y.; Hou, S.-C.; Wan, X.-S.; Kou, C.-H.; Feng, C.-L. 2,5-Bis(3-pyridyl)-1,3,4-thiadiazole. Acta Crystallogr. E 2007, 63, o2146–o2147. [Google Scholar] [CrossRef]
- Niu, C.; Ning, A.; Dang, Y.; Wan, X.; Kou, C. Crystal structure of cis-2,5-bis(3-pyridyl)-1,3,4-thiadiazole, C12H8N4S. Z. Kristallogr. NCS 2008, 223, 277–278. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- CrysAlisPro Software System, Version 1.171.38.41r; Rigaku Oxford Diffraction: Oxford, UK, 2015.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |
|---|---|---|
| CCDC deposition number | 2,162,326 | 2,162,328 |
| Empirical formula | C18H8F4I2N4S | C18H8F3I3N4S |
| Formula weight | 642.14 | 750.04 |
| Temperature/K | 293 (2) | 293 (2) |
| Crystal system | monoclinic | monoclinic |
| Space group | Cc | P21/n |
| a/Å | 22.0520 (5) | 7.5248 (2) |
| b/Å | 7.84693 (17) | 19.6708 (5) |
| c/Å | 11.8297 (2) | 14.6179 (5) |
| β/° | 99.301 (2) | 103.947 (3) |
| Volume/Å3 | 2020.10 (8) | 2099.94 (11) |
| Z | 4 | 4 |
| ρcalc/g‧cm−3 | 2.111 | 2.372 |
| Color | colorless | colorless |
| Crystal size/mm3 | 0.29 × 0.25 × 0.21 | 0.31 × 0.28 × 0.12 |
| Reflections collected | 20,134 | 11,182 |
| Independent reflections | 3542 | 3854 |
| Rint | 0.033 | 0.042 |
| Number of refined parameters | 263 | 263 |
| Goodness-of-fit on F2 | 1.074 | 1.047 |
| Final R1 index [I ≥ 2σ(I)] | 0.0224 | 0.0327 |
| Final wR2 index [I ≥ 2σ(I)] | 0.0521 | 0.0668 |
| Final R1 index [all data] | 0.0235 | 0.0394 |
| Final wR2 index [all data] | 0.0529 | 0.0699 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Wang, W.; Li, H.-B. Tuning of the Electrostatic Potentials on the Surface of the Sulfur Atom in Organic Molecules: Theoretical Design and Experimental Assessment. Molecules 2023, 28, 3919. https://doi.org/10.3390/molecules28093919
Wang Z, Wang W, Li H-B. Tuning of the Electrostatic Potentials on the Surface of the Sulfur Atom in Organic Molecules: Theoretical Design and Experimental Assessment. Molecules. 2023; 28(9):3919. https://doi.org/10.3390/molecules28093919
Chicago/Turabian StyleWang, Ziyu, Weizhou Wang, and Hai-Bei Li. 2023. "Tuning of the Electrostatic Potentials on the Surface of the Sulfur Atom in Organic Molecules: Theoretical Design and Experimental Assessment" Molecules 28, no. 9: 3919. https://doi.org/10.3390/molecules28093919
APA StyleWang, Z., Wang, W., & Li, H.-B. (2023). Tuning of the Electrostatic Potentials on the Surface of the Sulfur Atom in Organic Molecules: Theoretical Design and Experimental Assessment. Molecules, 28(9), 3919. https://doi.org/10.3390/molecules28093919