3.5. Synthetic Procedures for the Synthesis of Compound 3
A 10 mL Schlenk tube was charged with iodobenzene (2, 0.4 mmol), transient directing groups (TDG1 or TDG2, 0.08 mmol), 2-pyridone ligands (L1 or L2, 0.12 mmol), Pd(OAc)2 (4.53 mg, 0.02 mmol), and AgTFA (66.27 mg, 0.3 mmol). The tube was evacuated and filled with N2 three times. Next, aldehyde (1, 0.2 mmol) and the mixture of HFIP (1.5 mL) and HOAc (0.5 mL) were added into the tube quickly. The reaction was then stirred vigorously at room temperature for 20 min before being heated to 100 °C for 24 or 72 h. After cooling to room temperature, the reaction mixture was diluted with EtOAc (15 mL), filtered through a pad of celite, and the filtrate was then concentrated in vacuo; the residue was purified by flash chromatography on silica gel using petroleum ether/EtOAc as the eluent to yield the product 3.
Methyl 4-(2,2-dimethyl-3-oxopropyl)benzoate (
3a-mono). The title compound was prepared by using
TDG1 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 10:1). Colorless oil, 22.5 mg, yield: 51% (known compound [
21]).
1H NMR (300 MHz, CDCl
3) δ 9.50 (s, 1H), 7.88 (d,
J = 8.3 Hz, 2H), 7.10 (d,
J = 8.2 Hz, 2H), 3.83 (s, 3H), 2.76 (s, 2H), 0.98 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 205.41, 167.00, 142.50, 130.33, 129.46, 128.53, 52.12, 46.95, 42.90, 21.45.
Dimethyl 4,4′-(2-formyl-2-methylpropane-1,3-diyl)dibenzoate (3a-di). The title compound was prepared by using TDG1 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 10:1). White solid, 22.7 mg, yield: 32%. 1H NMR (300 MHz, CDCl3) δ 9.58 (s, 1H), 7.88 (d, J = 8.3 Hz, 4H), 7.09 (d, J = 8.3 Hz, 4H), 3.84 (s, 6H), 3.00 (d, J = 13.5 Hz, 2H), 2.69 (d, J = 13.5 Hz, 2H), 0.93 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 200.17, 161.91, 136.75, 125.44, 124.60, 123.79, 47.18, 46.15, 37.59, 13.36. HRMS (ESI, m/z): calcd. For C21H22NaO5 [M + Na]+: 377.1358, found: 377.1358.
Ethyl 4-(2,2-dimethyl-3-oxopropyl)benzoate (
3b-mono). The title compound was prepared by using
TDG1 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 10:1). Colorless oil, 23.4 mg, yield: 50% (known compound [
21]).
1H NMR (400 MHz, CDCl
3) δ 9.50 (s, 1H), 7.93–7.84 (m, 2H), 7.10 (d,
J = 8.3 Hz, 2H), 4.32–4.26 (m, 2H), 2.76 (s, 2H), 1.31 (t,
J = 7.1 Hz, 3H), 0.98 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 204.34, 165.47, 141.30, 129.21, 128.36, 127.84, 59.87, 45.87, 41.87, 20.39, 13.29.
Diethyl 4,4′-(2-formyl-2-methylpropane-1,3-diyl)dibenzoate (3b-di). The title compound was prepared by using TDG1 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 10:1). White solid, 24.5 mg, yield: 32%. 1H NMR (400 MHz, CDCl3) δ 9.58 (s, 1H), 7.88 (d, J = 8.3 Hz, 4H), 7.08 (d, J = 8.3 Hz, 4H), 4.32–4.26 (m, 4H), 2.99 (d, J = 13.5 Hz, 2H), 2.69 (d, J = 13.5 Hz, 2H), 1.31 (t, J = 7.1 Hz, 6H), 0.92 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 205.16, 166.41, 141.62, 130.37, 129.55, 129.14, 60.98, 51.13, 42.58, 18.35, 14.35. HRMS (ESI, m/z): calcd. For C23H27O5 [M + H]+: 383.1853, found: 383.1838.
4-(2,2-Dimethyl-3-oxopropyl)benzonitrile (
3c-mono). The title compound was prepared by using
TDG1 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 50:1). Colorless oil, 22.8 mg, yield: 61% (known compound [
39]).
1H NMR (300 MHz, CDCl
3) δ 9.56 (s, 1H), 7.57 (d,
J = 8.0 Hz, 2H), 7.23 (d,
J = 8.0 Hz, 2H), 2.85 (s, 2H), 1.06 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 204.84, 142.86, 131.93, 131.06, 118.79, 110.57, 46.92, 42.82, 21.52.
4,4′-(2-Formyl-2-methylpropane-1,3-diyl)dibenzonitrile (3c-di). The title compound was prepared by using TDG1 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 50:1). Colorless oil, 8.1 mg, yield: 14%. 1H NMR (300 MHz, CDCl3) δ 9.60 (s, 1H), 7.58 (d, J = 8.0 Hz, 4H), 7.20 (d, J = 8.0 Hz, 4H), 3.07 (d, J = 13.5 Hz, 2H), 2.75 (d, J = 13.5 Hz, 2H), 1.02 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 204.25, 141.68, 132.14, 131.14, 118.58, 111.07, 51.09, 42.60, 18.47. HRMS (ESI, m/z): calcd. For C19H16N2NaO [M + Na]+: 311.1155, found: 311.1151.
2,2-Dimethyl-3-(4-nitrophenyl)propanal (
3d-mono). The title compound was prepared by using
TDG1 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 50:1). Colorless oil, 31.1 mg, yield: 75% (known compound [
21]).
1H NMR (300 MHz, CDCl
3) δ 9.57 (s, 1H), 8.14 (d,
J = 8.7 Hz, 2H), 7.29 (d,
J = 8.7 Hz, 2H), 2.91 (s, 2H), 1.09 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 204.78, 146.84, 145.06, 131.15, 123.39, 46.99, 42.46, 21.57.
2-Methyl-2-(4-nitrobenzyl)-3-(4-nitrophenyl)propanal (
3d-di). The title compound was prepared by using
TDG1 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 50:1). Colorless oil, 6.6 mg, yield: 10% (known compound [
18]).
1H NMR (300 MHz, CDCl
3) δ 9.56 (s, 1H), 8.09 (d,
J = 8.4 Hz, 4H), 7.20 (d,
J = 5.6 Hz, 4H), 3.07 (d,
J = 13.4 Hz, 2H), 2.75 (d,
J = 13.5 Hz, 2H), 0.99 (s, 3H).
13C NMR (75 MHz, CDCl
3) δ 204.10, 147.09, 143.74, 131.26, 123.61, 51.15, 42.30, 18.54.
2,2-Dimethyl-3-(4-(trifluoromethyl)phenyl)propanal (
3e-mono). The title compound was prepared by using
TDG1 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 50:1). Colorless oil, 22.6 mg, yield: 49% (known compound [
40]).
1H NMR (300 MHz, CDCl
3) δ 9.50 (s, 1H), 7.45 (d,
J = 7.9 Hz, 2H), 7.14 (d,
J = 7.9 Hz, 2H), 2.77 (s, 2H), 0.99 (s, 6H).
13C NMR (101 MHz, CDCl
3) δ 205.28, 141.24, 130.59, 128.90 (q,
J = 32.5 Hz), 125.09 (q,
J = 3.8 Hz), 124.24 (q,
J = 272.0 Hz), 46.91, 42.64, 21.44.
2-Methyl-2-(4-(trifluoromethyl)benzyl)-3-(4-(trifluoromethyl)phenyl)propanal (
3e-di). The title compound was prepared by using
TDG1 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 50:1). Colorless oil, 16.5 mg, yield: 22% (known compound [
40]).
1H NMR (300 MHz, CDCl
3) δ 9.57 (s, 1H), 7.46 (d,
J = 8.0 Hz, 4H), 7.13 (d,
J = 8.0 Hz, 4H), 3.00 (d,
J = 13.6 Hz, 2H), 2.69 (d,
J = 13.6 Hz, 2H), 0.94 (s, 3H).
13C NMR (101 MHz, CDCl
3) δ 204.97, 140.39, 130.70, 129.22 (q,
J = 32.6 Hz), 125.28 (q,
J = 3.7 Hz), 124.14 (q,
J = 271.9 Hz), 51.05, 42.38, 18.32.
Methyl 3-(2,2-dimethyl-3-oxopropyl)benzoate (
3f-mono). The title compound was prepared by using
TDG1 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 10:1). Colorless oil, 20.3 mg, yield: 46% (known compound [
21]).
1H NMR (300 MHz, CDCl
3) δ 9.52 (s, 1H), 7.84 (d,
J = 7.4 Hz, 1H), 7.72 (s, 1H), 7.31–7.21 (m, 2H), 3.84 (s, 3H), 2.77 (s, 2H), 0.99 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 205.49, 167.06, 137.35, 134.79, 131.26, 130.10, 128.27, 127.88, 52.18, 46.90, 42.71, 21.39.
Dimethyl 3,3′-(2-formyl-2-methylpropane-1,3-diyl)dibenzoate (3f-di). The title compound was prepared by using TDG1 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 10:1). Colorless oil, 22.7 mg, yield: 32%. 1H NMR (300 MHz, CDCl3) δ 9.61 (s, 1H), 7.84 (d, J = 7.6 Hz, 2H), 7.70 (s, 2H), 7.32–7.19 (m, 4H), 3.84 (s, 6H), 2.99 (d, J = 13.6 Hz, 2H), 2.70 (d, J = 13.6 Hz, 2H), 0.94 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 205.23, 166.94, 136.73, 134.85, 131.38, 130.24, 128.41, 128.09, 52.17, 51.07, 42.31, 18.25. HRMS (ESI, m/z): calcd. For C21H22NaO5 [M + Na]+: 377.1354, found: 377.1358.
2,2-Dimethyl-3-(3-nitrophenyl)propanal (3g-mono). The title compound was prepared by using TDG1 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 50:1). Colorless oil, 22.0 mg, yield: 53%. 1H NMR (300 MHz, CDCl3) δ 9.50 (s, 1H), 8.02–8.00 (m, 1H), 7.92 (s, 1H), 7.38 (d, J = 5.0 Hz, 2H), 2.84 (s, 2H), 1.01 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 204.78, 148.11, 139.21, 136.47, 129.10, 124.98, 121.76, 46.88, 42.26, 21.47. HRMS (ESI, m/z): calcd. For C11H14NO3 [M + H]+: 208.0968, found: 208.0968.
2-Methyl-2-(3-nitrobenzyl)-3-(3-nitrophenyl)propanal (3g-di). The title compound was prepared by using TDG1 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 50:1). Yellow solid, 11.8 mg, yield: 18%. 1H NMR (300 MHz, CDCl3) δ 9.59 (s, 1H), 8.06 (d, J = 7.6 Hz, 2H), 7.91 (s, 2H), 7.47–7.31 (m, 4H), 3.07 (d, J = 13.7 Hz, 2H), 2.77 (d, J = 13.7 Hz, 2H), 1.02 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 204.05, 148.21, 138.07, 136.47, 129.40, 125.11, 122.18, 51.04, 41.98, 18.47. HRMS (ESI, m/z): calcd. For C17H16N2NaO5 [M + Na]+: 351.0951, found: 351.0933.
2,2-Dimethyl-3-(6-(trifluoromethyl)yridine-3-yl)propanal (
3h). The title compound was prepared by using
TDG1 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 10:1). Yellow oil, 23.1 mg, yield: 50% (known compound [
40]).
1H NMR (400 MHz, CDCl
3) δ 9.47 (s, 1H), 8.44 (s, 1H), 7.60–7.53 (m, 2H), 2.81 (s, 2H), 1.02 (s, 6H).
13C NMR (101 MHz, CDCl
3) δ 204.47, 151.43, 146.49 (q,
J = 34.7 Hz), 139.02, 136.33, 121.60 (q,
J = 273.8 Hz), 119.96 (q,
J = 2.7 Hz), 46.81, 39.27, 21.47.
(1S,2S,5S)-2-Isopropyl-5-methylcyclohexyl 4-(2,2-dimethyl-3-oxopropyl)benzoate (3i-mono). The title compound was prepared by using TDG1 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 10:1). Colorless oil, 26.2 mg, yield: 38%. 1H NMR (300 MHz, CDCl3) δ 9.51 (s, 1H), 7.88 (d, J = 8.1 Hz, 2H), 7.10 (d, J = 8.1 Hz, 2H), 4.89–4.80 (m, 1H), 2.76 (s, 2H), 2.06–2.02 (m, 1H), 1.91–1.86 (m, 1H), 1.67–1.64 (m, 2H), 1.54–1.43 (m, 2H), 1.21–0.96 (m, 9H), 0.86–0.83 (m, 6H), 0.72 (d, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 205.38, 165.98, 142.22, 130.24, 129.43, 129.27, 74.79, 47.29, 46.93, 42.95, 40.99, 34.34, 31.45, 26.47, 23.62, 22.05, 21.48, 21.44, 20.80, 16.50. HRMS (ESI, m/z): calcd. For C22H32NaO3 [M + Na]+: 367.2244, found: 367.2249.
(1S,2S,5S)-2-isopropyl-5-methylcyclohexyl 4-(2-formyl-3-(4-((((1R,2R,5R)-2-isopropyl-5-methylcyclohexyl)oxy)carbonyl)phenyl)-2-methylpropyl)benzoate (3i-di). The title compound was prepared by using TDG1 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 10:1). Colorless oil, 29.0 mg, yield: 24%. 1H NMR (300 MHz, CDCl3) δ 9.59 (s, 1H), 7.88 (d, J = 8.0 Hz, 4H), 7.08 (d, J = 8.0 Hz, 4H), 4.89–4.80 (m, 2H), 3.00 (dd, J = 13.5, 2.0 Hz, 2H), 2.70 (dd, J = 13.5, 2.6 Hz, 2H), 2.06–2.02 (m, 2H), 1.90–1.86 (m, 2H), 1.67–1.64 (m, 4H), 1.53–1.43 (m, 4H), 1.21–0.99 (m, 6H), 0.94 (s, 3H), 0.96–0.83 (m, 12H), 0.72 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 205.25, 165.91, 141.51, 130.36, 129.57, 129.49, 74.86, 51.20, 47.27, 42.66, 40.98, 34.32, 31.46, 26.45, 23.57, 22.08, 20.83, 18.34, 16.49. HRMS (ESI, m/z): calcd. For C39H55O5 [M+H]+: 603.4044, found: 603.4031.
(1R,4S)-1,3,3-Trimethylbicyclo[2.2.1]heptan-2-yl 4-(2,2-dimethyl-3-oxopropyl)benzoate (
3j-mono). The title compound was prepared by using
TDG1 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 10:1). Colorless oil, 34.2 mg, yield: 50% (known compound [
21]).
1H NMR (300 MHz, CDCl
3) δ 9.51 (s, 1H), 7.90 (d,
J = 8.0 Hz, 2H), 7.11 (d,
J = 8.0 Hz, 2H), 4.53 (s, 1H), 2.77 (s, 2H), 1.90–1.81 (m, 1H), 1.74–1.67 (m, 2H), 1.61–1.54 (m, 1H), 1.50–1.39 (m, 1H), 1.17 (d,
J = 8.2 Hz, 2H), 1.11 (s, 3H), 1.03 (s, 3H), 1.00 (s, 6H), 0.77 (s, 3H).
13C NMR (75 MHz, CDCl
3) δ 205.38, 166.75, 142.32, 130.32, 129.41, 129.10, 86.65, 48.64, 48.43, 46.95, 42.96, 41.47, 39.85, 29.76, 26.89, 25.92, 21.47, 20.33, 19.50.
(1S,4R)-1,3,3-Trimethylbicyclo[2.2.1]heptan-2-yl 4-(2-formyl-2-methyl-3-(4-((((1R,4S)-1,3,3-trimethylbicyclo[2.2.1]heptan-2-yl)oxy)carbonyl)phenyl)propyl)benzoate (3j-di). The title compound was prepared by using TDG1 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 10:1). White solid, 36.0 mg, yield: 30%. 1H NMR (300 MHz, CDCl3) δ 9.60 (s, 1H), 7.90 (d, J = 8.0 Hz, 4H), 7.10 (d, J = 8.0 Hz, 4H), 4.53 (s, 2H), 3.01 (d, J = 13.5 Hz, 2H), 2.71 (d, J = 13.5 Hz, 2H), 1.89–1.80 (m, 2H), 1.74–1.67 (m, 4H), 1.60–1.58 (m, 2H), 1.50–1.40 (m, 2H), 1.17 (d, J = 10.0 Hz, 4H), 1.10 (s, 6H), 1.03 (s, 6H), 0.95 (s, 3H), 0.77 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 205.18, 166.65, 141.61, 130.43, 129.54, 129.34, 86.71, 51.20, 48.64, 48.43, 42.65, 41.47, 39.85, 29.76, 26.89, 25.92, 20.35, 19.51, 18.38. HRMS (ESI, m/z): calcd. For C39H50NaO5 [M + Na]+: 621.3550, found: 621.3543.
Compound
3k is a mixture of
2,2-dimethyl-3-phenylpropanal (
3k-mono), 2-benzyl-2-methyl-3-phenylpropanal (
3k-di), and 2,2-dibenzyl-3-phenylpropanal (
3k-tri).
3k-mono:3k-di:3k-tri = 1.0:0.76:0.35. The compound
3k was prepared by using
TDG2 and
L2, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 100:1). Colorless oil, 28.5 mg, yield: 66% (known compound [
18]).
2,2-Dimethyl-3-phenylpropanal (
3k-mono):
1H NMR (300 MHz, CDCl
3) δ 9.51 (s, 1H), 7.22–7.00 (m, 5H), 2.70 (s, 2H), 0.98 (s, 6H).
2-Benzyl-2-methyl-3-phenylpropanal (
3k-di):
1H NMR (300 MHz, CDCl
3) δ 9.61 (s, 1H), 7.22–7.00 (m, 5H), 2.95 (d,
J = 13.6 Hz, 2H), 2.64 (d,
J = 13.6 Hz, 2H), 0.91 (s, 3H).
2,2-Dibenzyl-3-phenylpropanal (
3k-tri):
1H NMR (300 MHz, CDCl
3) δ 9.70 (s, 1H), 7.22–7.00 (m, 5H), 2.85 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 206.86, 206.22, 206.01, 136.92, 136.62, 136.59, 130.59, 130.37, 130.26, 128.30, 128.26, 128.17, 126.66, 126.55, 53.56, 51.24, 46.96, 43.23, 42.84, 40.22, 21.40, 18.17.
3-(4-Methoxyphenyl)-2,2-dimethylpropanal (
3l-mono). The title compound was prepared by using
TDG2 and
L2, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 50:1). Colorless oil, 11.5 mg, yield: 30% (known compound [
18]).
1H NMR (300 MHz, CDCl
3) δ 9.50 (s, 1H), 6.93 (d,
J = 8.1 Hz, 2H), 6.73 (d,
J = 8.1 Hz, 2H), 3.71 (s, 3H), 2.65 (s, 2H), 0.96 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 206.24, 158.32, 131.18, 128.90, 113.58, 55.22, 47.04, 42.38, 21.33.
2-(4-Methoxybenzyl)-3-(4-methoxyphenyl)-2-methylpropanal (
3l-di). The title compound was prepared by using
TDG2 and
L2, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 50:1). Colorless oil, 13.1 mg, yield: 22% (known compound [
18]).
1H NMR (300 MHz, CDCl
3) δ 9.58 (s, 1H), 6.92 (d,
J = 8.1 Hz, 4H), 6.72 (d,
J = 8.3 Hz, 4H), 3.70 (s, 6H), 2.88 (d,
J = 13.8 Hz, 2H), 2.56 (d,
J = 13.8 Hz, 2H), 0.88 (s, 3H).
13C NMR (75 MHz, CDCl
3) δ 206.66, 158.35, 131.30, 128.62, 113.64, 55.22, 51.46, 41.91, 18.06.
2,2-Bis(4-methoxybenzyl)-3-(4-methoxyphenyl)propanal (
3l-tri). The title compound was prepared by using
TDG2 and
L2, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 50:1). Colorless oil, 8.1 mg, yield: 10% (known compound [
18]).
1H NMR (300 MHz, CDCl
3) δ 9.74 (s, 1H), 7.02 (d,
J = 8.3 Hz, 6H), 6.80 (d,
J = 8.4 Hz, 6H), 3.78 (s, 9H), 2.84 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 207.40, 158.28, 131.53, 128.62, 113.64, 55.22, 53.81, 39.23.
Compound
3m is a mixture of
2,2-dimethyl-3-(p-tolyl)propanal (
3m-mono),
2-methyl-
2-(4-methylbenzyl)-3-(p-tolyl)propanal (
3m-di), and
2,2-bis(4-methylbenzyl)-3-(p-tolyl)propanal (
3m-tri).
3m-mono:3m-di:3m-tri = 1.0:0.71:0.42. The compound
3m was prepared by using
TDG2 and
L2, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 100:1). Colorless oil, 33.0 mg, yield:68% (known compound [
21]).
2,2-Dimethyl-3-(p-tolyl)propanal (
3m-mono):
1H NMR (300 MHz, CDCl
3) δ 9.51 (s, 1H), 7.18–6.88 (m, 4H), 2.66 (s, 2H), 2.23 (s, 3H), 0.97 (s, 6H).
2-Methyl-2-(4-methylbenzyl)-3-(p-tolyl)propanal (
3m-di):
1H NMR (300 MHz, CDCl
3) δ 9.60 (s, 1H), 7.18–6.88 (m, 4H), 2.90 (d,
J = 13.6 Hz, 2H), 2.58 (d,
J = 13.6 Hz, 2H), 2.23 (s, 6H), 0.88 (s, 3H).
2,2-bis(4-methylbenzyl)-3-(p-tolyl)propanal (
3m-tri):
1H NMR (300 MHz, CDCl
3) δ 9.67 (s, 1H), 7.18–6.88 (m, 4H), 2.79 (s, 6H), 2.23 (s, 9H).
13C NMR (75 MHz, CDCl
3) δ 207.30, 206.50, 206.16, 136.16, 136.08, 133.75, 133.58, 133.51, 130.48, 130.22, 130.12, 128.93, 128.86, 53.59, 51.30, 46.95, 42.87, 42.43, 39.71, 21.37, 21.01, 18.10.
3-(4-Bromophenyl)-2,2-dimethylpropanal (
3n-mono). The title compound was prepared by using
TDG2 and
L2, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 100:1). Colorless oil, 24.0 mg, yield: 50% (known compound [
18]).
1H NMR (400 MHz, CDCl
3) δ 9.48 (s, 1H), 7.32 (d,
J = 8.4 Hz, 2H), 6.90 (d,
J = 8.3 Hz, 2H), 2.66 (s, 2H), 0.97 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 205.52, 135.97, 131.95, 131.27, 120.57, 46.80, 42.39, 21.39.
2-(4-Bromobenzyl)-3-(4-bromophenyl)-2-methylpropanal (
3n-di). The title compound was prepared by using
TDG2 and
L2, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 100:1). Colorless oil, 19.8 mg, yield: 25% (known compound [
18]).
1H NMR (400 MHz, CDCl
3) δ 9.54 (s, 1H), 7.32 (d,
J = 8.3 Hz, 4H), 6.87 (d,
J = 8.4 Hz, 4H), 2.87 (d,
J = 13.7 Hz, 2H), 2.56 (d,
J = 13.7 Hz, 2H), 0.89 (s, 3H).
13C NMR (75 MHz, CDCl
3) δ 205.44, 135.33, 132.03, 131.42, 120.84, 50.91, 41.99, 18.23.
Compound
3o is a mixture of
3-(2-fluorophenyl)-2,2-dimethylpropanal (
3o-mono), 2-(2-fluorobenzyl)-3-(2-fluorophenyl)-2-methylpropanal (
3o-di), and 2,2-bis(2-fluorobenzyl)-3-(2-fluorophenyl)propanal (
3o-tri). The compound
3o was prepared by using
TDG2 and
L2, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 100:1).
3o-mono:3o-di:3o-tri = 1.0:0.77:0.28. Colorless oil, 29.0 mg, yield: 60% (known compound [
18]).
3-(2-Fluorophenyl)-2,2-dimethylpropanal (
3o-mono):
1H NMR (400 MHz, CDCl
3) δ 9.51 (d,
J = 1.3 Hz, 1H), 7.17–6.92 (m, 4H), 2.76 (d,
J = 1.1 Hz, 2H), 1.00 (s, 6H).
2-(2-Fluorobenzyl)-3-(2-fluorophenyl)-2-methylpropanal (
3o-di):
1H NMR (400 MHz, CDCl
3) δ 9.61 (t,
J = 1.9 Hz, 1H), 7.17–6.92 (m, 4H), 3.00 (d,
J = 13.7 Hz, 2H), 2.76 (d,
J = 13.7 Hz, 2H), 0.91 (s, 3H).
2,2-Bis(2-fluorobenzyl)-3-(2-fluorophenyl)propanal (
3o-tri):
1H NMR (400 MHz, CDCl
3) δ 9.57 (d,
J = 1.7 Hz, 1H), 7.17–6.92 (m, 4H), 2.91 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 204.44, 203.86, 203.16, 161.99, 161.87, 161.83, 158.75, 158.63, 158.58, 131.92, 131.86, 131.63, 131.58, 131.52, 127.76, 127.65, 127.55, 127.44, 122.99, 122.92, 122.87, 122.80, 122.75, 122.65, 122.44, 114.62, 114.54, 114.31, 114.23, 52.75, 50.58, 46.09, 34.71, 34.42, 31.66, 20.23, 16.27.
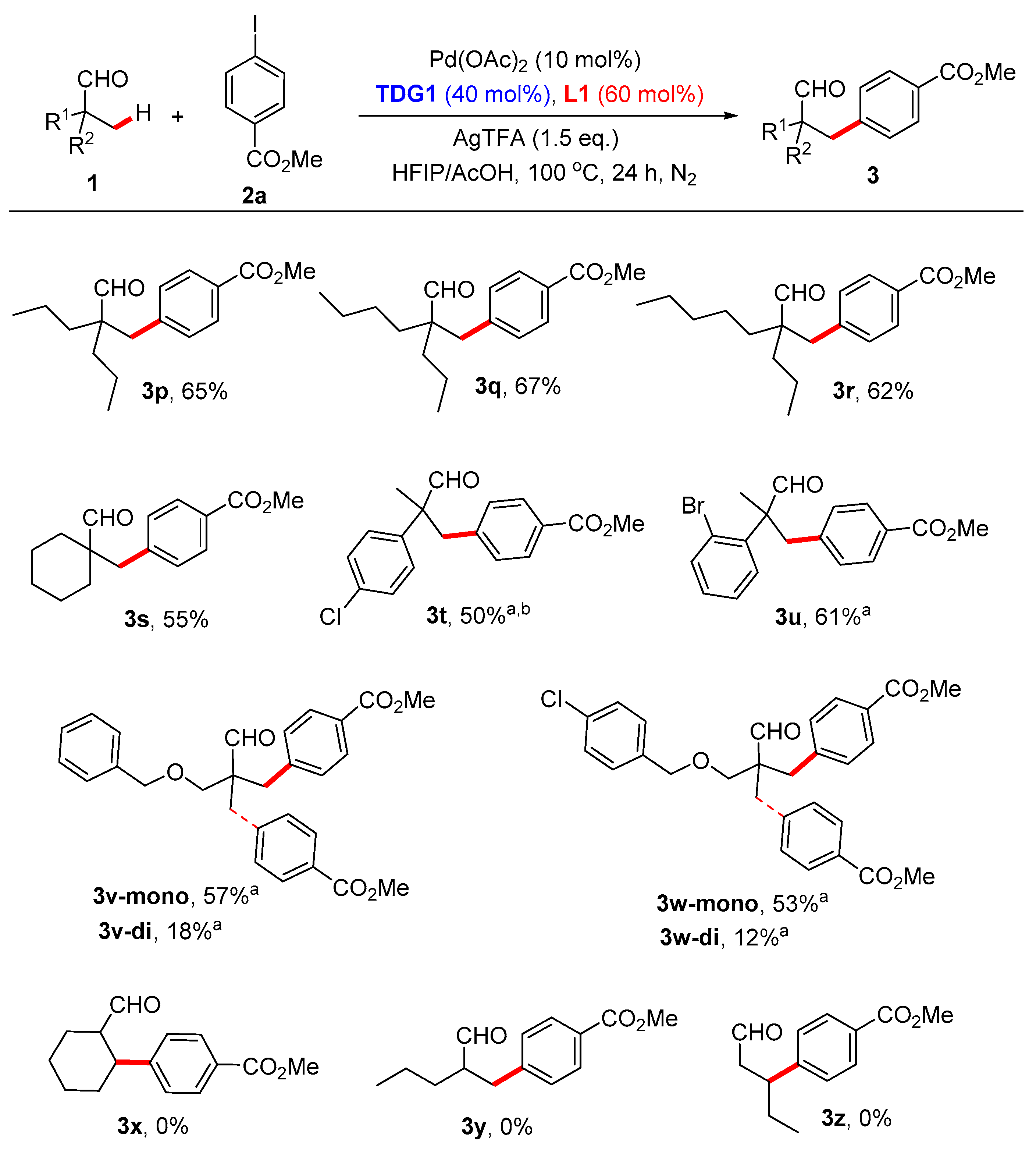
Methyl 4-(2-formyl-2-propylpentyl)benzoate (3p). The title compound was prepared by using TDG1 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 10:1). Colorless oil, 36.0 mg, yield: 65%. 1H NMR (400 MHz, CDCl3) δ 9.47 (s, 1H), 7.86 (d, J = 7.9 Hz, 2H), 7.07 (d, J = 7.9 Hz, 2H), 3.83 (s, 3H), 2.81 (s, 2H), 1.41–1.31 (m, 4H), 1.24–1.18 (m, 4H), 0.83 (t, J = 7.1 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 206.57, 166.99, 142.77, 130.06, 129.51, 128.43, 53.59, 52.09, 38.51, 34.32, 31.75, 25.77, 23.25, 16.98, 14.62, 13.97, 1.04. HRMS (ESI, m/z): calcd. for C17H25O3 [M + H]+: 277.1798, found: 277.1789.
Methyl 4-(2-formyl-2-propylhexyl)benzoate (3q). The title compound was prepared by using TDG1 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 10:1). Colorless oil, 39.0 mg, yield: 67%. 1H NMR (300 MHz, CDCl3) δ 9.54 (s, 1H), 7.94 (d, J = 7.9 Hz, 2H), 7.14 (d, J = 7.9 Hz, 2H), 3.90 (s, 3H), 2.88 (s, 2H), 1.52–1.27 (m, 10H), 0.90 (t, J = 6.9 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 205.50, 165.92, 141.71, 128.99, 128.45, 127.36, 52.53, 51.02, 37.43, 33.24, 30.67, 24.70, 22.18, 15.91, 13.56, 12.90. HRMS (ESI, m/z): calcd. for C18H27O3 [M + H]+: 291.1955, found: 291.1948.
Methyl 4-(2-formyl-2-propylheptyl)benzoate (3r). The title compound was prepared by using TDG1 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 10:1). Colorless oil, 37.7 mg, yield: 62%. 1H NMR (300 MHz, CDCl3) δ 9.47 (s, 1H), 7.86 (d, J = 8.2 Hz, 2H), 7.07 (d, J = 8.2 Hz, 2H), 3.83 (s, 3H), 2.81 (s, 2H), 1.45–1.18 (m, 12H), 0.85–0.79 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 205.52, 165.96, 141.76, 129.02, 128.47, 127.39, 52.60, 51.04, 37.45, 33.30, 31.32, 31.02, 22.25, 21.44, 15.95, 13.58, 13.00. HRMS (ESI, m/z): calcd. for C19H28NaO3 [M + Na]+: 327.1931, found: 327.1921.
Methyl 4-((1-formylcyclohexyl)methyl)benzoate (
3s). The title compound was prepared by using
TDG1 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 10:1). Colorless oil, 28.6 mg, yield: 55% (known compound [
21]).
1H NMR (300 MHz, CDCl
3) δ 9.45 (s, 1H), 7.86 (d,
J = 8.3 Hz, 2H), 7.06 (d,
J = 8.3 Hz, 2H), 3.83 (s, 3H), 2.70 (s, 2H), 1.85–1.81 (m, 2H), 1.59–1.46 (m, 3H), 1.29–1.17 (m, 5H).
13C NMR (75 MHz, CDCl
3) δ 205.78, 165.94, 140.74, 129.25, 128.38, 127.52, 51.04, 49.67, 42.25, 30.14, 24.47, 21.58.
Methyl 4-(2-(4-chlorophenyl)-2-methyl-3-oxopropyl)benzoate (
3t). The title compound was prepared by using
TDG2 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 10:1). Colorless oil, 31.7 mg, yield: 50% (known compound [
20]).
1H NMR (300 MHz, CDCl
3) δ 9.49 (s, 1H), 7.74 (d,
J = 7.7 Hz, 2H), 7.26 (d,
J = 8.1 Hz, 2H), 6.98 (d,
J = 8.1 Hz, 2H), 6.76 (d,
J = 7.8 Hz, 2H), 3.80 (s, 3H), 3.12 (s, 2H), 1.30 (s, 3H).
13C NMR (75 MHz, CDCl3) δ 200.91, 166.98, 141.98, 137.10, 133.79, 130.43, 129.18, 129.03, 128.99, 128.51, 77.49, 77.06, 76.64, 54.65, 52.09, 42.70, 18.14.
Methyl 4-(2-(2-bromophenyl)-2-methyl-3-oxopropyl)benzoate (
3u). The title compound was prepared by using
TDG2 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 10:1). Yellow oil, 44.0 mg, yield: 61% (known compound [
20]).
1H NMR (300 MHz, CDCl
3) δ 9.89 (s, 1H), 7.76 (d,
J = 8.0 Hz, 2H), 7.70–7.67 (m, 1H), 7.23–7.15 (m, 2H), 6.82–6.79 (m, 1H), 6.73 (d,
J = 8.0 Hz, 2H), 3.87 (s, 3H), 3.71 (d,
J = 13.5 Hz, 1H), 3.29 (d,
J = 13.5 Hz, 1H), 1.32 (s, 3H).
13C NMR (75 MHz, CDCl
3) δ 202.20, 167.05, 142.29, 139.00, 134.43, 130.63, 130.52, 129.63, 128.95, 128.31, 127.57, 123.51, 55.95, 52.03, 39.13, 20.74.
Methyl 4-(3-(benzyloxy)-2-formyl-2-methylpropyl)benzoate (
3v-mono). The title compound was prepared by using
TDG1 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 10:1). Colorless oil, 37.2 mg, yield: 57% (known compound [
21]).
1H NMR (300 MHz, CDCl
3) δ 9.58 (s, 1H), 7.84 (d,
J = 8.2 Hz, 2H), 7.34–7.20 (m, 5H), 7.09 (d,
J = 8.2 Hz, 2H), 4.42 (s, 2H), 3.82 (s, 3H), 3.29 (dd,
J = 27.8, 9.3 Hz, 2H), 2.88 (dd,
J = 52.1, 13.4 Hz, 2H), 0.90 (s, 3H).
13C NMR (75 MHz, CDCl3) δ 204.63, 167.01, 142.22, 137.72, 130.47, 129.46, 128.50, 127.88, 127.74, 73.38, 72.08, 52.10, 51.13, 37.70, 16.68.
Dimethyl 4,4′-(2-((benzyloxy)methyl)-2-formylpropane-1,3-diyl)dibenzoate (3v-di). The title compound was prepared by using TDG1 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 10:1). White solid, 16.6 mg, yield: 18%. 1H NMR (400 MHz, CDCl3) δ 9.67 (s, 1H), 7.91 (d, J = 8.2 Hz, 4H), 7.42–7.32 (m, 5H), 7.14 (d, J = 8.2 Hz, 4H), 4.45 (s, 2H), 3.90 (s, 6H), 3.29 (s, 2H), 3.11 (d, J = 13.6 Hz, 2H), 2.97 (d, J = 13.6 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 204.31, 166.90, 141.49, 137.51, 130.31, 129.65, 128.80, 128.56, 128.03, 73.49, 68.02, 55.57, 52.14, 38.53. HRMS (ESI, m/z): calcd. for C28H29O6 [M + H]+: 461.1959, found: 461.1949.
Methyl 4-(3-((4-chlorobenzyl)oxy)-2-formyl-2-methylpropyl)benzoate (
3w-mono). The title compound was prepared by using
TDG2 and
L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (
v/
v = 10:1). White solid, 38.2 mg, yield: 53% (known compound [
21]).
1H NMR (300 MHz, CDCl
3) δ 9.58 (s, 1H), 7.86 (d,
J = 8.1 Hz, 2H), 7.27 (d,
J = 8.3 Hz, 2H), 7.18 (d,
J = 8.3 Hz, 2H), 7.10 (d,
J = 8.1 Hz, 2H), 4.38 (s, 2H), 3.84 (s, 3H), 3.29 (dd,
J = 25.7, 9.3 Hz, 2H), 2.88 (dd,
J = 49.0, 13.4 Hz, 2H), 0.92 (s, 3H).
13C NMR (75 MHz, CDCl
3) δ 204.45, 166.99, 142.04, 136.20, 133.63, 130.40, 129.49, 128.95, 128.66, 128.60, 72.61, 72.28, 52.11, 51.11, 37.87, 16.76.
Dimethyl 4,4′-(2-(((4-chlorobenzyl)oxy)methyl)-2-formylpropane-1,3-diyl)dibenzoate (3w-di). The title compound was prepared by using TDG2 and L1, then isolated by flash chromatography on the silica gel by using mixed petroleum ether and ethyl acetate (v/v = 10:1). White solid, 12.0 mg, yield: 12%. 1H NMR (400 MHz, CDCl3) δ 9.68 (s, 1H), 7.92 (d, J = 8.2 Hz, 4H), 7.36 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 7.1 Hz, 2H), 7.14 (d, J = 8.2 Hz, 4H), 4.40 (s, 2H), 3.91 (s, 6H), 3.28 (s, 2H), 3.12 (d, J = 13.6 Hz, 2H), 2.97 (d, J = 13.6 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 204.12, 166.88, 141.37, 135.98, 133.81, 130.25, 129.68, 129.23, 128.86, 128.73, 72.66, 68.21, 55.58, 52.19, 38.52. HRMS (ESI, m/z): calcd. for C28H27ClNaO6 [M + H]+: 495.1569, found: 495.1561.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}