

Recent Advances in Manganese(III)-Assisted Radical Cyclization for the Synthesis of Natural Products: A Comprehensive Review



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Intermolecular Radical Cyclization
3. Intramolecular Radical Cyclizations
3.1. Mono Cyclizations
3.2. Tandem Cyclizations
3.3. Intramolecular Rearrangement Reactions
4. Oxidation Reactions
5. Aromatization
6. Acetoxidation
7. Halogen Transfer
8. Polymerization
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Melikyan, G.G.; Sargsyan, A.B.; Badanyan, S.O. Synthesis of 5-substituted furans on the basis of the reactions of 1,3-alkadiynes with β-dicarbonyl compounds. Chem. Heterocycl. Compd. 1989, 25, 606. [Google Scholar] [CrossRef]
- Melikyan, G.G. Manganese(III) mediated reactions of unsaturated systems. Synthesis 1993, 9, 833–850. [Google Scholar] [CrossRef]
- Iqbal, J.; Bhatia, B.; Nayyar, N.K. Transition metal-promoted free-radical reactions in organic synthesis: The formation of carbon-carbon bonds. Chem. Rev. 1994, 94, 519. [Google Scholar] [CrossRef]
- Snider, B.B. Manganese(III)-based oxidative free-radical cyclizations. Chem. Rev. 1996, 96, 339. [Google Scholar] [CrossRef] [PubMed]
- Melikyan, G.G. Carbon-carbon bond-forming reactions promoted by trivalent manganese. Org. React. 1997, 49, 427. [Google Scholar]
- Nishino, H. Manganese(III)-based peroxidation of alkenes to heterocycles. Top. Heterocycl. Chem. 2006, 6, 39. [Google Scholar]
- Demir, A.S.; Emrullahoglu, M. Manganese(III) acetate: A versatile reagent in organic chemistry. Manganese(III)-based peroxidation of alkenes to heterocycles. Curr. Org. Synth. 2007, 4, 321. [Google Scholar] [CrossRef]
- Snider, B.B. Mechanisms of Mn(OAc)3-based oxidative free-radical additions and cyclizations. Tetrahedron 2009, 65, 10738. [Google Scholar] [CrossRef]
- Mondal, M.; Bora, U. Recent advances in Manganese(III) acetate mediated organic synthesis. RSC Adv. 2013, 3, 18716. [Google Scholar] [CrossRef]
- Jasperse, C.P.; Curran, D.P.; Fevig, T.L. Radical reactions in natural product synthesis. Chem. Rev. 1991, 91, 1237. [Google Scholar] [CrossRef]
- Snider, B.B. Transition Metals for Organic Synthesis; Wiley-VCH: Weinheim, Germany, 1998; pp. 439–446. [Google Scholar]
- Burton, J.W. Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons: New York, NY, USA, 2012; pp. 1–41. [Google Scholar]
- Bush, J.B.; Finkbeiner, H. Oxidation reactions of manganese(III) acetate. II. Formation of γ-lactones from olefins and acetic acid. J. Am. Chem. Soc. 1968, 90, 5903. [Google Scholar]
- Heiba, E.I.; Dessau, R.M.; Koehl, W.J. Oxidation by metal salts. IV. A new method for the preparation of -lactones by the reaction of manganic acetate with olefins. J. Am. Chem. Soc. 1968, 90, 5905. [Google Scholar] [CrossRef]
- Melikyan, G.G. Manganese-based organic and bioinorganic transformations. Aldr. Acta 1998, 31, 50. [Google Scholar]
- Yılmaz, M.; Biçer, E.; Pekel, A.T. Manganese(III) acetate mediated free radical cyclization of 1,3-dicarbonyl compounds with sterically hindered olefins. Turk. J. Chem. 2005, 29, 579. [Google Scholar]
- Yılmaz, M.; Uzunalioğlu, N.; Yakut, M.; Pekel, A.T. Oxidative cyclisation of 3-oxopropanenitriles mediated manganese(iii) acetate with 2-thienyl substituted alkenes. Turk. J. Chem. 2008, 32, 411–422. [Google Scholar]
- Biçer, E.; Yılmaz, M.; Karataş, M.; Pekel, A.T. Radical cyclization reactions via manganese(iii) acetate leading to 2-thienyl-substituted dihydrofuran compounds. Helv. Chim. Acta 2012, 95, 795. [Google Scholar] [CrossRef]
- Biçer, E.; Yılmaz, M. Synthesis of trifluoromethylated dihydrofurans by addition of 1,3-dicarbonyl compounds to alkenes promoted by manganese(III) acetate. Arkivoc 2013, 3, 304. [Google Scholar] [CrossRef]
- Biçer, E.; Yılmaz, M.; Burgaz, E.V.; Pekel, A.T. Synthesis of thienyl-substituted dihydrofuran compounds promoted by manganese(iii) acetate. Helv. Chim. Acta 2013, 96, 135. [Google Scholar] [CrossRef]
- Biçer, E.; Yılmaz, M.; Ustalar, A.; Pekel, A.T. Synthesis of furan-substituted dihydrofuran compounds by radical cyclization reactions mediated by manganese(III) acetate. Arkivoc 2014, 5, 225. [Google Scholar]
- Yılmaz, M.; Burgaz, E.V.; Yakut, M.; Bicer, E. Synthesis of 4,5-dihydrofuran-3-carbonitrile derivatives with electron-rich alkenes in the presence of manganese(III) acetate. J. Chin. Chem. Soc. 2014, 61, 1101. [Google Scholar] [CrossRef]
- Nishino, H.; Kumabe, R.; Hamada, R.; Yakut, M. Mn(III)-based reaction of alkenes with quinolinones. Formation of peroxyquinolinones and quinoline-related derivatives. Tetrahedron 2014, 70, 1437. [Google Scholar] [CrossRef]
- Alagöz, O.; Yilmaz, M.; Pekel, A.T.; Graiff, C.; Maggi, R. Synthesis of dihydrofuro- and C-alkenylated naphthoquinones catalyzed by manganese(III) acetate. RSC Adv. 2014, 4, 14644–14654. [Google Scholar] [CrossRef]
- Ustalar, A.; Yılmaz, M. Microwave assisted synthesisof 2,3-dihydro-4H-benzo[4,5]thiazolo[3,2-a]furo[2,3-d]pyrimidin-4-ones and 6,7-Dihydro-5H-furo[2,3-d]thiazolo[3,2-a]pyrimidin-5-ones using Mn(OAc)3. Tetrahedron Lett. 2017, 58, 516. [Google Scholar] [CrossRef]
- Ustalar, A.; Yilmaz, M.; Osmani, A.; Keçeli, S.A. Synthesis and antifungal activity of new dihydrofurocoumarins and dihydrofuroquinolines. Turk. J. Chem. 2017, 41, 80–88. [Google Scholar] [CrossRef]
- Özgür, M.; Yılmaz, M.; Nishino, H.; Avar, E.Ç.; Dal, H.; Pekel, A.T.; Hökelek, T. Efficient syntheses and antimicrobial activities of new thiophene containing pyranone and quinolinone derivatives using manganese(III) acetate: The effect of thiophene on ring closure–opening reactions. N. J. Chem. 2019, 43, 5737. [Google Scholar] [CrossRef]
- Sari, S.; Yılmaz, M. Synthesis, characterization, acetylcholinesterase inhibition, and molecular docking studies of new piperazine substituted dihydrofuran compounds. Med. Chem. Res. 2020, 29, 1804–1818. [Google Scholar] [CrossRef]
- Yakut, M.; Yılmaz, M.; Pekel, A.T.; Erkan, S.; Katırcı, R.; Biçer, E. Manganese(III) acetate-based radical cyclization reactions for pyranocoumarin and pyranoquinoline compounds: Synthesis, DFT and molecular docking studies. ChemistrySelect 2022, 7, e202202787. [Google Scholar] [CrossRef]
- Williams, G.J.; Hunter, N.R. Situselective α’-acetoxylation of some α,β-enones by manganic acetate oxidation. Can. J. Chem. 1976, 54, 3830. [Google Scholar] [CrossRef]
- Dunlap, N.K.; Sabol, M.R.; Watt, D.S. Oxidation of enones to α’-acetoxyenones using manganese triacetate. Tetrahedron Lett. 1984, 25, 5839. [Google Scholar] [CrossRef]
- Demir, A.S.; Jeganathan, A.; Watt, D.S. Synthesis of α’-acyloxy enones from enones using manganese(III) acetate in combination with manganese(II) carboxylates or carboxylic acids. J. Org. Chem. 1989, 54, 4020. [Google Scholar] [CrossRef]
- Demir, A.S.; Sayraç, T.; Watt, D.S. The oxidation of β-alkoxycyclopentenones and β-alkoxycyclohexenones to α’-acyloxy derivatives using manganese(III) acetate in combination with carboxylic acids. Synthesis 1990, 1119. [Google Scholar] [CrossRef]
- Demir, A.S.; Camkerten, N.; Gercek, Z.; Duygu, N.; Reis, O.; Arikan, E. Butenolide annelation using a manganese(III) oxidation. A synthesis of 4-arylfuran-2(5H)-ones. Tetrahedron 1999, 56, 2441. [Google Scholar] [CrossRef]
- Tsuruta, T.; Harada, T.; Nishino, H.; Kurosawa, K. The α-chlorination of aryl ketones with manganese(III) acetate in the presence of chloride ion. Bull. Chem. Soc. Jpn. 1985, 58, 142. [Google Scholar] [CrossRef]
- Yonemura, H.; Nishino, H.; Kurosawa, K. Reaction of α,β-unsaturated carboxylic acids with manganese(III) acetate in the presence of chloride ion. Bull. Chem. Soc. Jpn. 1986, 59, 3153. [Google Scholar] [CrossRef]
- Ketcha, D.M. The manganese(III) acetate oxidation of N-protected indolines. Tetr. Lett. 1988, 29, 2151. [Google Scholar] [CrossRef]
- Ketcha, D.M.; Zhou, Q.; Grossie, D. Manganese(III) acetate oxidation of alkyl substituted 1-(phenylsulfonyl)indoles. Synth. Commun. 1994, 24, 565. [Google Scholar] [CrossRef]
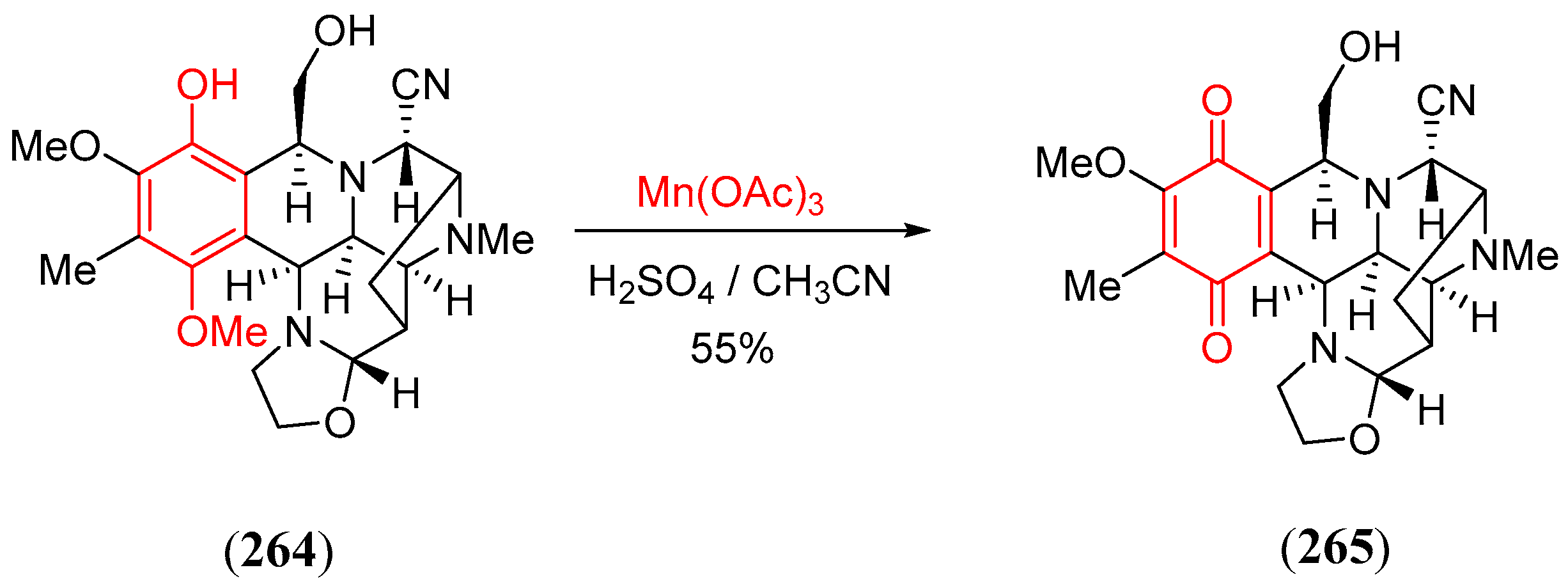
- Mitasev, B.; Porco, J.A. Manganese(III)-mediated transformations of phloroglucinols: A formal oxidative [4 + 2] cycloaddition leading to bicyclo[2.2.2]octadiones. Org. Lett. 2009, 11, 2285. [Google Scholar] [CrossRef] [PubMed]
- Cossy, J.; Bouzide, A. Manganese(III) acetate and copper(II) acetate promoted oxidation of β-enaminoamides and β-enaminoesters. Tetrahedron 1999, 55, 6483. [Google Scholar] [CrossRef]
- Singh, O.V.; Muthukrishnan, M.; Raj, G. Manganese(III) acetate mediated oxidation of flavanones: A facile synthesis of flavones. Synth. Commun. 2005, 35, 2723. [Google Scholar] [CrossRef]
- Biçer, E. A review on C-P bonding reactions assisted by manganese(III) acetate. Phosphorus Sulfur Silicon Relat. Elem. 2021, 196, 791–808. [Google Scholar] [CrossRef]
- Berger, O.; Montchamp, J.-L. Manganese-Mediated Homolytic Aromatic Substitution with Phosphinylidenes. Chem. Rec. 2017, 17, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, K.S.; Barbier, F.; Bégué, J.P.; Bonnet-Delpon, D. Manganese (III) acetate dihydrate catalyzed aerobic epoxidation of unfunctionalized olefins in fluorous solvents. Tetrahedron 1998, 54, 7457. [Google Scholar] [CrossRef]
- Hwang, S.; Lee, Y.W.; Lee, C.H.; Ahn, I.S. Manganese(III) acetate-catalyzed synthesis of polyguaiacol. J. Poly. Sci. Part A Poly. Chem. 2008, 46, 6009. [Google Scholar] [CrossRef]
- Hessel, L.W.; Romers, C. The crystal structure of anhydrous manganic acetate. Recl. Trav. Chim. Pays-Bas 1969, 88, 545. [Google Scholar] [CrossRef]
- Baikie, A.R.E.; Hursthouse, M.B.; New, D.B.; Thornton, P. Preparation, crystal structure, and magnetic properties of a trinuclear mixed-valence manganese carboxylate. J. Chem. Soc. Chem. Comm. 1978, 2, 62. [Google Scholar] [CrossRef]
- Güvenç, A.; Karabacakoğlu, B.; Koçkar, Ö.M.; Pekel, A.T. The synthesis of manganese(III) acetate in bipolar packed-bed and trickle-bed electrode cells. Turk. J. Chem. 2000, 24, 101. [Google Scholar]
- Güvenç, A.; Pekel, A.T.; Koçkar, Ö.M. The experimental optimization of the electrosynthesis of manganese(III) acetate in a bipolar packed-bed reactor. Chem. Eng. J. 2004, 99, 257. [Google Scholar] [CrossRef]
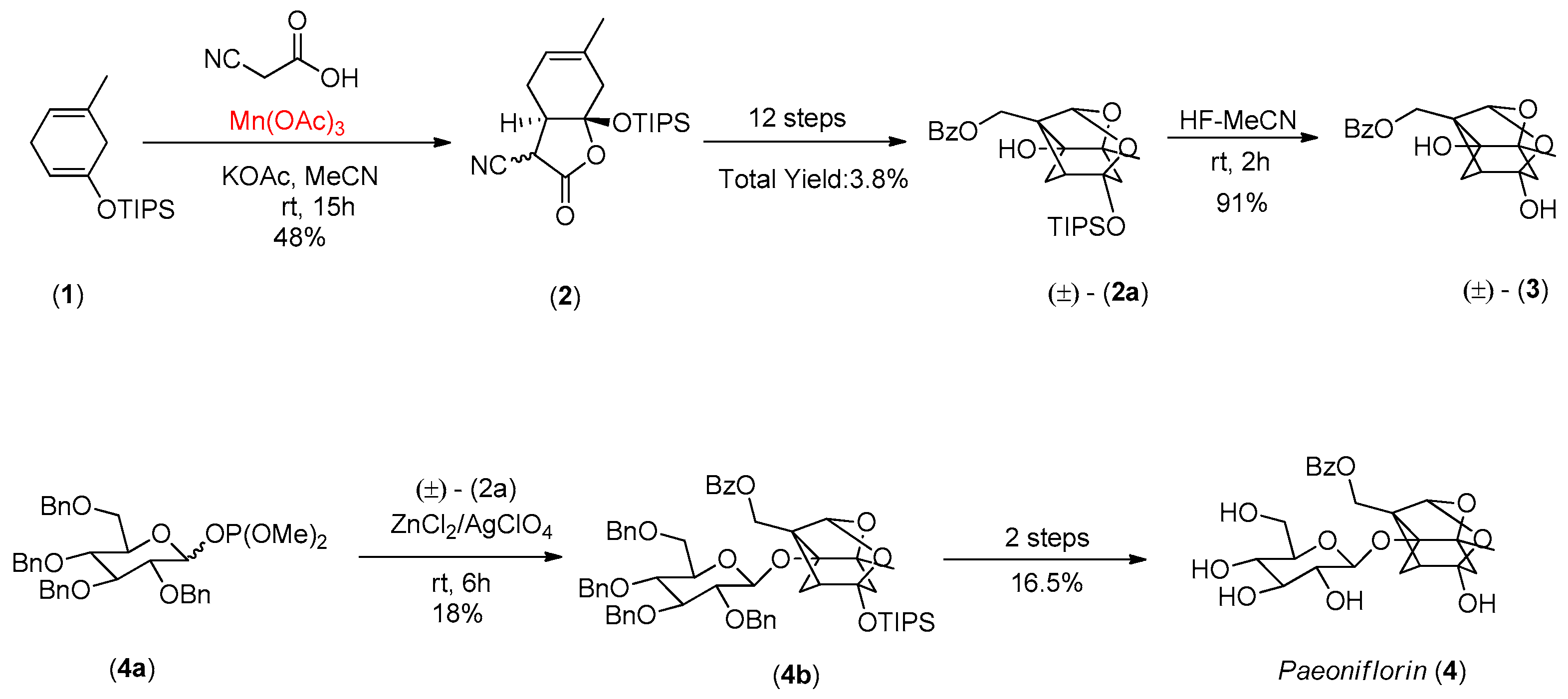
- Corey, E.J.; Wu, Y.J. Total synthesis of (±)-paeoniflorigenin and paeoniflorin. J. Am. Chem. Soc. 1993, 115, 8871. [Google Scholar] [CrossRef]
- Martinet, S.; Méou, A.; Brun, P. Access to enantiopure 2,5-diaryltetrahydrofurans–application to the synthesis of (–)-virgatusin and (+)-urinaligran. Eur. J. Org. Chem. 2009, 2009, 2306. [Google Scholar] [CrossRef]
- Snider, B.B.; Han, L.; Xie, C. Synthesis of 2,3-dihydrobenzofurans by Mn(OAc)3-based oxidative cycloaddition of 2-cyclohexenones with alkenes. Synthesis of (±)-conocarpan. J. Org. Chem. 1997, 62, 6978. [Google Scholar] [CrossRef]
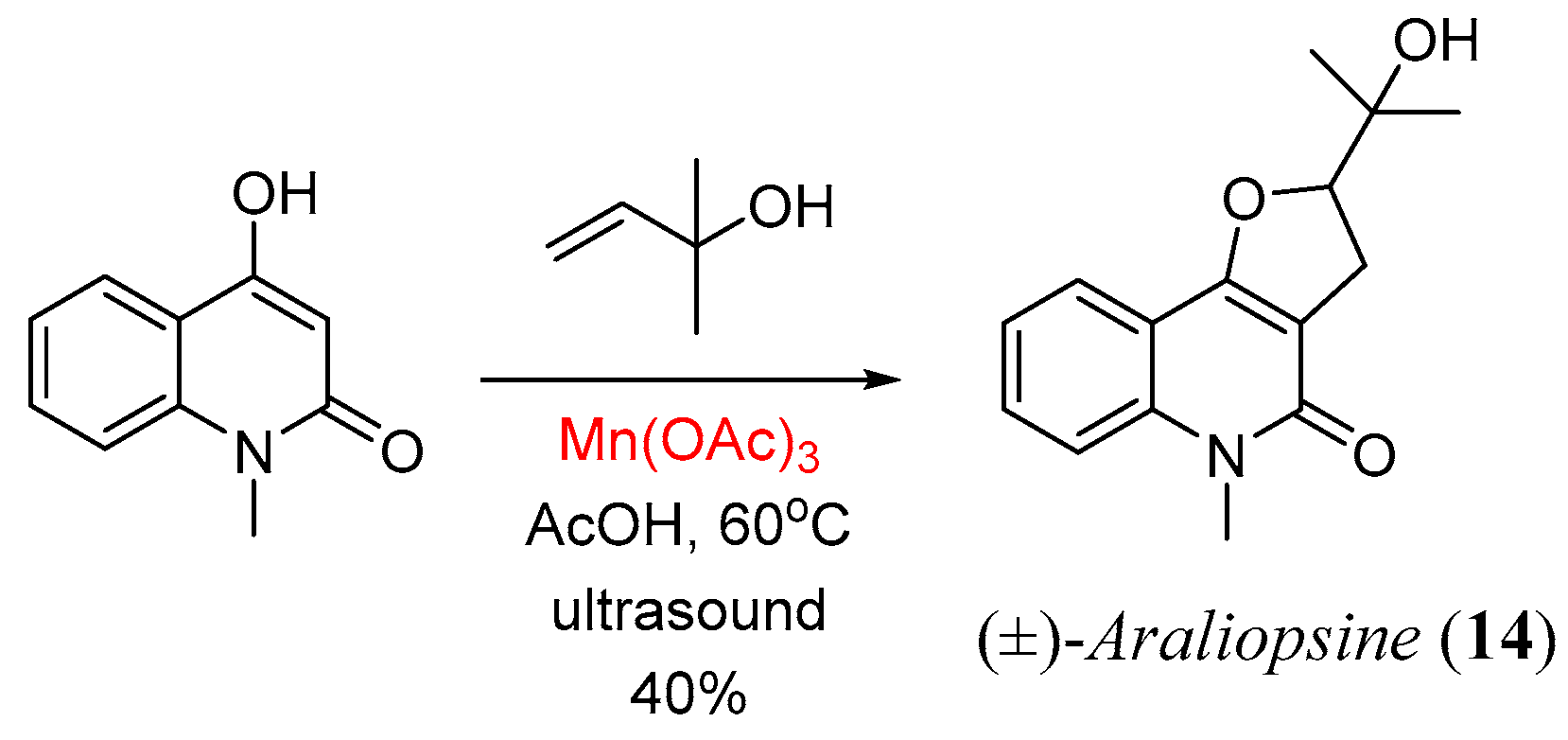
- Bar, G.; Parsons, A.F.; Thomas, C.B. A radical approach to araliopsine and related quinoline alkaloids using manganese(III) acetate. Tetrahedron Lett. 2000, 41, 7751. [Google Scholar]
- Bar, G.; Parsons, A.F.; Thomas, C.B. Manganese(III) acetate mediated radical reactions leading to araliopsine and related quinoline alkaloids. Tetrahedron 2001, 57, 4719. [Google Scholar] [CrossRef]
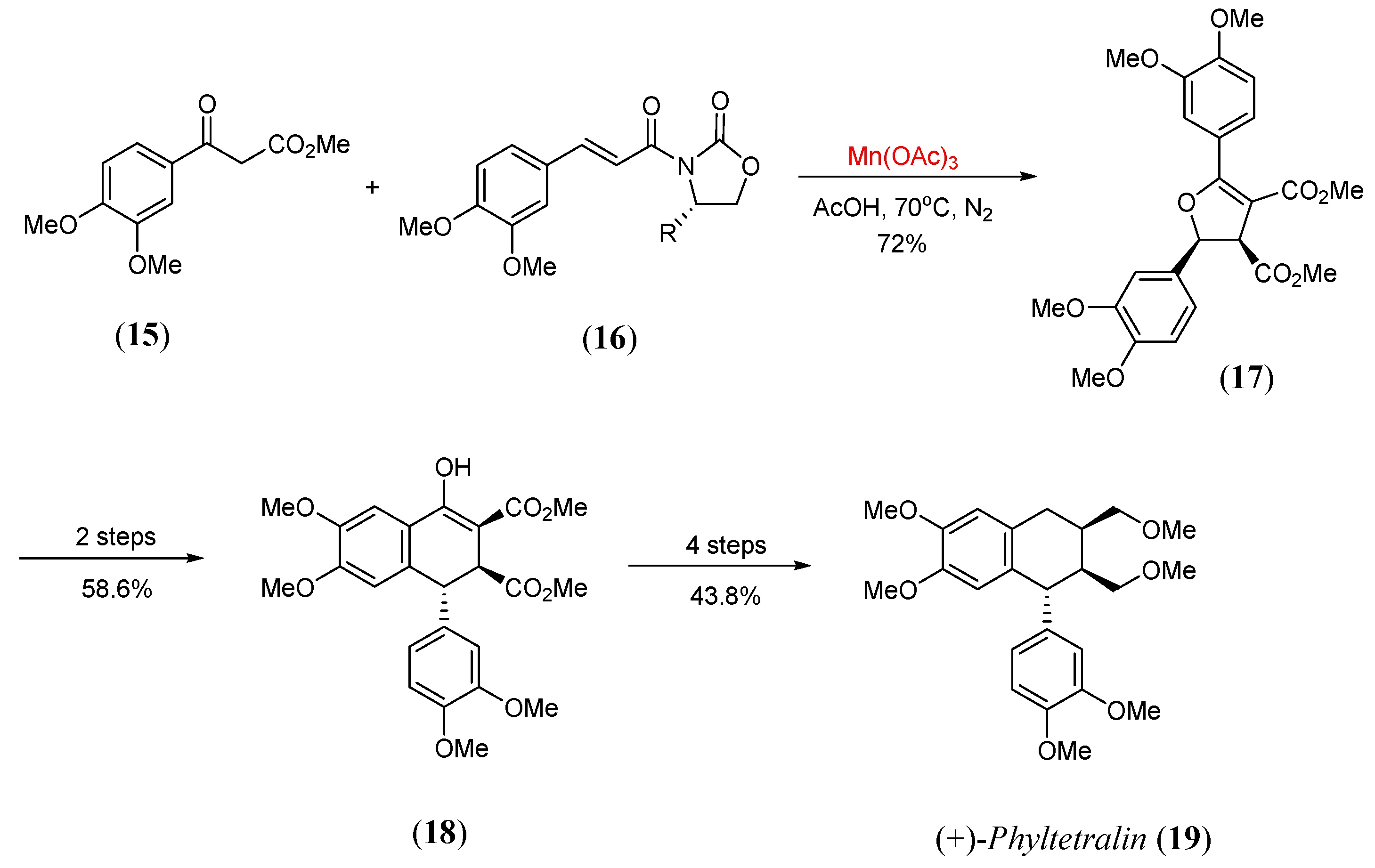
- Garzino, F.; Meou, A.; Brun, P. Asymmetric radical synthesis of 2,5-diaryl-2,3-dihydrofurans–Application to the preparation of (+)-phyltetralin. Eur. J. Org. Chem. 2003, 2003, 1410. [Google Scholar] [CrossRef]
- Garzino, F.; Meou, A.; Brun, P. A new approach toward the synthesis of heterolignans. Tetr. Lett. 2002, 43, 5049. [Google Scholar] [CrossRef]
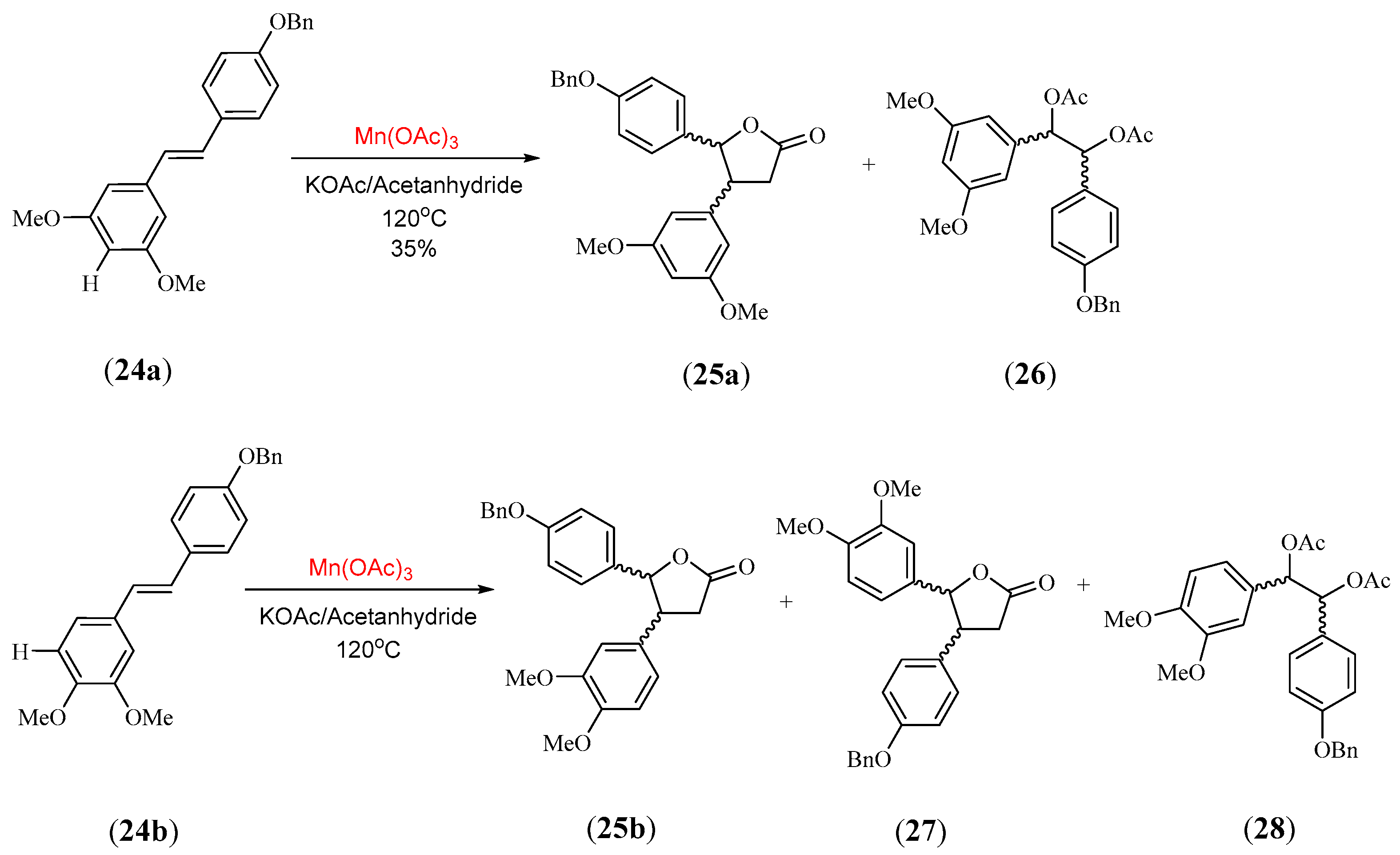
- Thomas, N.F.; Lee, K.C.; Paraidathathu, T.; Weber, J.F.F.; Awang, K. Manganese triacetate oxidative lactonisation of electron-rich stilbenes possessing catechol and resorcinol substitution (resveratrol analogues). Tetrahedron Lett. 2002, 43, 3151. [Google Scholar] [CrossRef]
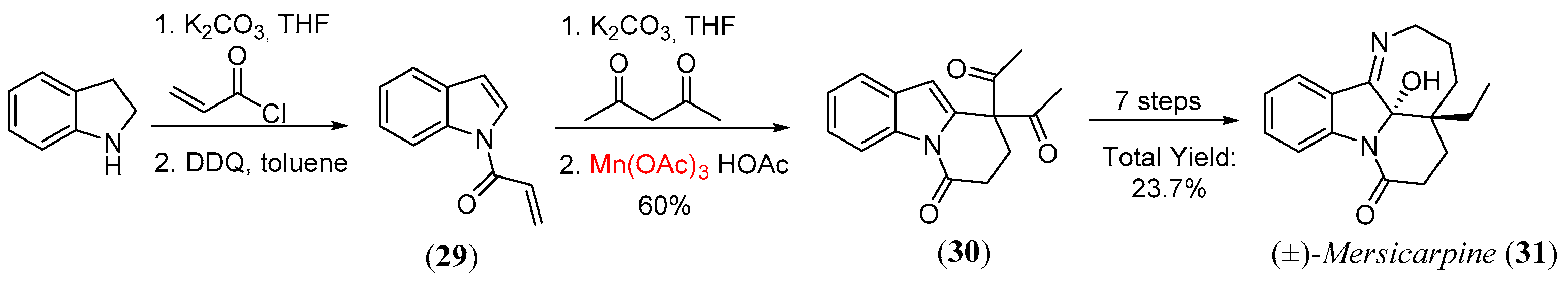
- Magolan, J.; Carson, C.A.; Kerr, M.A. Total synthesis of (±)-mersicarpine. Org. Lett. 2008, 10, 1437. [Google Scholar] [CrossRef]
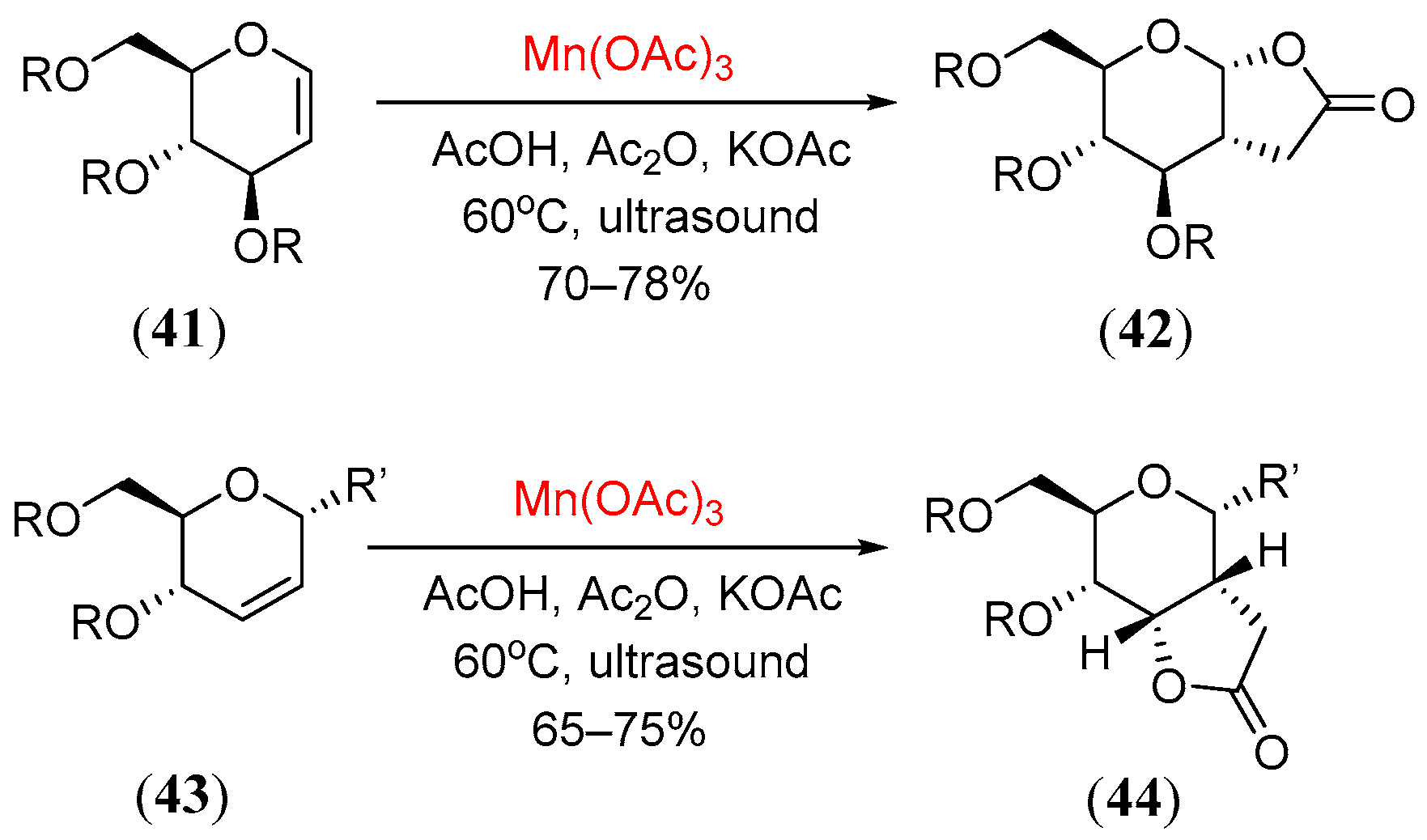
- Hartmann, K.; Kim, B.G.; Linker, T. Manganese(III)-mediated radical reactions in carbohydrate chemistry: A new route to 3-deoxy-D-manno-oct-2-ulosonic acid (KDO). Synlett 2004, 15, 2728. [Google Scholar]
- Kim, B.G.; Schilde, U.; Linker, T. New radical approaches to 3-deoxy-D-oct-2-ulosonic acids (KDO). Synthesis 2005, 9, 1507. [Google Scholar]
- Chuang, C.P.; Wang, S.F. Manganese(IIl) acetate initiated oxidative free radical reaction between 1,4-naphthoquinones and α-alkylmalonates. Tetrahedron 1998, 54, 10043. [Google Scholar] [CrossRef]
- Murphy, W.S.; Neville, D.; Ferguson, G. Regiospecific free radical annulation reactions of diethylbenzylmalonate with unsymmetrical 1,4-naphthoquinones: A direct approach to ring an aromatic angucyclinones. Tetrahedron Lett. 1996, 37, 7615. [Google Scholar] [CrossRef]
- Carreno, M.C.; Urbano, A. Recent advances in the synthesis of angucyclines. Synlett 2005, 1. [Google Scholar]
- Yousuf, S.H.; Mukherjee, D.; Mallikharjunrao, L.; Taneja, S.C. Highly regio- and stereoselective one-pot synthesis of carbohydrate-based butyrolactones. Org. Lett. 2001, 13, 576. [Google Scholar]
- Altun, Y.; Dogan, S.D.; Balcı, M. Synthesis of branched carbasugars via photooxygenation and manganese(III) acetate free radical cyclization. Tetrahedron 2014, 70, 4884. [Google Scholar]
- McDonald, J.W.; Miller, J.E.; Kim, M.; Velu, S.E. An expedient synthesis of murrayaquinone A via a novel oxidative free radical reaction. Tetrahedron Lett. 2018, 59, 550–553. [Google Scholar] [CrossRef]
- White, J.D.; Somers, T.C.; Yager, K.M. Synthesis of dihydropallescensin d via manganese mediated cyclization of an olefinic β-keto ester. Tetrahedron Lett. 1990, 31, 59. [Google Scholar]
- Iwasawa, N.; Funahashi, M.; Narasaka, K. Total synthesis of 10-isothiocyanaguaia-6-ene. Chem. Lett. 1994, 1697. [Google Scholar] [CrossRef]
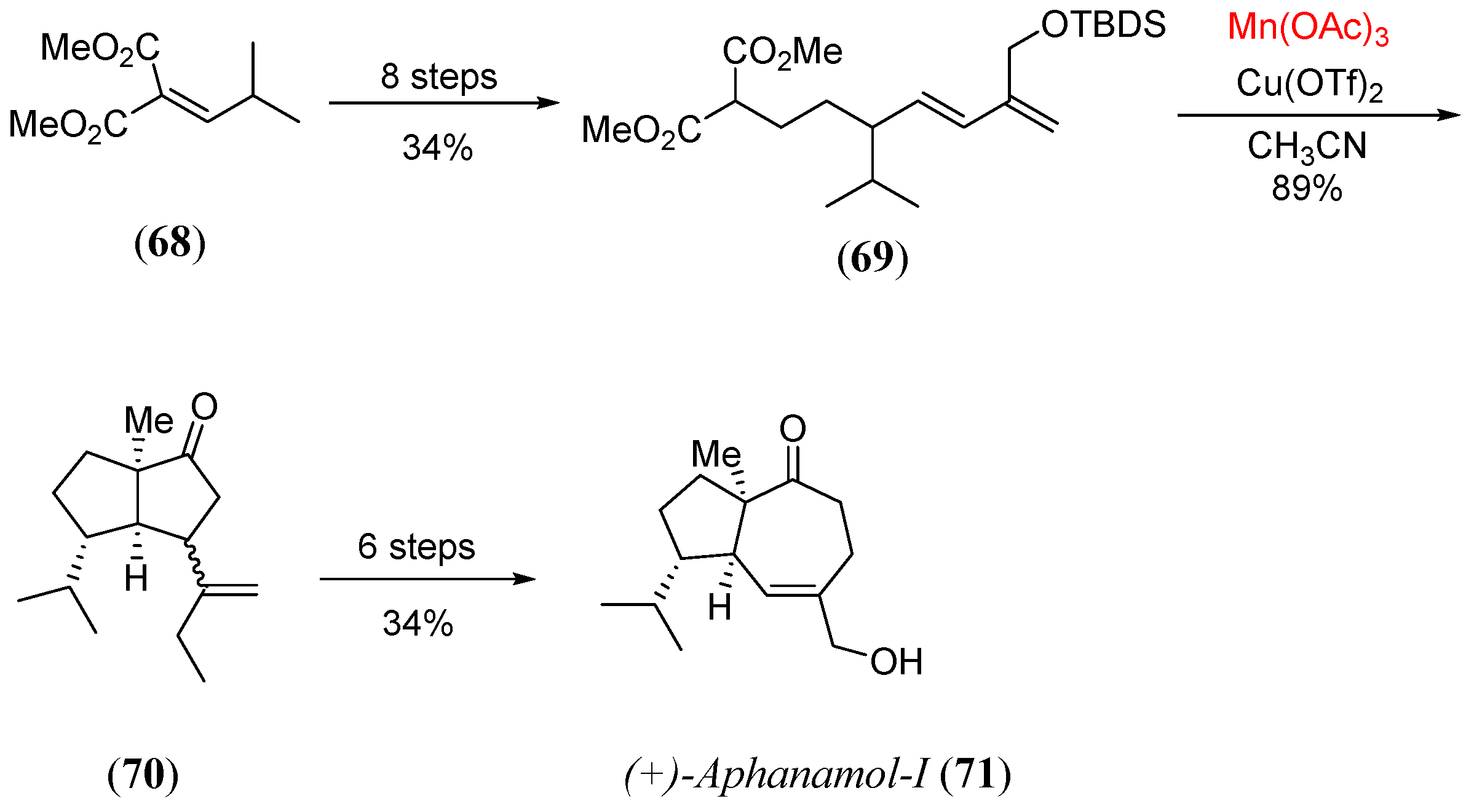
- Ferrara, S.J.; Burton, J.W. A short synthesis of aphanamol I in both racemic and enantiopure forms. Chem. Eur. J. 2016, 22, 11597. [Google Scholar] [CrossRef]
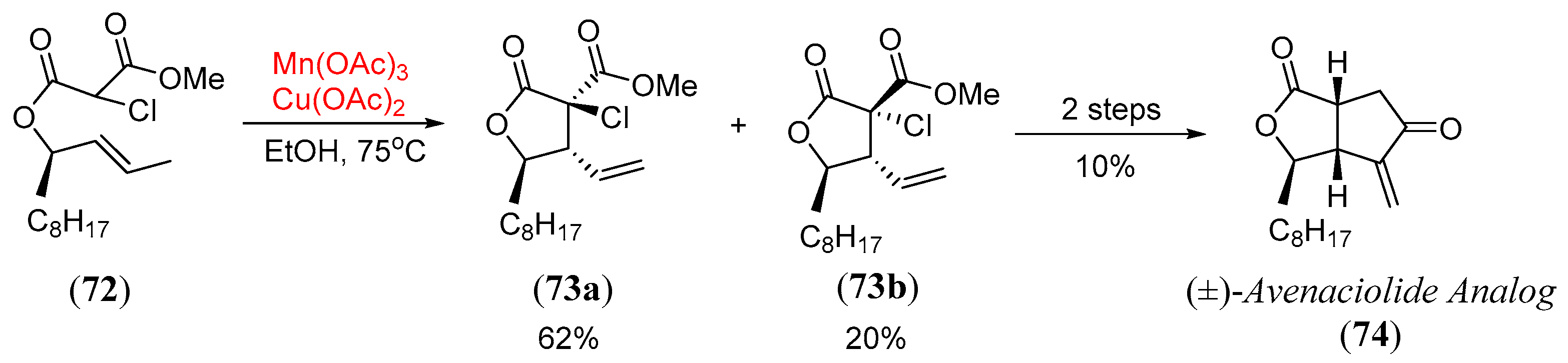
- Snider, B.B.; McCarthy, B.A. Oxidative free-radical cyclization of allylic α-chloromalonates. Synthesis of (±)-avenaciolide. Tetrahedron 1993, 49, 9447. [Google Scholar] [CrossRef]
- Paker, W.; Johnson, F. The total synthesis of dl-avenaciolide. J. Org. Chem. 1973, 38, 2489–2496. [Google Scholar] [CrossRef]
- Martinez, S.A.; Gillard, M.; Chany, A.C.; Burton, J.W. Short total syntheses of the avenaciolide family of natural products. Tetrahedron 2018, 74, 5012. [Google Scholar]
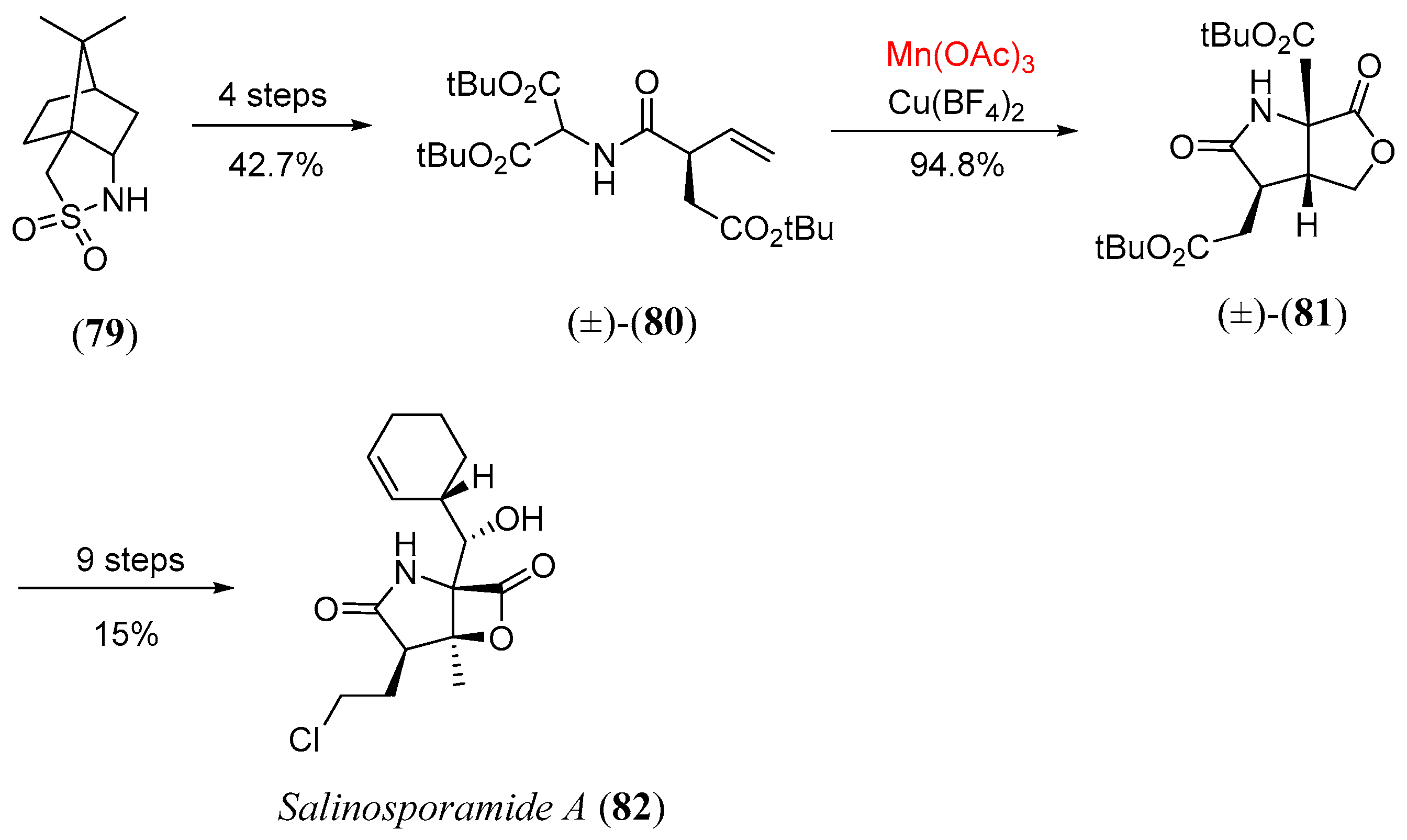
- Logan, A.W.J.; Sprague, S.J.; Foster, R.W.; Marx, L.B.; Garzya, V.; Hallside, M.S.; Thompson, A.L.; Burton, J.W. Diastereoselective synthesis of fused lactone-pyrrolidinones; Application to a formal synthesis of (-)-salinosporamide A. Org. Lett. 2014, 16, 4078. [Google Scholar] [CrossRef] [PubMed]
- Marx, L.B.; Burton, J.W. A total synthesis of salinosporamide A. Chem. Eur. J. 2018, 24, 6747. [Google Scholar] [CrossRef] [PubMed]
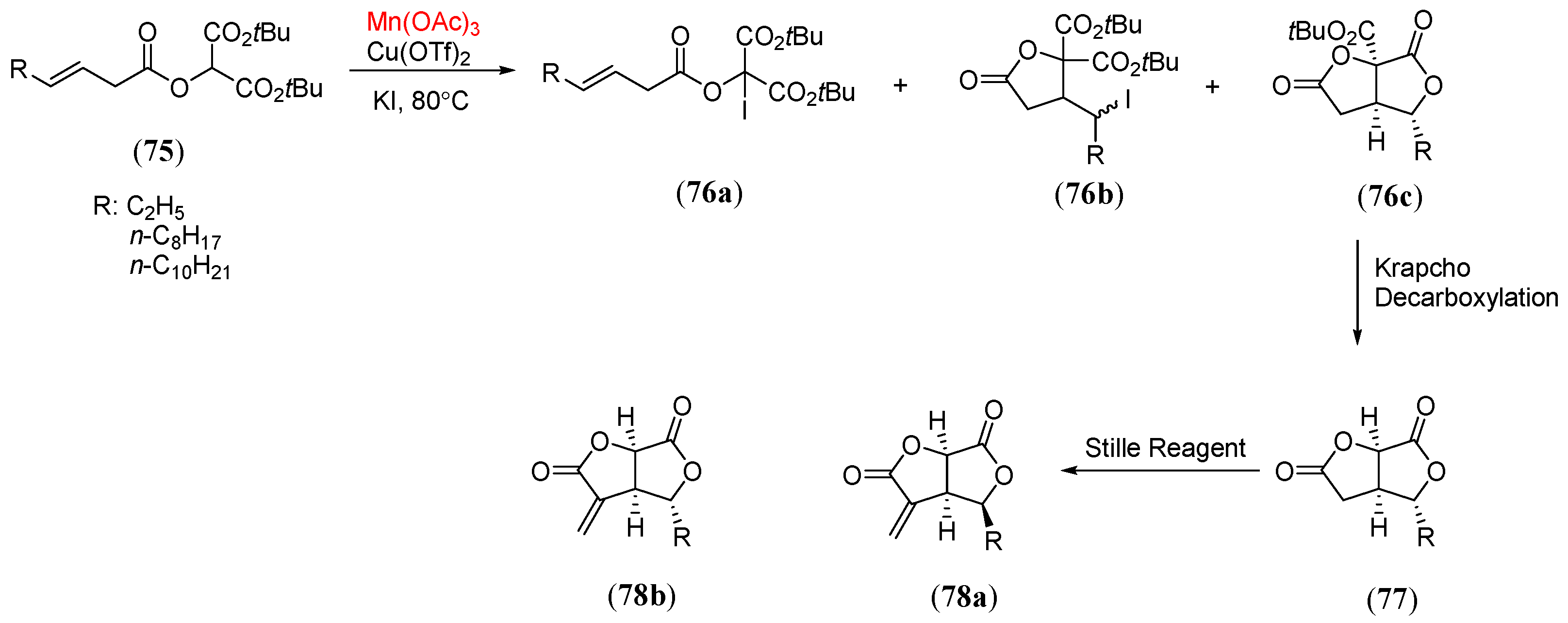
- Logan, A.W.J.; Parker, J.S.; Hallside, M.S.; Burton, J.W. Manganese(III) acetate mediated oxidative radical cyclizations. Toward vicinal all-carbon quaternary stereocenters. Org. Lett. 2012, 14, 2940. [Google Scholar] [CrossRef] [PubMed]
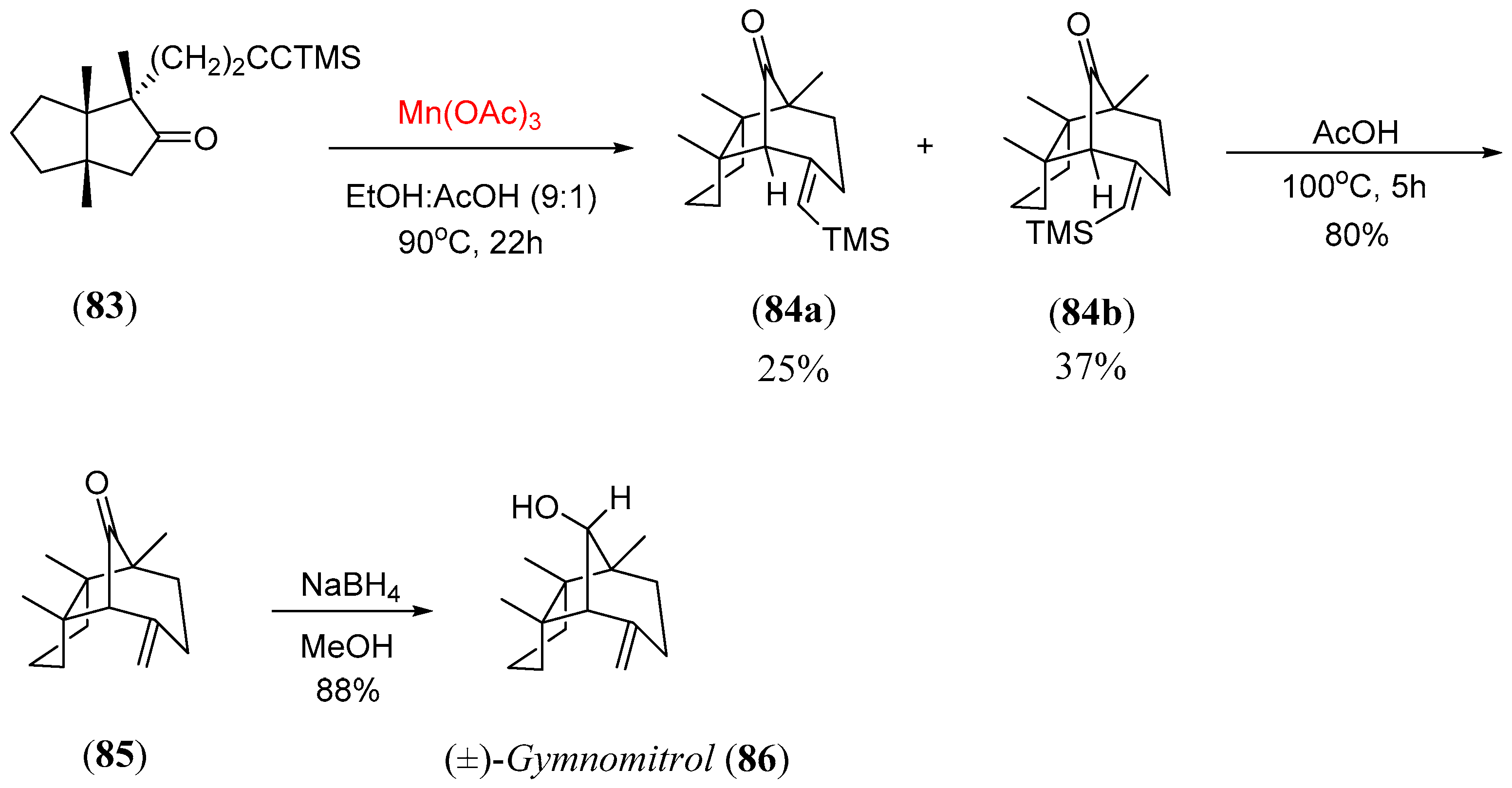
- O’Neil, S.V.; Quickley, C.A.; Snider, B.B. Synthesis of (±)-gymnomitrol. Mn(OAc)3 -Initiated free-radical cyclization of alkynyl ketones. J. Org. Chem. 1997, 62, 1970. [Google Scholar] [CrossRef] [PubMed]
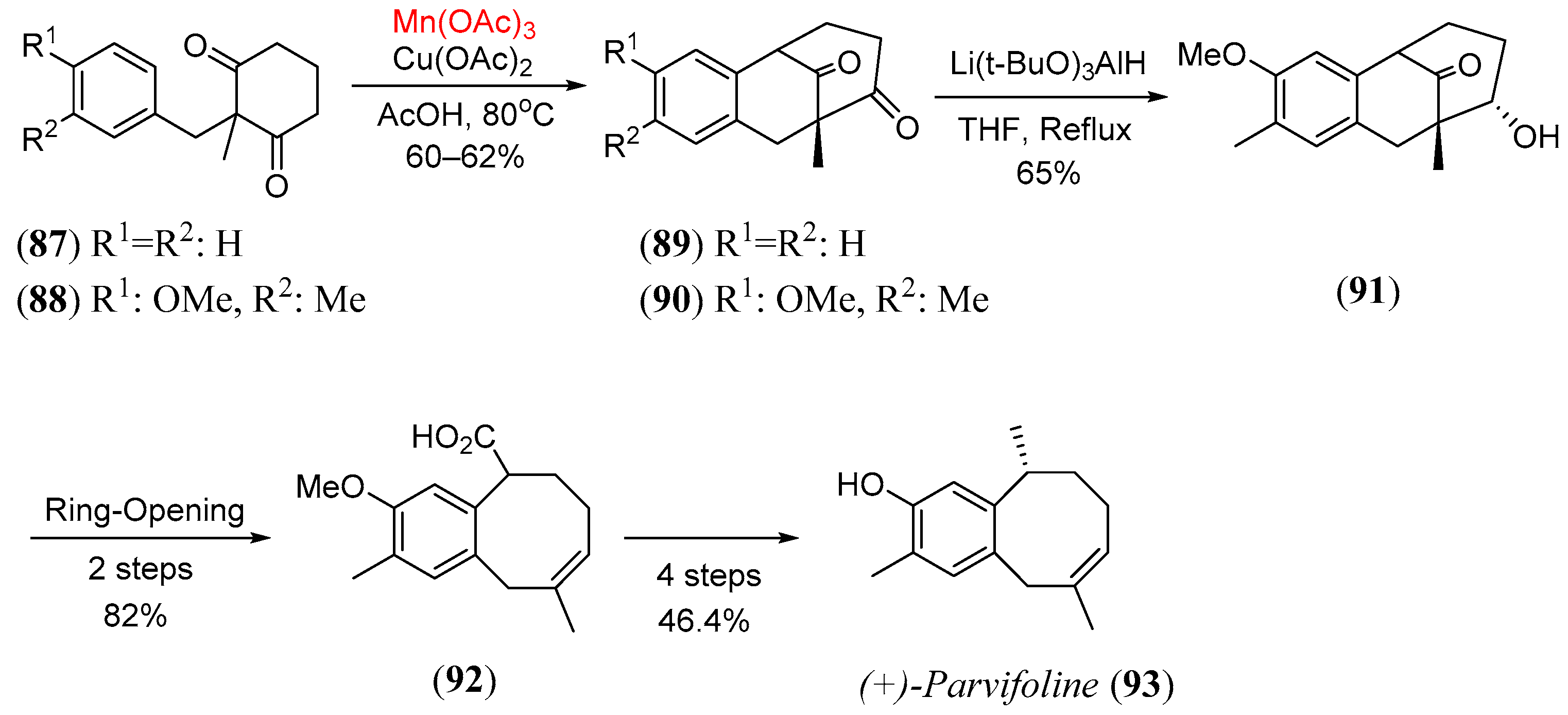
- Bhowmik, D.R.; Venkateswaran, R.V. A formal synthesis of (±) parvifoline by manganese(III)-based oxidative arylation of ketones. Tetr. Lett. 1999, 40, 7431. [Google Scholar] [CrossRef]
- Covarrubias-Zúñiga, A.; Cantú, F.; Maldonado, L.A. A total synthesis of the racemic sesquiterpene parvifoline. J. Org. Chem. 1998, 63, 2918–2921. [Google Scholar] [CrossRef]
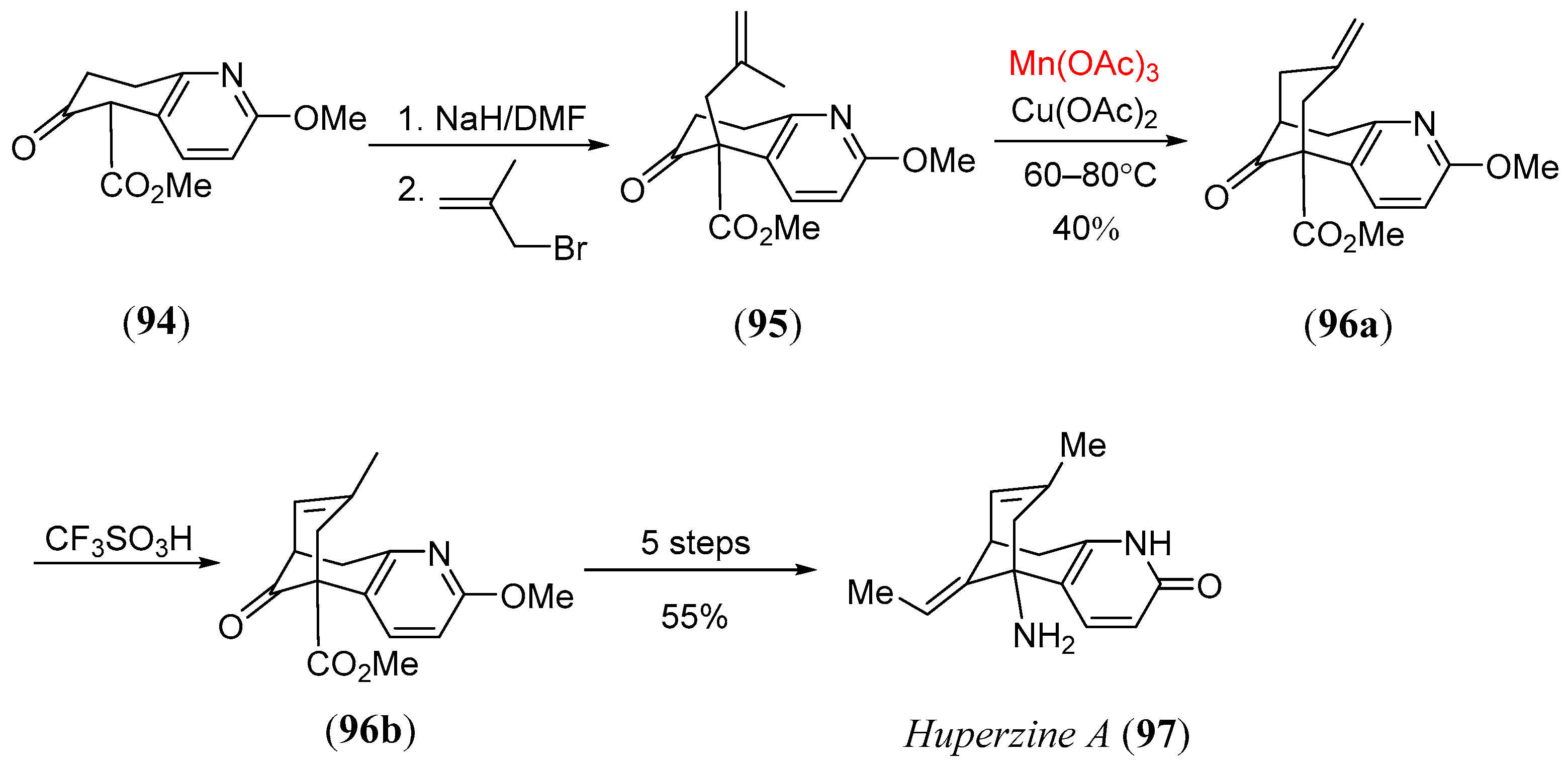
- Lee, I.Y.C.; Jung, M.H.H.; Lee, W.; Yang, J.Y. Synthesis of huperzine intermediates via Mn(III)-mediated radical cyclization. Tetr. Lett. 2002, 43, 2407. [Google Scholar] [CrossRef]
- Campiani, G.; Sun, L.Q.; Kozikowski, A.P.; Aagaard, P.; McKinney, M. A palladium-catalyzed route to huperzine a and its analogues and their anticholinesterase activity. J. Org. Chem. 1993, 58, 7660–7669. [Google Scholar] [CrossRef]
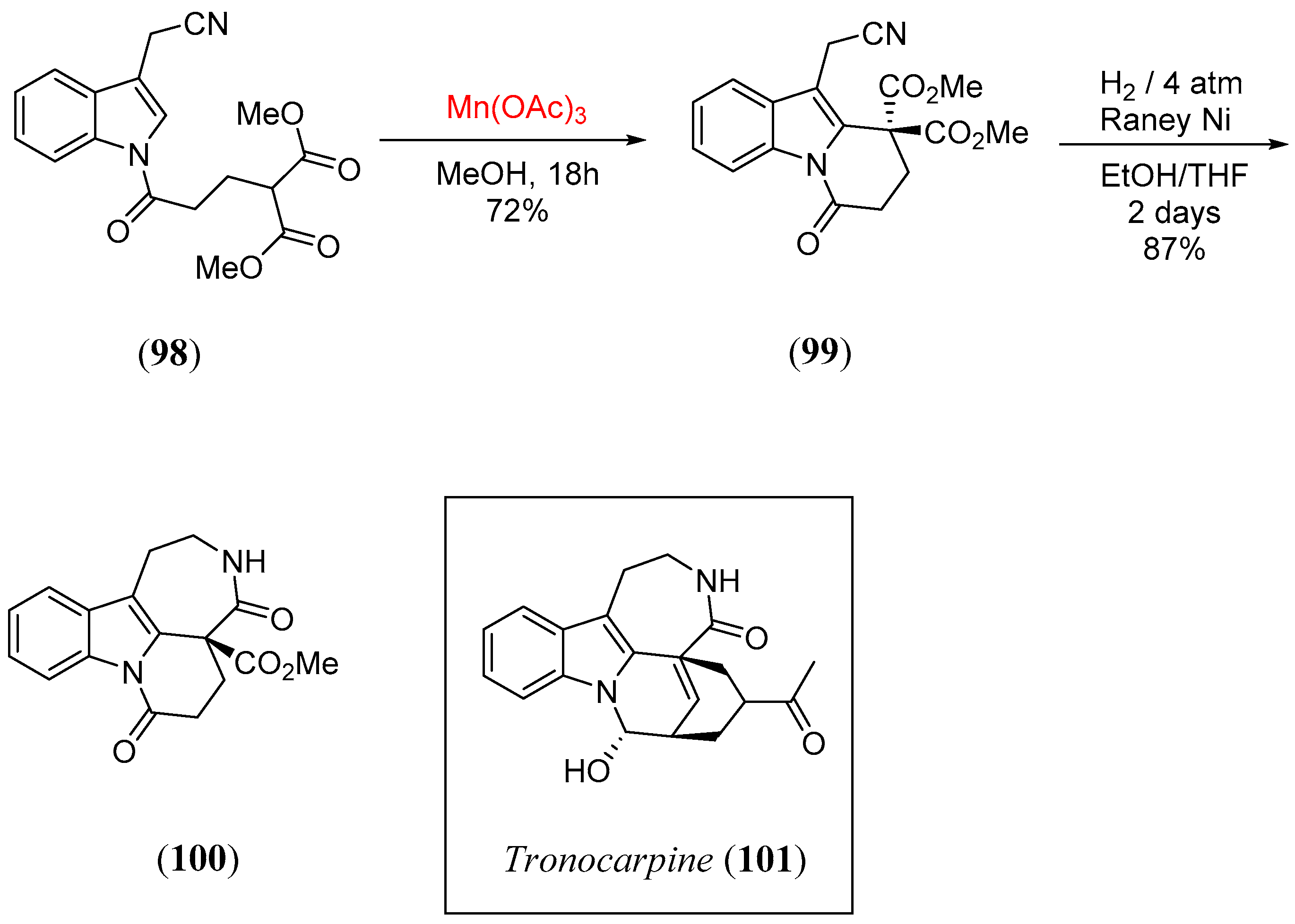
- Magolan, J.; Kerr, M.A. Expanding the scope of Mn(OAc)3-mediated cyclizations: Synthesis of the tetracyclic core of tronocarpine. Org. Lett. 2006, 8, 4561. [Google Scholar] [CrossRef]
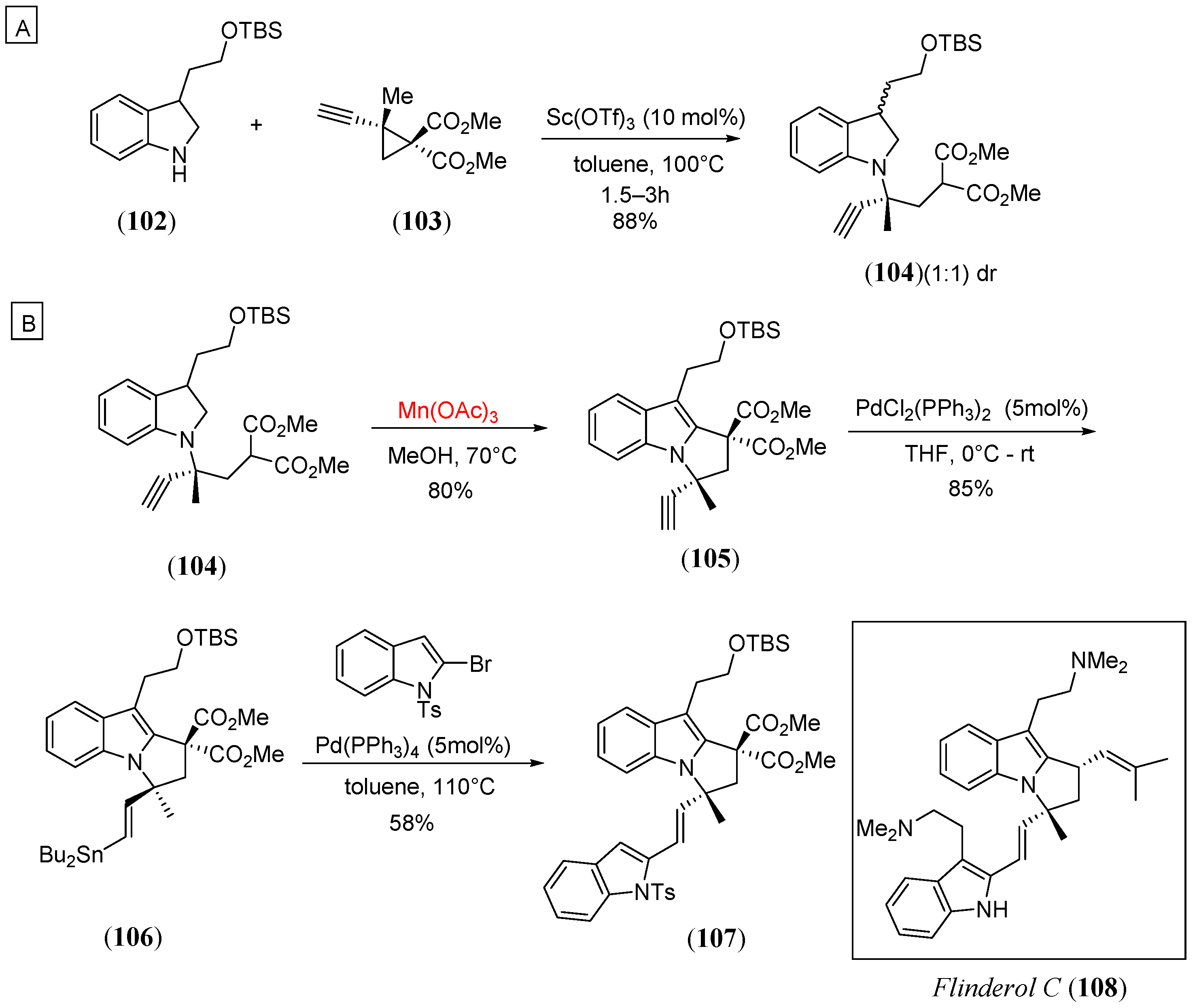
- Tejeda, J.E.C.; Landschoot, B.K.; Kerr, M.A. Radical cyclizations for the synthesis of pyrroloindoles: Progress toward the flinderoles. Org. Lett. 2016, 18, 2142. [Google Scholar] [CrossRef]
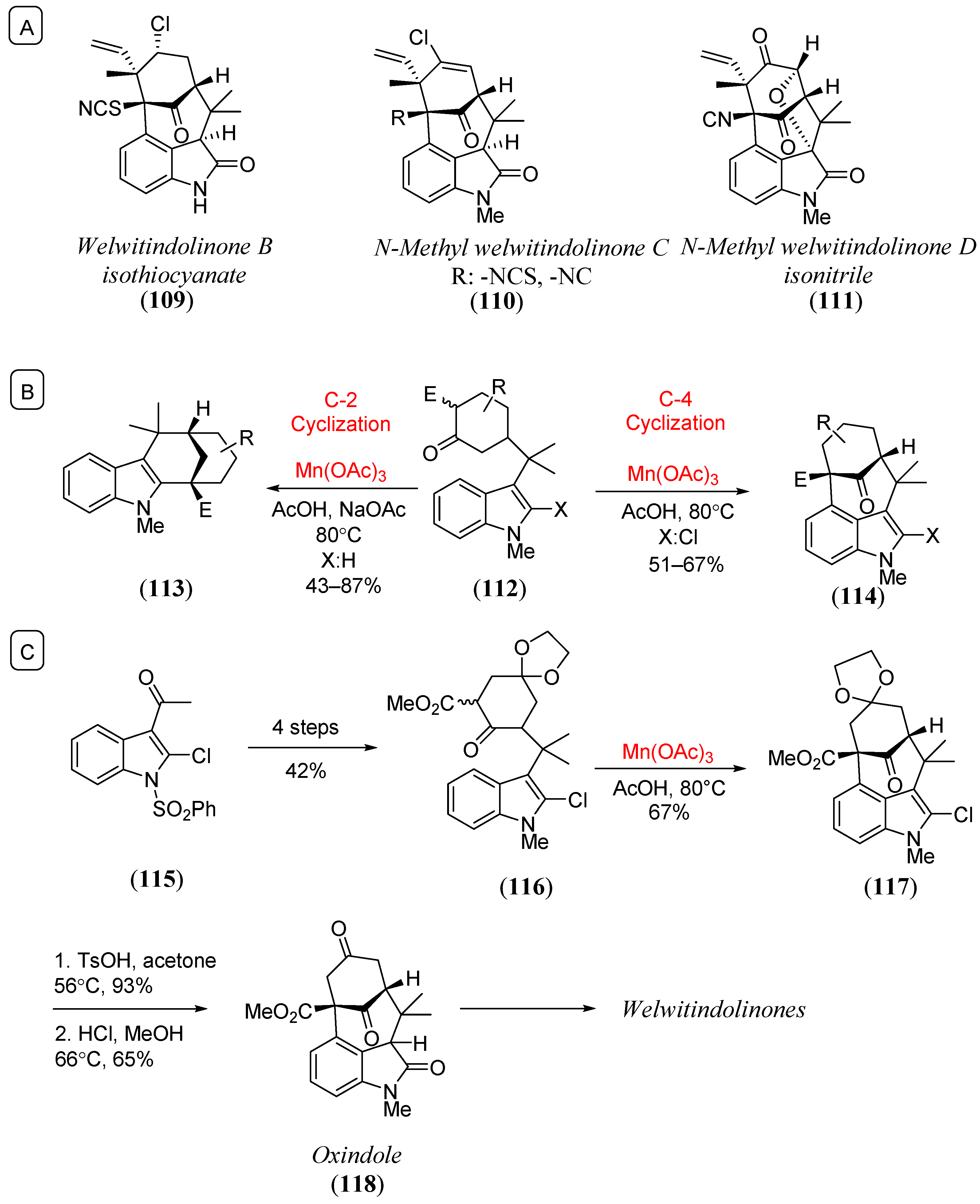
- Bhat, V.; MacKay, J.A.; Rawal, V.H. Directed oxidative cyclizations to C2- or C4-positions of indole: Efficient construction of the bicyclo[4.3.1]decane core of welwitindolinones. Org. Lett. 2011, 13, 3214. [Google Scholar] [CrossRef] [PubMed]
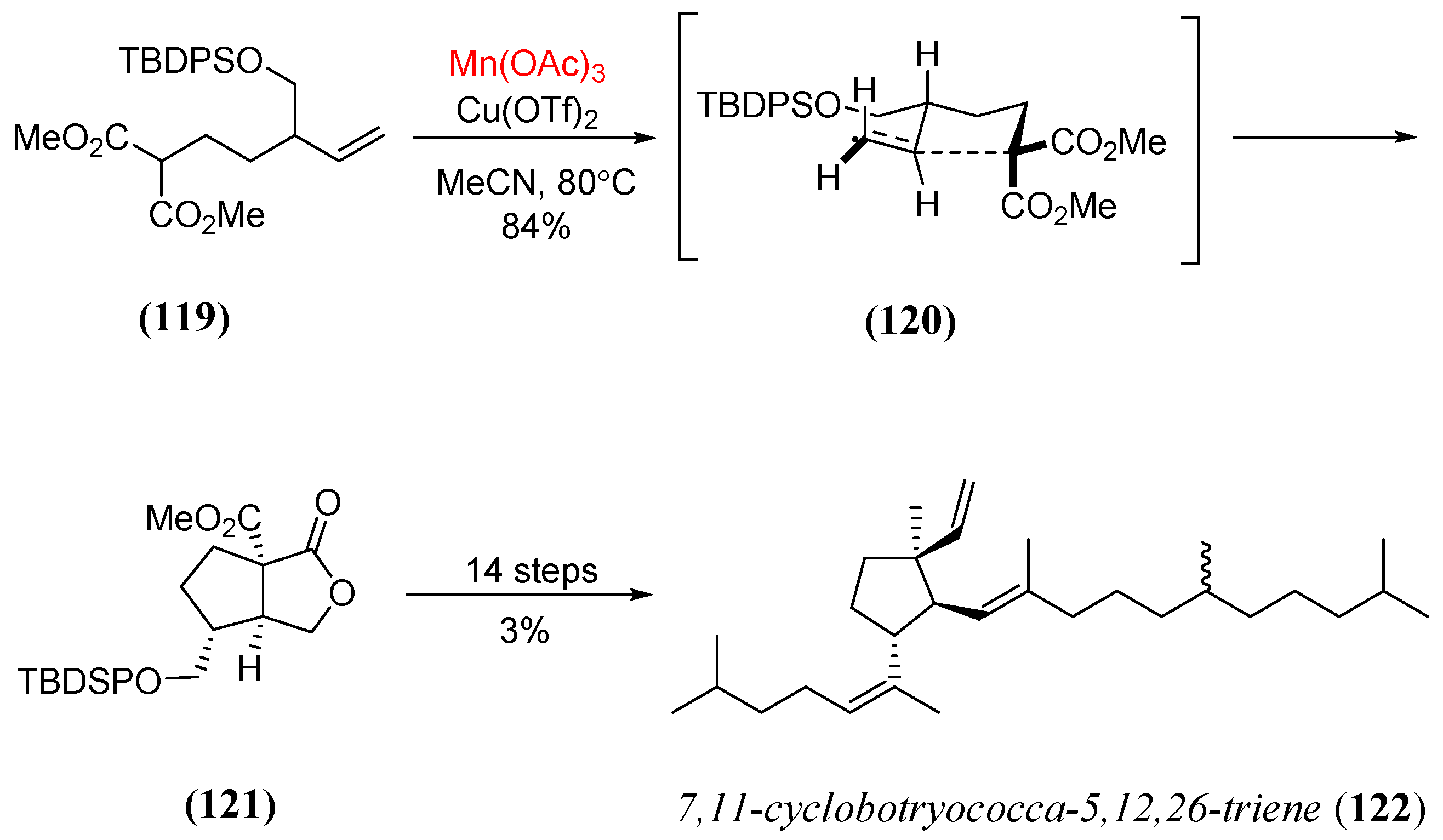
- Davies, J.J.; Krulle, T.M.; Burton, J.W. Total synthesis of 7,11-cyclobotryococca-5,12,26-triene using an oxidative radical cyclization as a key step. Org. Lett. 2010, 12, 2738. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.H.; Docherty, P.H.; Hulcoop, D.G.; Kemmitt, P.D.; Burton, J.W. Oxidative radical cyclisations for the synthesis of γ-lactones. Chem. Commun. 2008, 2559. [Google Scholar] [CrossRef] [PubMed]
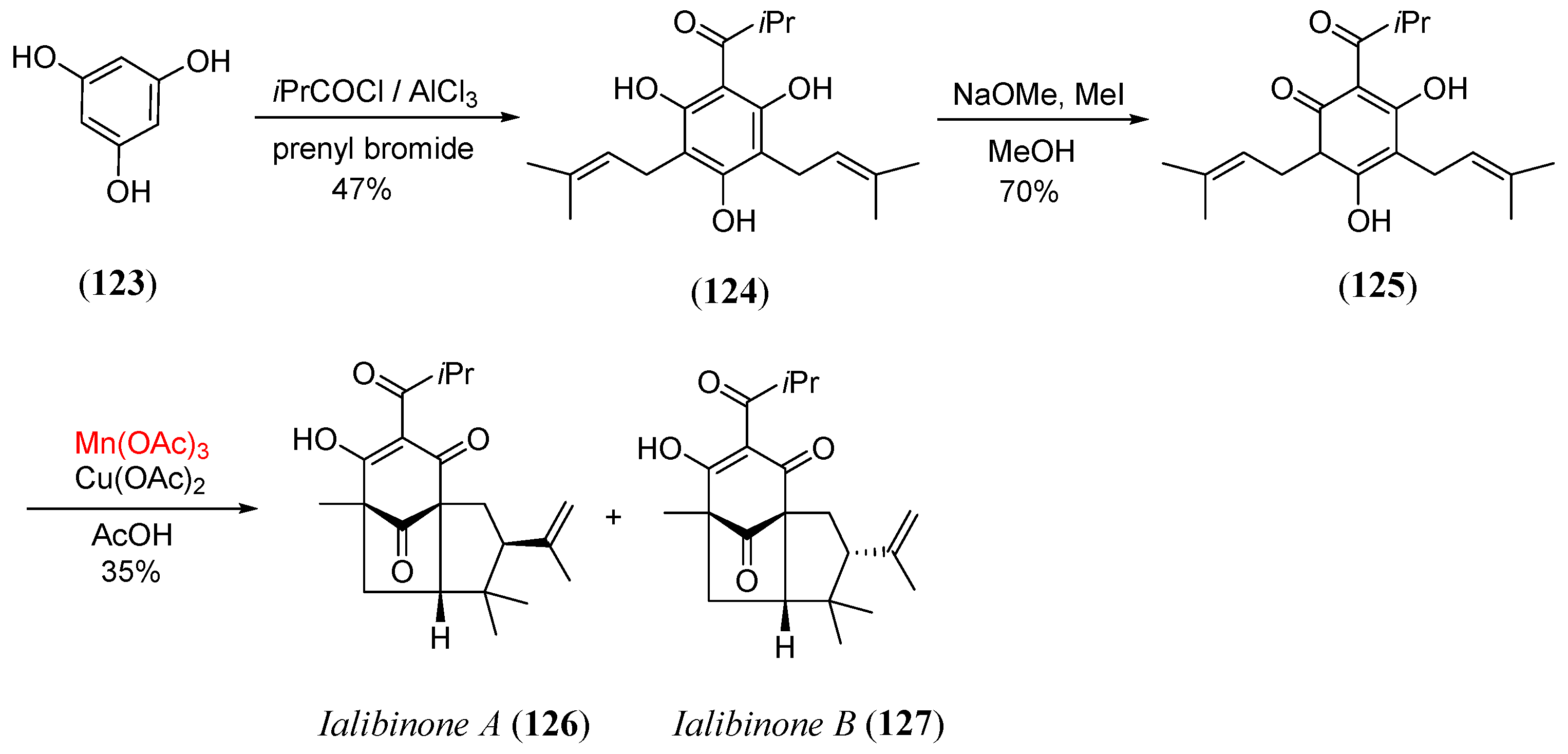
- Simpkins, N.S.; Weller, M.D. Expedient synthesis of ialibinones A and B by manganese(III)-mediated oxidative free radical cyclisation. Tetr. Lett. 2010, 51, 4823. [Google Scholar] [CrossRef]
- Lin, H.Y.; Snider, B.B. Mn(OAc)3-based α’-oxidative acetoxylation of N-trifluoroacetyl vinylogous amides. Heterocycles 2012, 86, 1391. [Google Scholar]
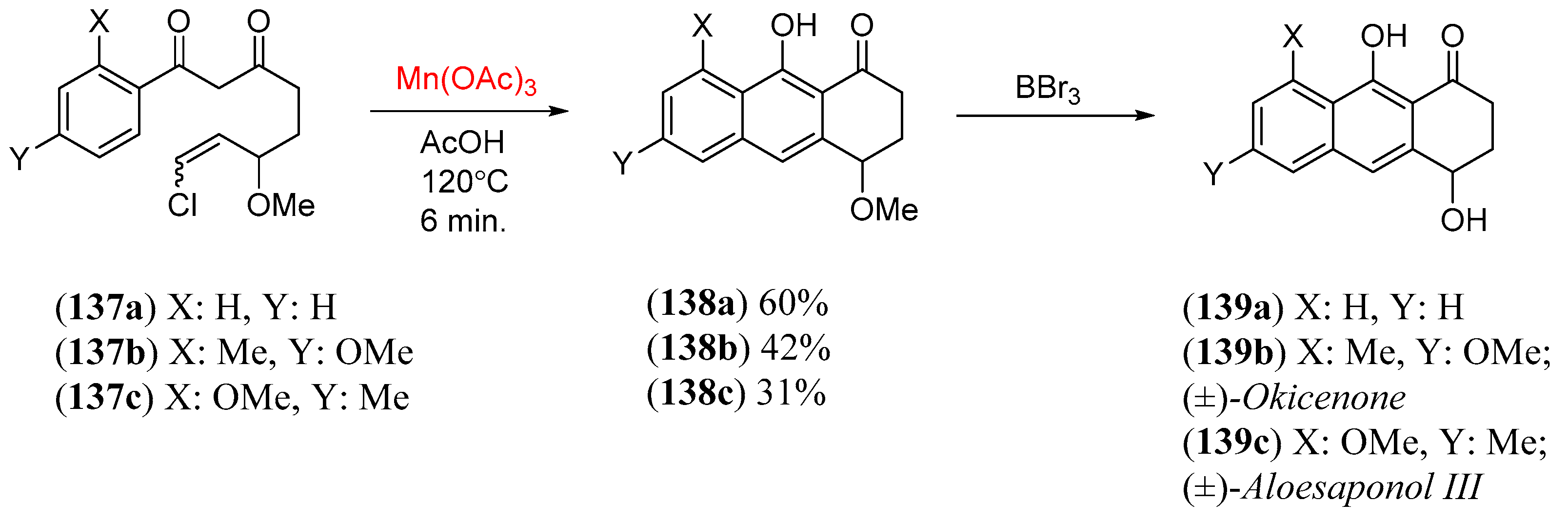
- Snider, B.B.; Zhang, Q. Synthesis of (±)-Okicenone and (±)-Aloesaponol III. J. Org. Chem. 1993, 58, 3185. [Google Scholar] [CrossRef]
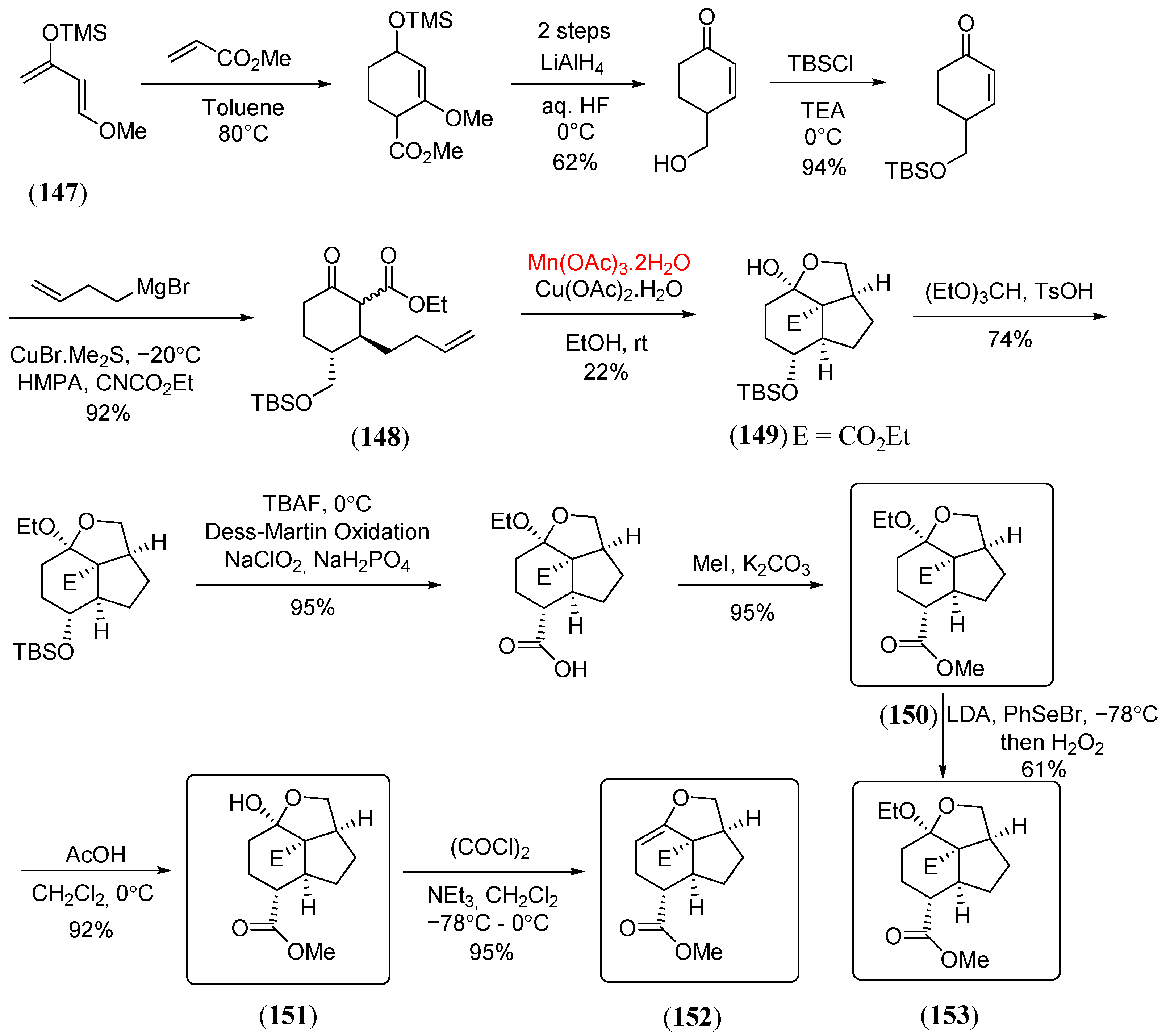
- Shepherd, E.D.; Hallside, M.S.; Sutro, J.L.; Thompson, A.; Hutchings, M.; Burton, J.W. Synthesis of the cyclopentane core of pepluanin A. Tetrahedron 2020, 76, 130981. [Google Scholar] [CrossRef]
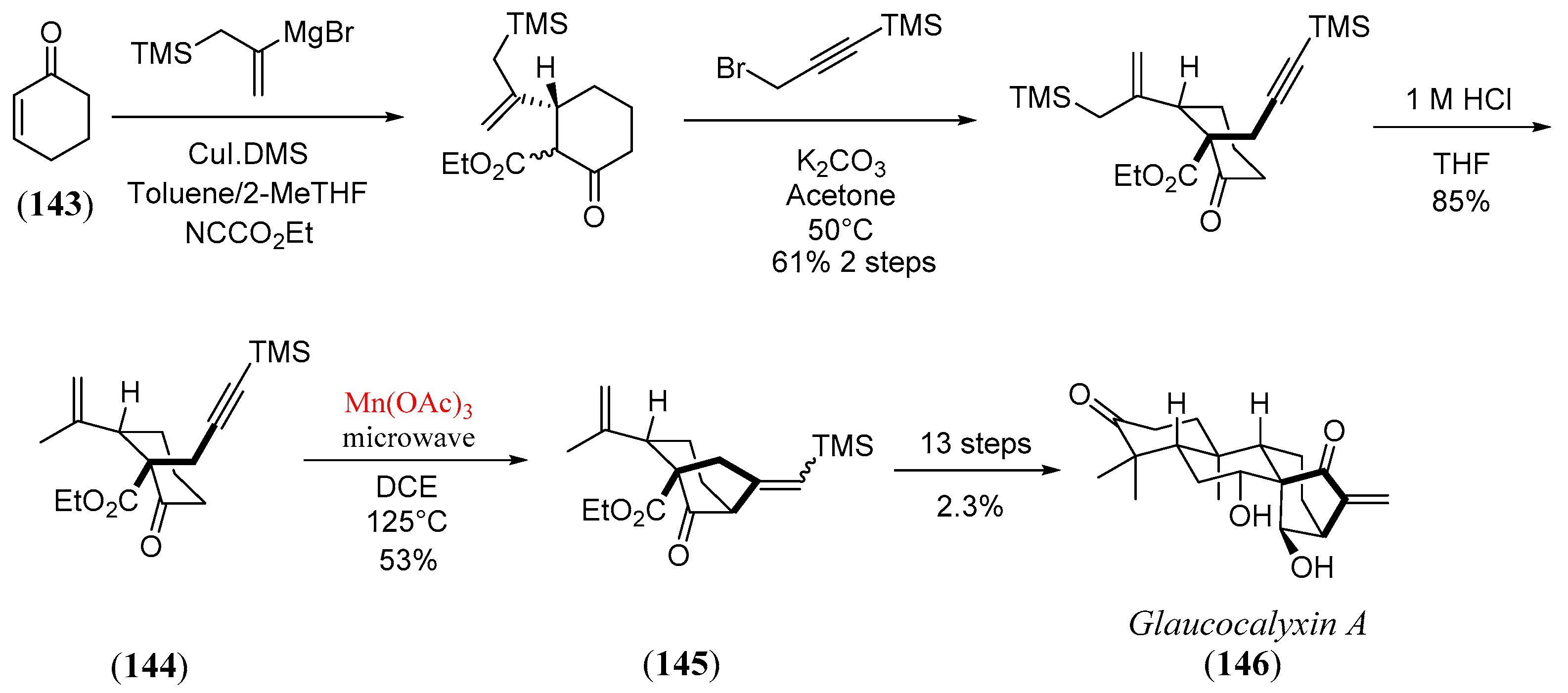
- Guo, J.; Li, B.; Ma, W.; Pitchakuntla, M.; Jia, Y. Total synthesis of (-)-glaucocalyxin A. Angew. Chem. Int. Ed. 2020, 59, 15195–15198. [Google Scholar] [CrossRef]
- Cao, J.; Thor, W.; Yang, S.; Zhang, M.; Bao, W.; Zhu, L.; Yang, W.; Cheng, Y.-K.; Lee, C.-S. Synthesis of the tricyclic picrotoxane motif by an oxidative cascade cyclization. Org. Lett. 2019, 21, 4896–4899. [Google Scholar] [CrossRef]
- Kuramochi, A.; Usuda, H.; Yamatsugu, K.; Kanai, M.; Shibasaki, M. Total synthesis of (±)-garsubellin A. J. Am. Chem. Soc. 2005, 127, 14200. [Google Scholar] [CrossRef]
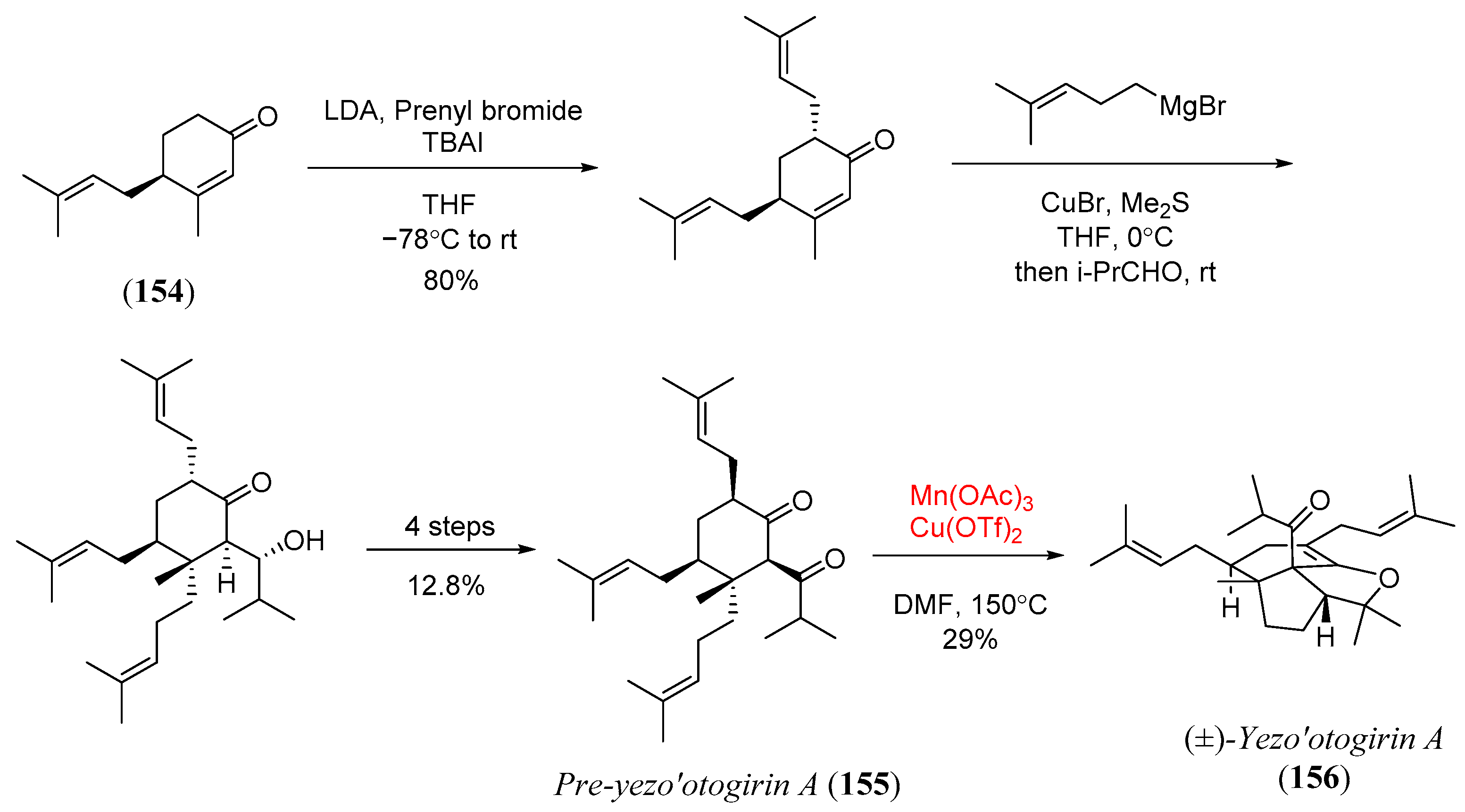
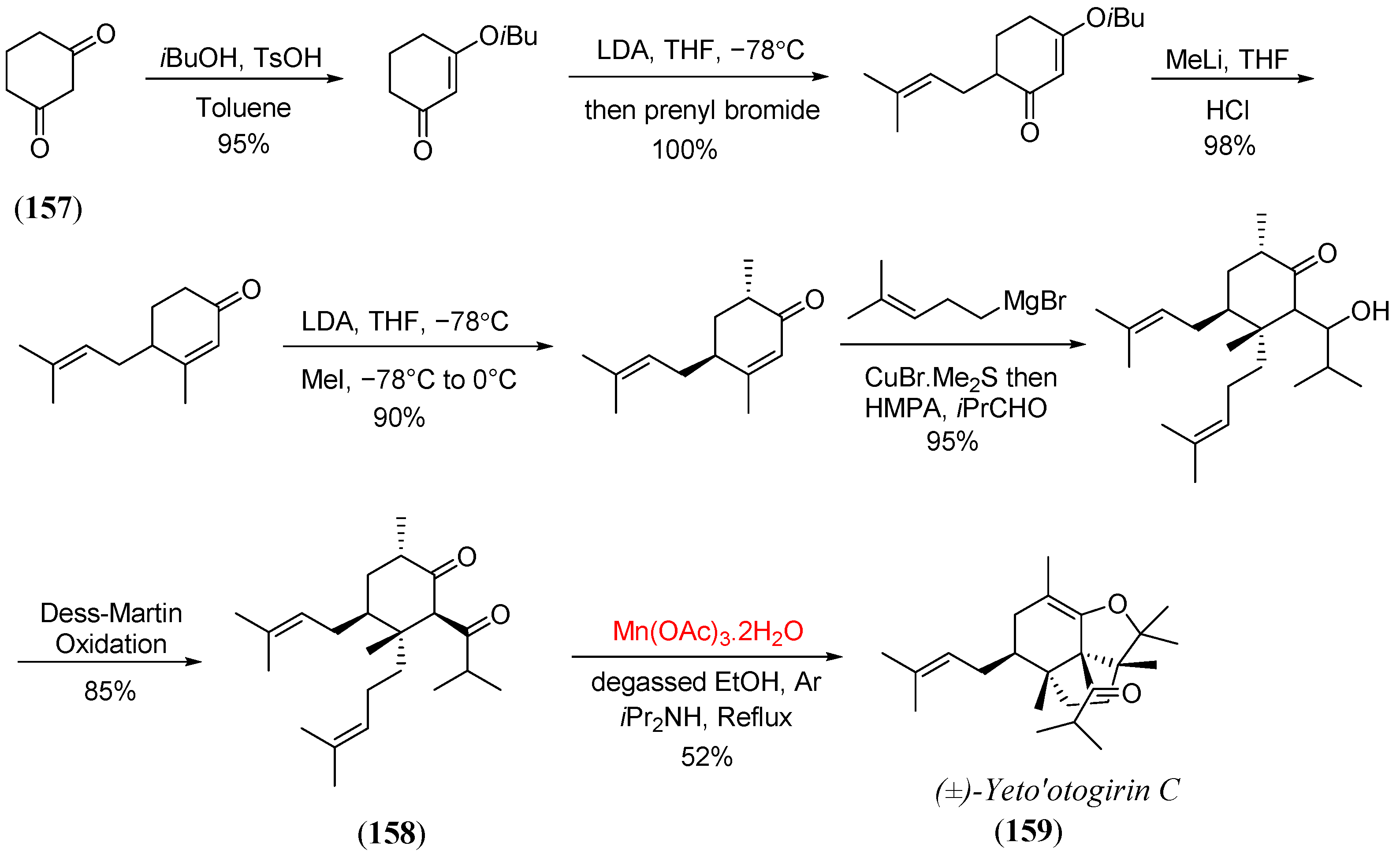
- Lam, H.C.; Kuan, K.K.W.; George, J.H. Biomimetic total synthesis of (±)-yezo’otogirin A. Org. Biomol. Chem. 2014, 12, 2519–2522. [Google Scholar] [CrossRef]
- He, S.; Yang, W.; Zhu, L.; Du, G.; Lee, C.-S. Bioinspired total synthesis of (±)-yezo’otogirin C. Org. Lett. 2014, 16, 496–499. [Google Scholar] [CrossRef]
- Yang, W.; Cao, J.; Zhang, M.; Lan, R.; Zhu, L.; Du, G.; He, S.; Lee, C.-S. Systemic study on the biogenic pathways of yezo’otogirins: Total synthesis and antitumor activities of (±)-yezo’otogirin C and its structural analogues. J. Org. Chem. 2015, 80, 836–846. [Google Scholar] [CrossRef]
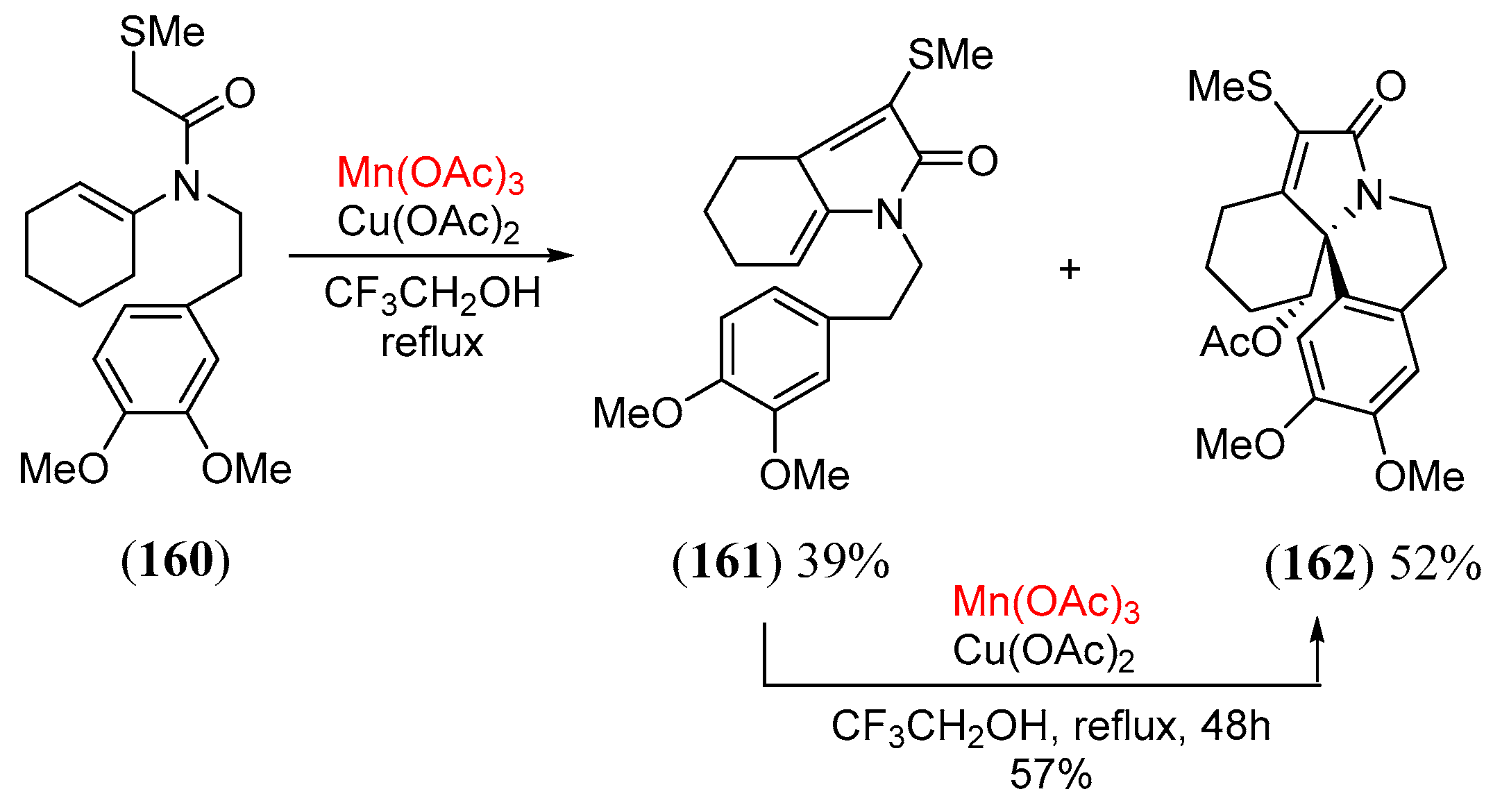
- Toyao, A.; Chikaoka, S.; Takeda, Y.; Tamura, O.; Muraoka, O.; Tanabe, G.; Ishibashi, H. A concise construction of an erythrinane skeleton using Mn(III)/Cu(II)-mediated oxidative radical cyclization of α-methylthio amides. Tetr. Lett. 2001, 42, 1729. [Google Scholar] [CrossRef]
- Chikaoka, S.; Toyao, A.; Ogasawara, M.; Tamura, O.; Ishibashi, H. Mn(III)/Cu(II)-mediated oxidative radical cyclization of α-(methylthio)acetamides leading to erythrinanes. J. Org. Chem. 2003, 68, 312. [Google Scholar] [CrossRef] [PubMed]
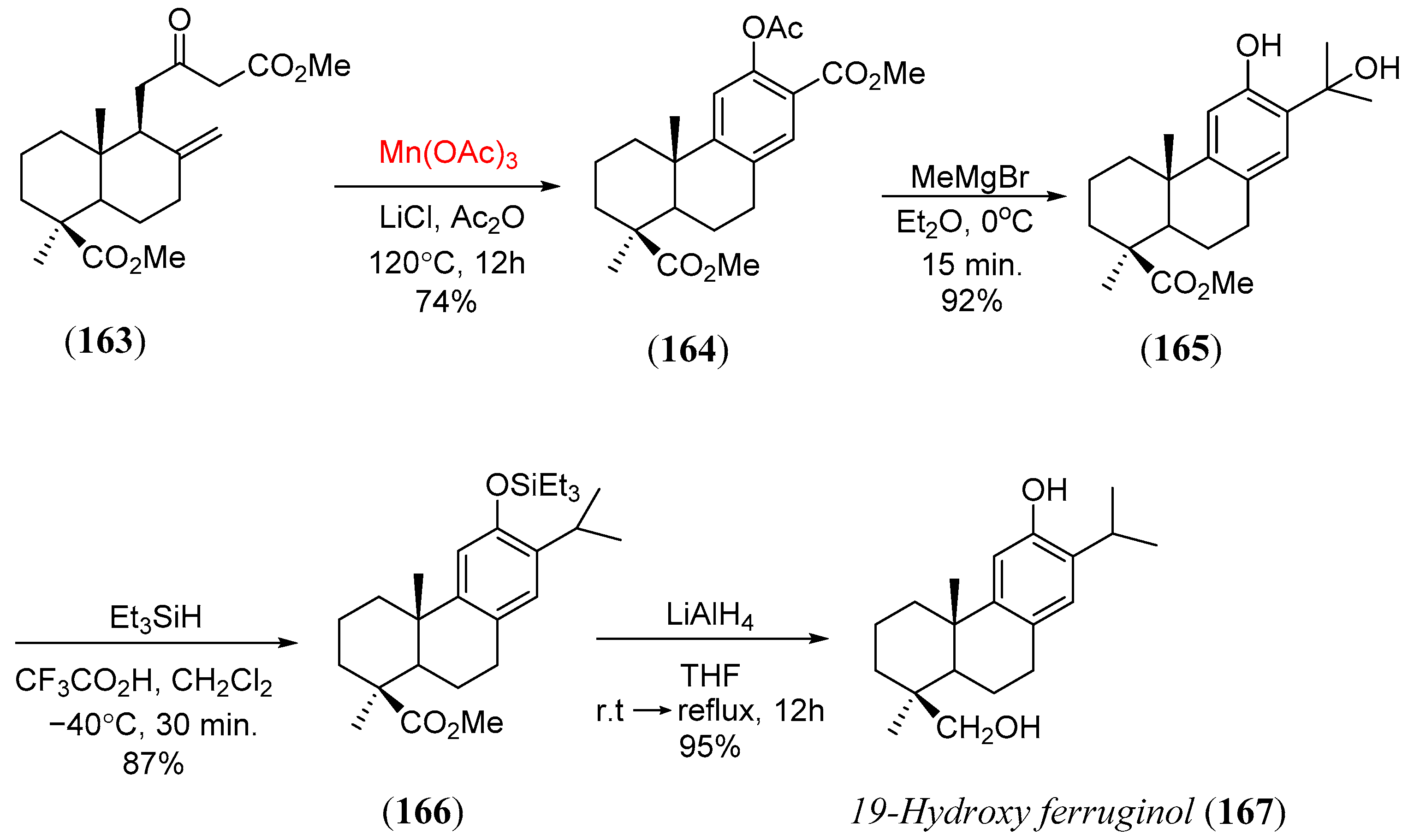
- Alvarez-Manzaneda, E.; Chahboun, R.; Cabrera, E.; Alvarez, E.; Alvarez-Manzaneda, R.; Lachkar, M.; Messouri, I. Synthesis of phenol abietane diterpenes based on the oxidative radical cyclization utilizing the Mn(OAc)3/Ac2O system. Synlett 2007, 2425. [Google Scholar] [CrossRef]
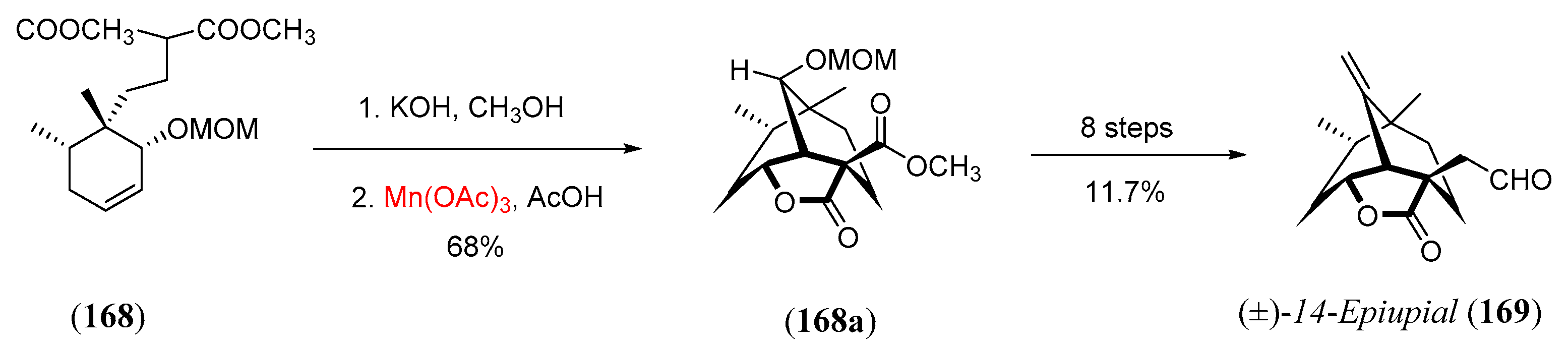
- Paquette, L.A.; Schaefer, A.G.; Springer, J.P. Synthesis of (±)-epiupial by manganese(III) γ-lactone annulation. Tetrahedron 1987, 43, 5567. [Google Scholar] [CrossRef]
- Takahashi, K.; Watanabe, M.; Honda, T. Highly efficient stereocontrolled total synthesis of (+)-upial. Angew. Chem. Int. Ed. 2008, 47, 131. [Google Scholar] [CrossRef] [PubMed]
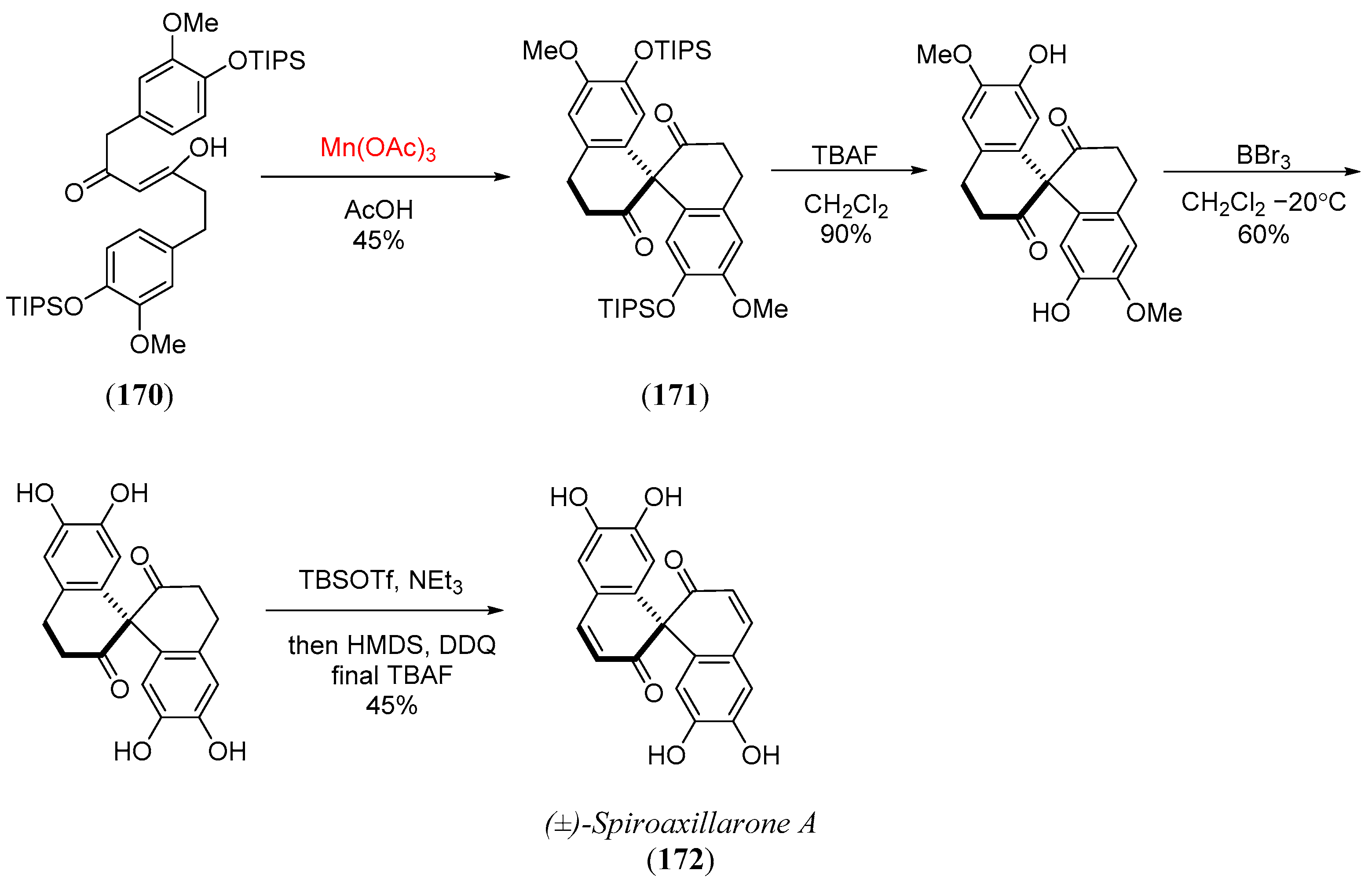
- Liao, M.; Li, X.; Zheng, Y.; Xie, Z. Total synthesis of (±)-spiroaxillarone A. J. Org. Chem. 2021, 86, 4835–4842. [Google Scholar] [CrossRef] [PubMed]
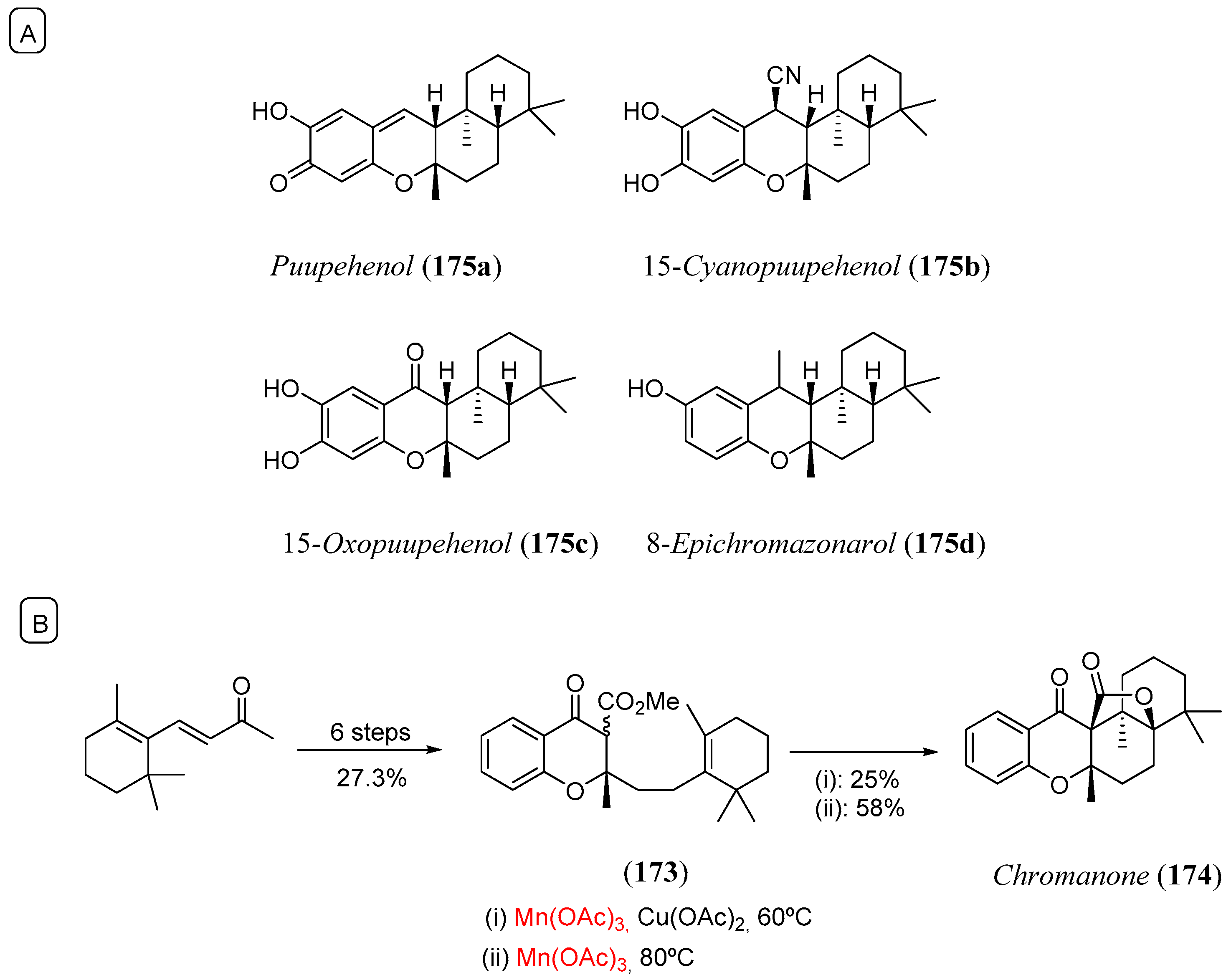
- Crombie, B.S.; Smith, C.; Varnavas, C.Z.; Wallace, T.W. A conjugate addition-radical cyclisation approach to sesquiterpenephenol natural products. J. Chem. Soc. Perkin Trans. 2001, 206. [Google Scholar] [CrossRef]
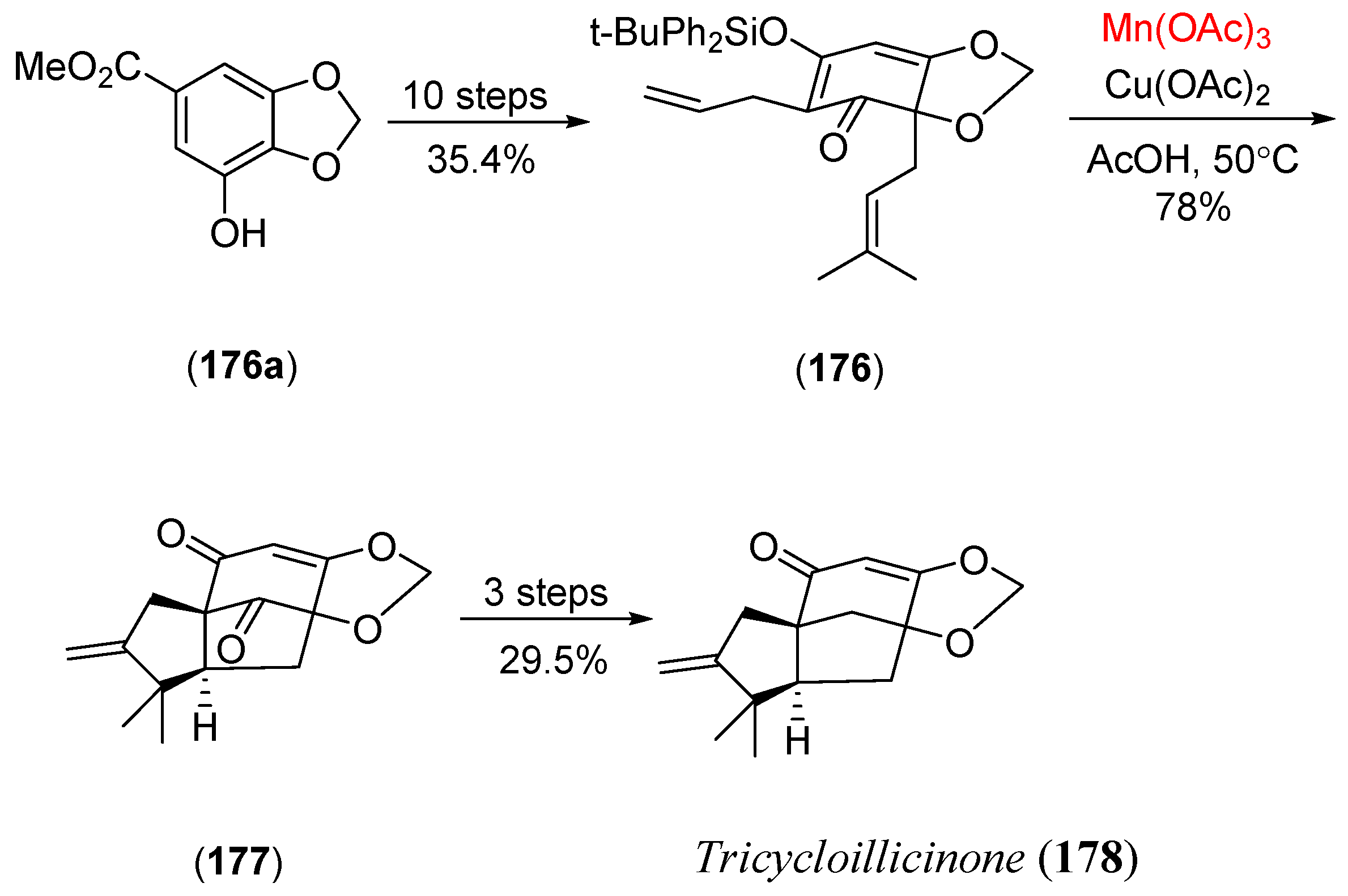
- Pettus, T.R.R.; Chen, X.T.; Danishefsky, S.J. A fully synthetic route to the neurotrophic illicinones by sequential aromatic claisen rearrangements. J. Am. Chem. Soc. 1998, 120, 12684. [Google Scholar] [CrossRef]
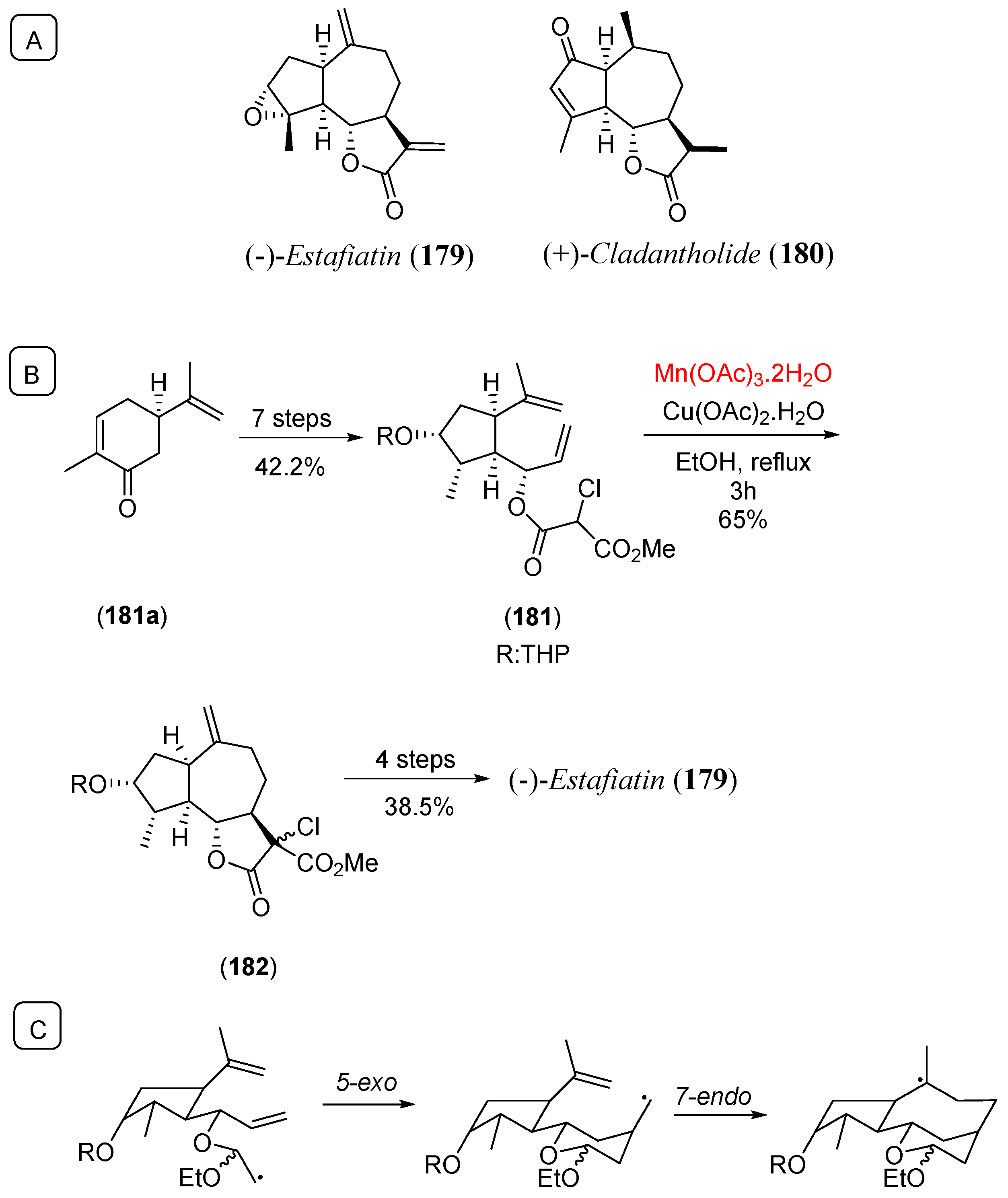
- Lee, E.; Lim, J.W.; Yoon, C.H.; Sung, Y.S.; Kim, Y.K. Total synthesis of (+)-cladantholide and (-)-estafiatin: 5-exo,7-endo radical cyclization strategy for the construction of guaianolide skeleton. J. Am. Chem. Soc. 1997, 119, 8391. [Google Scholar] [CrossRef]
- Snider, B.B.; Kiselgof, J.Y.; Foxman, B.M. Total syntheses of (±)-isosteviol and (±)-beyer-15-ene-3β,19-diol by manganese(III)-based oxidative quadruple free-radical cyclization. J. Org. Chem. 1998, 63, 7945. [Google Scholar] [CrossRef]
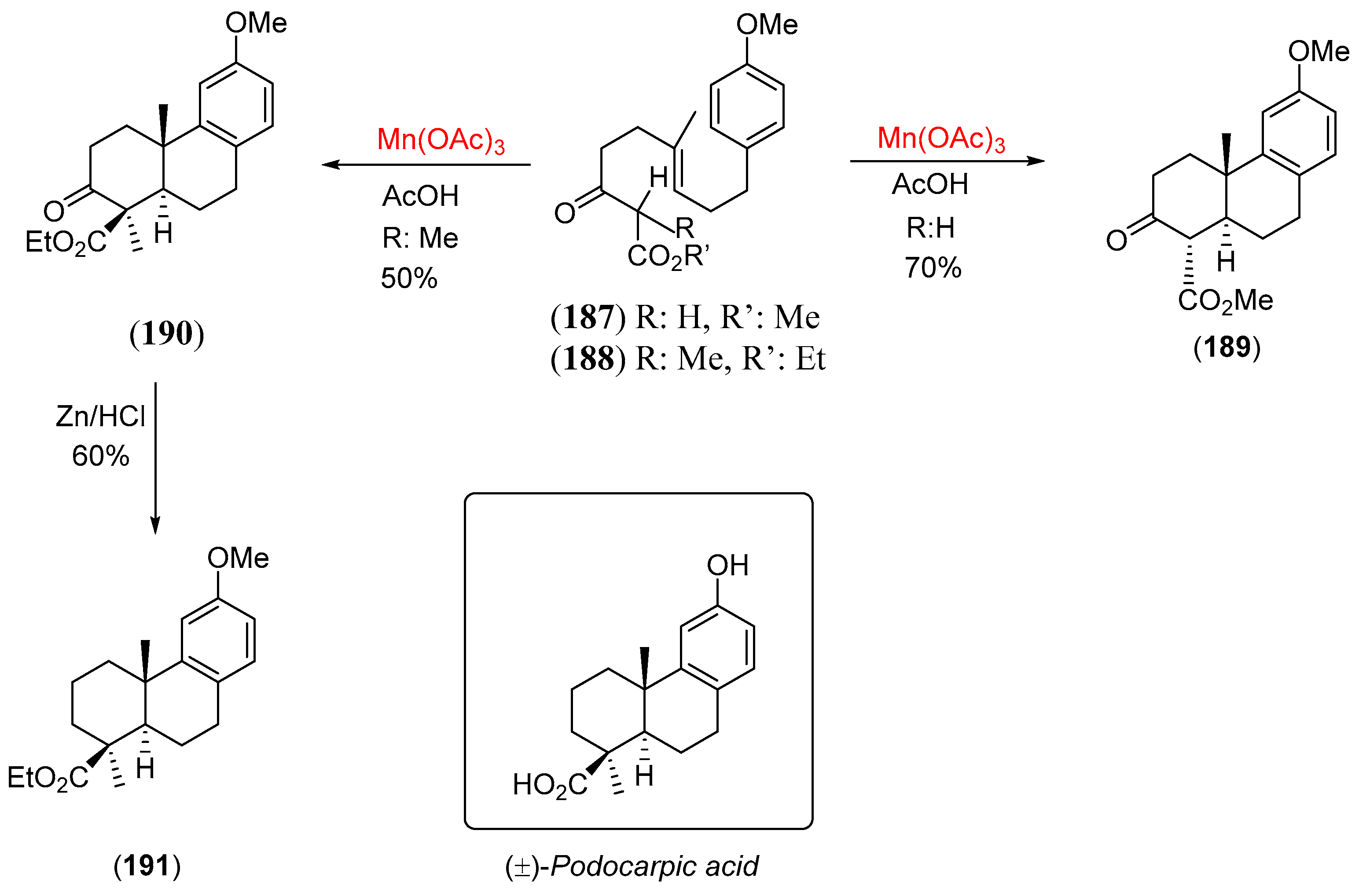
- Snider, B.B.; Mohan, R.; Kates, S.A. Manganese(III)-based oxidative free-radical cyclization. Synthesis of (±)-podocarpic acid. J. Org. Chem. 1985, 50, 3659. [Google Scholar] [CrossRef]
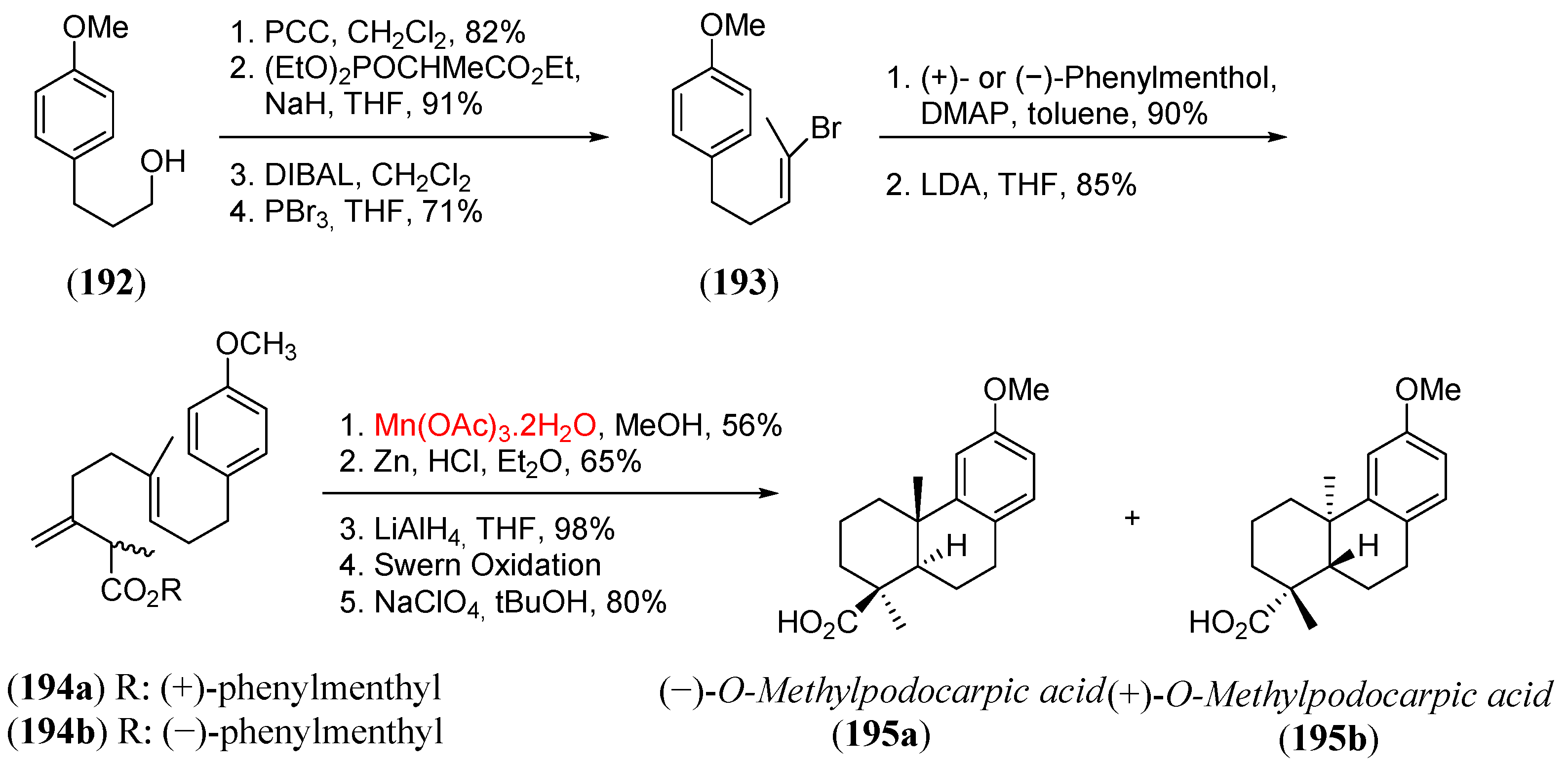
- Zhang, Q.; Mohan, R.M.; Cook, L.; Kazanis, S.; Peisach, D.; Foxman, B.M.; Snider, B.B. Asymmetric induction in Mn(III)-based oxidative free-radical cyclizations of phenylmenthyl acetoacetates and 2,5-dimethylpyrrolidine acetoacetamides. J. Org. Chem. 1993, 58, 7640. [Google Scholar] [CrossRef]
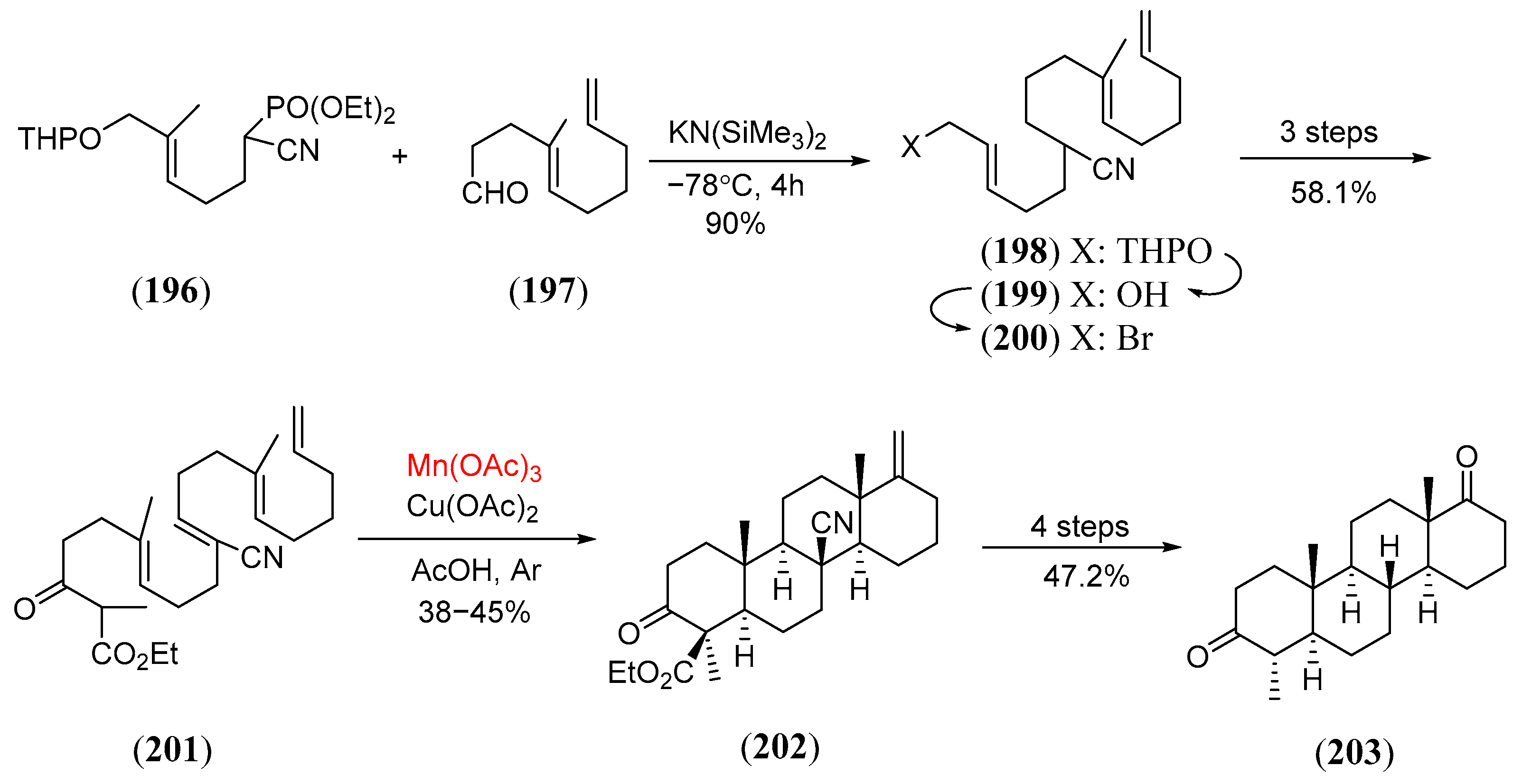
- Zoretic, P.A.; Chen, Z.; Zhang, Y. A radical prototype to steroids: Synthesis of d,l-5a-D-homoandrostane-4α-methyl-3,17a-dione. Tetr. Lett. 1996, 37, 7909. [Google Scholar] [CrossRef]
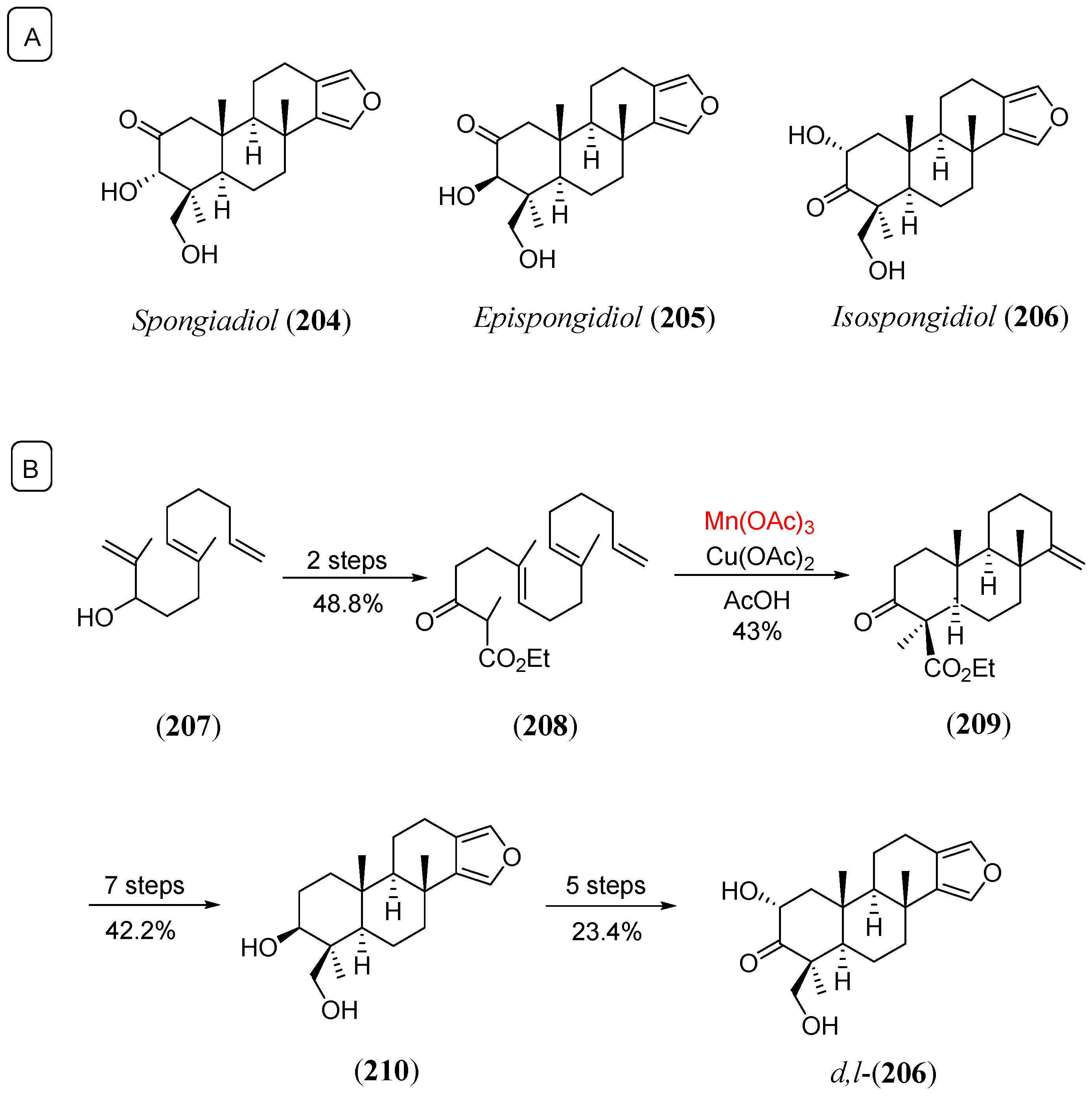
- Zoretic, P.A.; Wang, M.; Zhang, Y.; Shen, Z. Total synthesis of d,l-isospongiadiol: An intramolecular radical cascade approach to furanoditerpenes. J. Org. Chem. 1996, 61, 1806. [Google Scholar] [CrossRef]
- Zoretic, P.A.; Fang, H.; Ribeiro, A.A. Application of a radical methodology toward the synthesis of d,l-5α-pregnanes and related steroids: A stereoselective radical cascade approach. J. Org. Chem. 1998, 63, 7213. [Google Scholar] [CrossRef]
- Meng, Z.; Yu, H.; Li, L.; Tao, W.; Chen, H.; Wan, M.; Yang, P.; Edmonds, D.J.; Zhong, J.; Li, A. Total synthesis and antiviral activity of indolosesquiterpenoids from the xiamycin and oridamycin families. Nature Commun. 2015, 6, 6096. [Google Scholar] [CrossRef] [PubMed]
- Trotta, A.H. Toward a unified total synthesis of the xiamycin and oridamycin families of indolosesquiterpenes. J. Org. Chem. 2017, 82, 13500. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Ye, X.Y.; Gu, S.; Xu, M. Lanthanide triflates catalyze Mn(III)-based oxidative radical cyclization reactions. Enantioselective synthesis of (-)-triptolide, (-)-triptonide, and (+)-triptophenolide. J. Am. Chem. Soc. 1999, 121, 5579. [Google Scholar] [CrossRef]
- Yang, D.; Ye, X.Y.; Xu, M.; Pang, K.W.; Cheung, K.K. Investigation of Mn(III)-based oxidative free radical cyclization reactions toward the synthesis of triptolide: The effects of lanthanide triflates and substituents on stereoselectivity. J. Am. Chem. Soc. 2000, 122, 1658. [Google Scholar] [CrossRef]
- Yang, D.; Ye, X.-Y.; Xu, M.; Pang, K.-W.; Zou, N.; Letcher, R.M. A concise total synthesis of triptolide. J. Org. Chem. 1998, 63, 6446–6447. [Google Scholar] [CrossRef]
- Snider, B.B.; O’Hare, S.M. Tandem manganese(III)-based oxidative free-radical cyclizations terminated by addition to furans. Formal synthesis of 15-acetoxypallescensin-A. Synth. Commun. 2001, 31, 3753. [Google Scholar] [CrossRef]
- Zoretic, P.A.; Wang, M. The synthesis of d,l-15-acetoxypallescensin-A: An intramolecular oxidative free-radical approach. Synth. Commun. 1996, 26, 2783. [Google Scholar] [CrossRef]
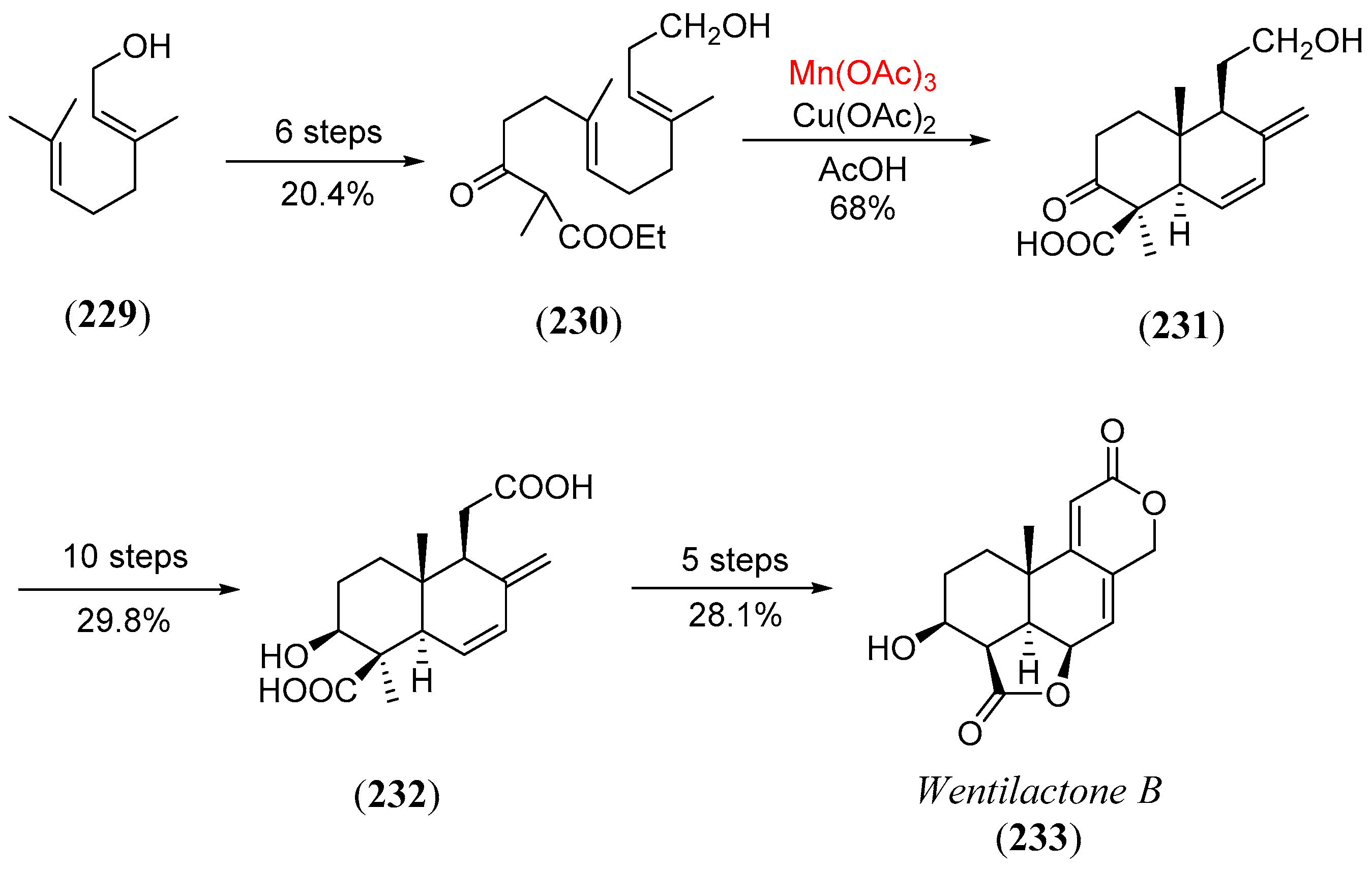
- Barrero, A.F.; Herrador, M.M.; Quilez del Moral, J.F.; Valdivia, M.V. A convenient synthesis of A-ring-functionalized podolactones. Revision of the structure of wentilactone B. Org. Lett. 2002, 4, 1379. [Google Scholar] [CrossRef] [PubMed]
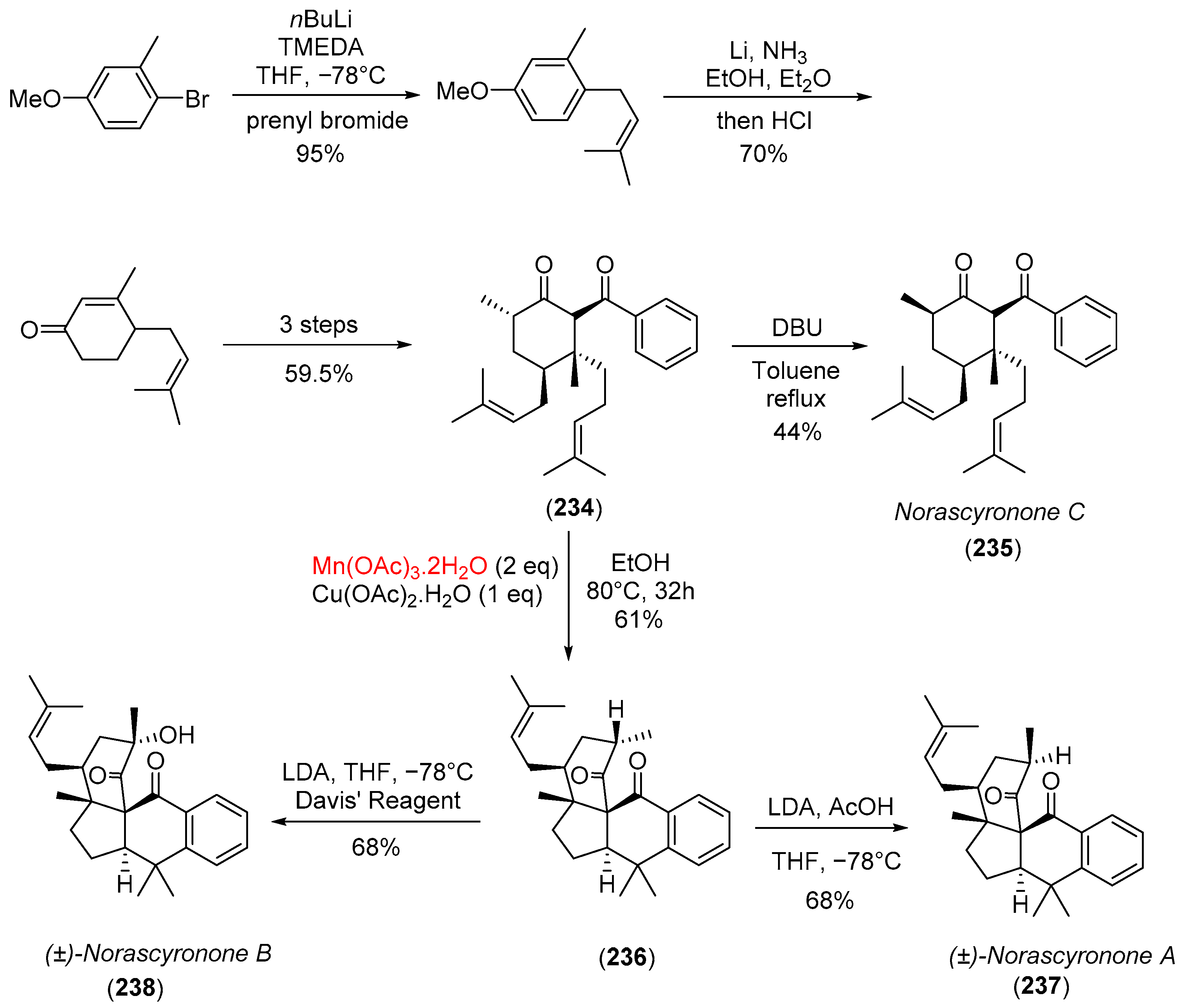
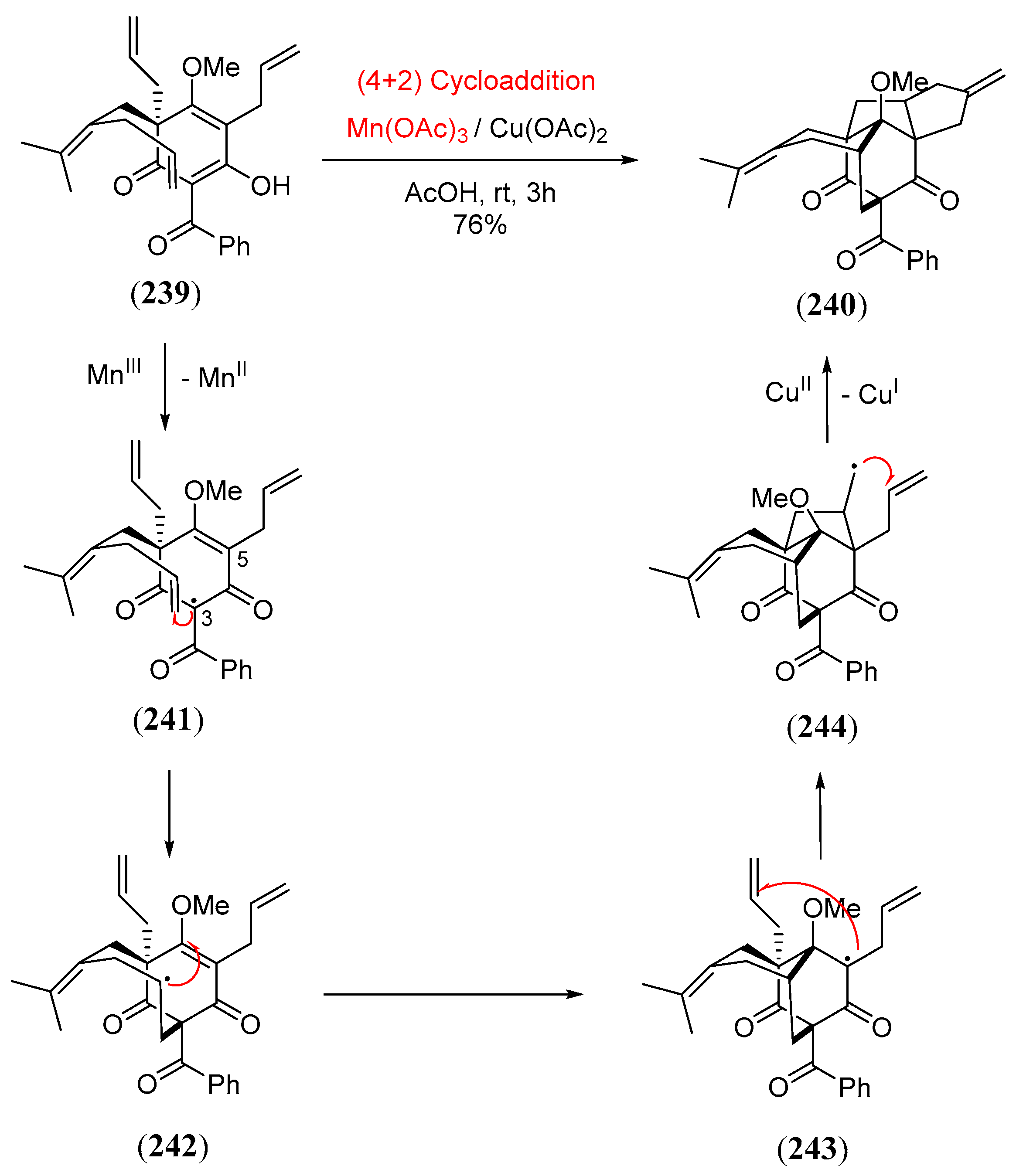
- Cao, T.; Zhu, L.; Lan, Y.; Huang, J.; Yang, Z. Protecting-group-free total syntheses of (±)-norascyronones A and B. Org. Lett. 2020, 22, 2517–2521. [Google Scholar] [CrossRef] [PubMed]
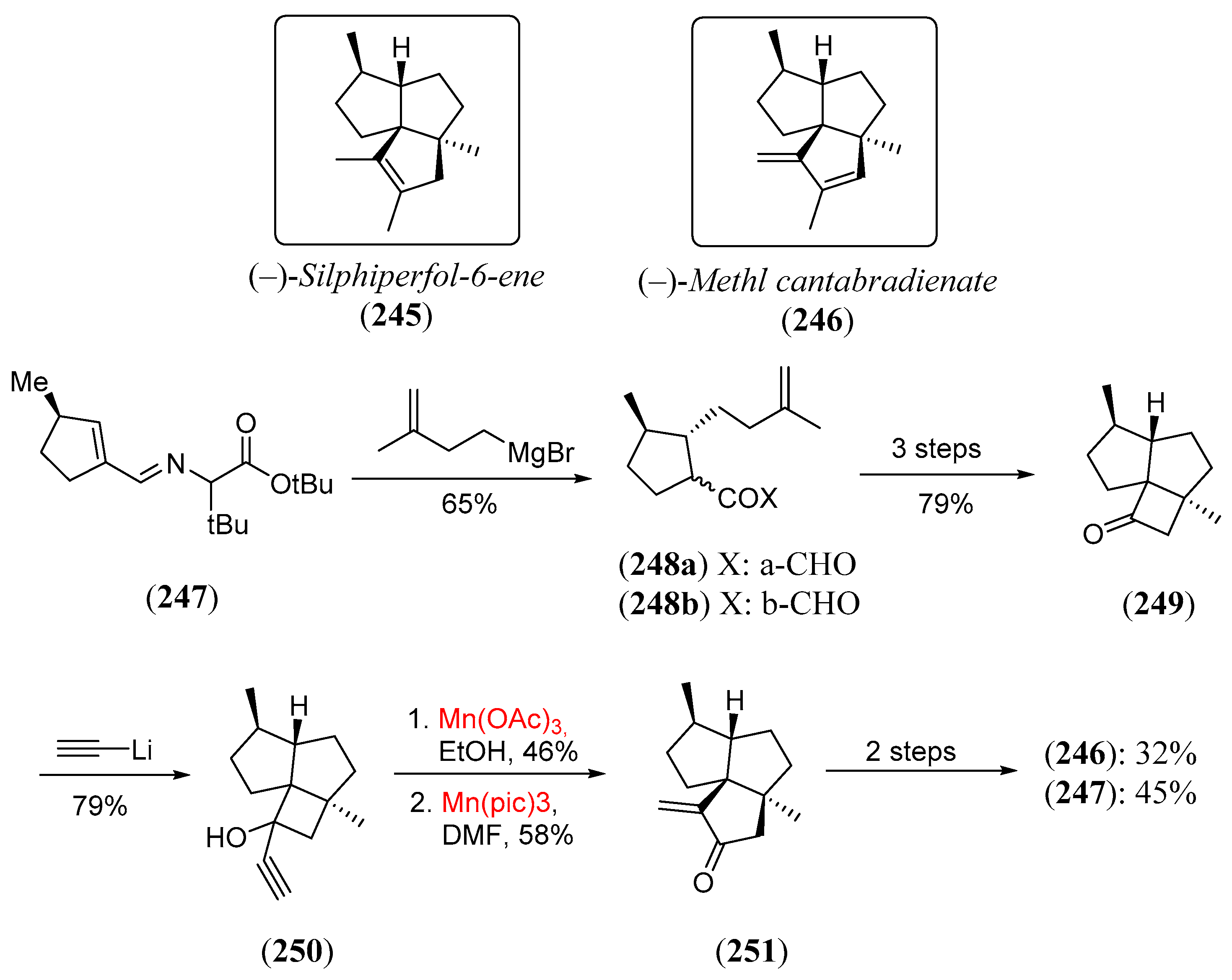
- Vo, N.H.; Snider, B.B. Total synthesis of (−)-silphiperfol-6-en and (−)-methyl cantabradienate. J. Org. Chem. 1994, 59, 5419. [Google Scholar] [CrossRef]
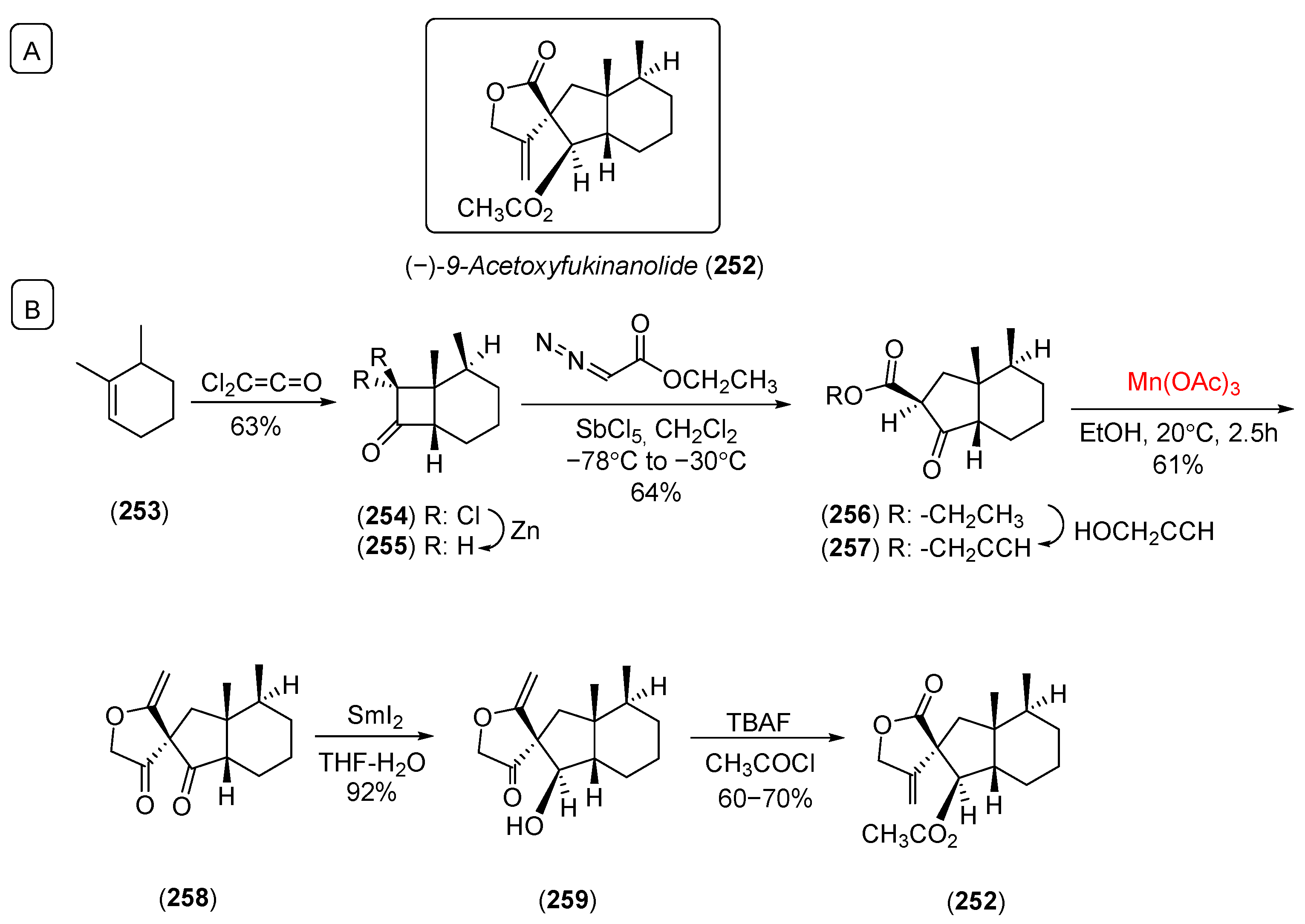
- Hamelin, O.; Depres, J.P.; Greene, A.E. Highly stereoselective first synthesis of an A-ring-functionalized bakkane: Novel free-radical approach to 9-acetoxyfukinanolide. J. Am. Chem. Soc. 1996, 118, 9992. [Google Scholar] [CrossRef]
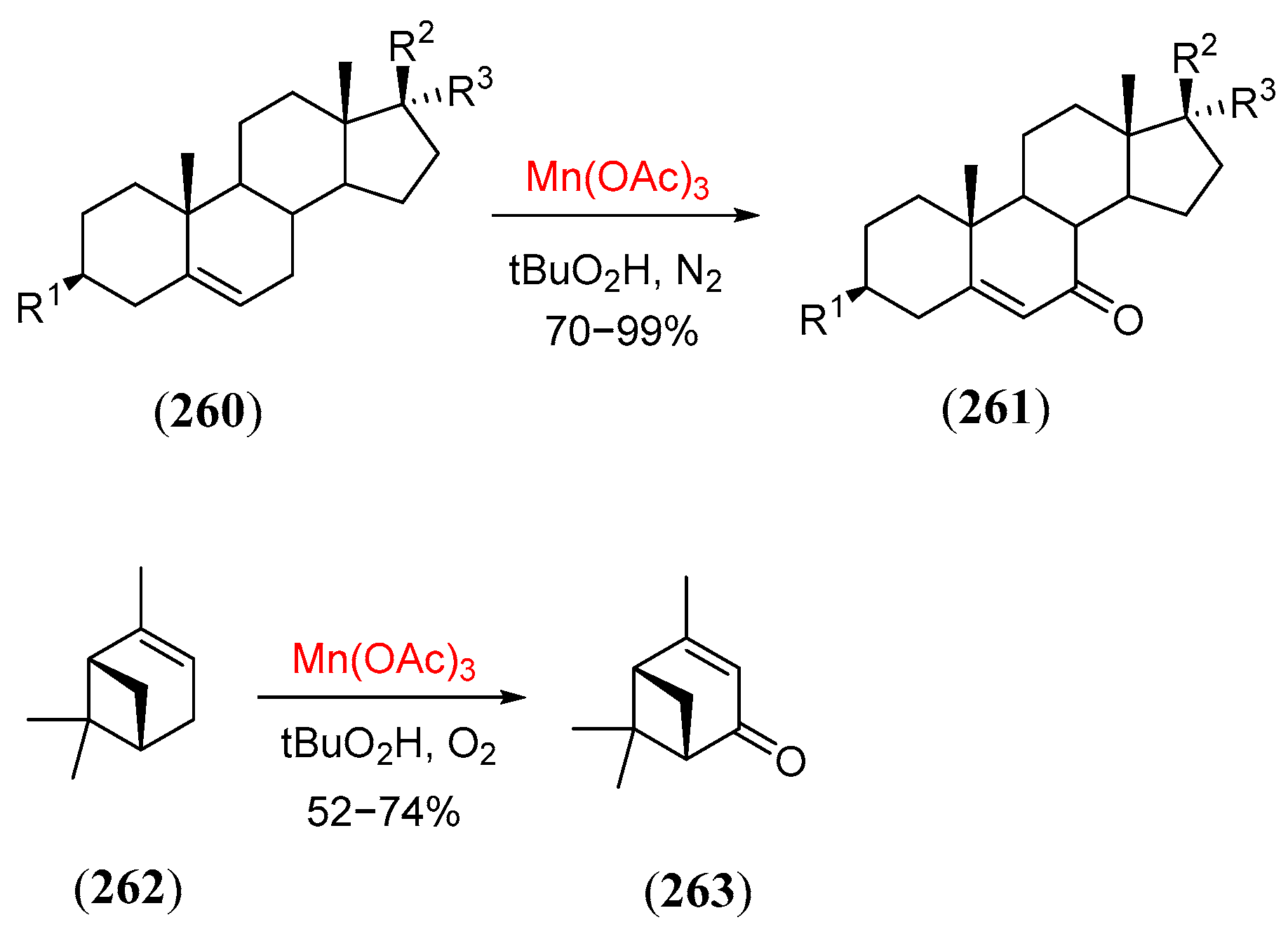
- Shing, T.K.M.; Yeung, Y.Y.; Su, P.L. Mild manganese(III) acetate catalyzed allylic oxidation: Application to simple and complex alkenes. Org. Lett. 2006, 8, 3149. [Google Scholar] [CrossRef]
- Fukuyama, T.; Li, L.; Laird, A.A.; Frank, R.K. Stereocontrolled total synthesis of (±)-cyanocycline A. J. Am. Chem. Soc. 1987, 109, 1587. [Google Scholar] [CrossRef]
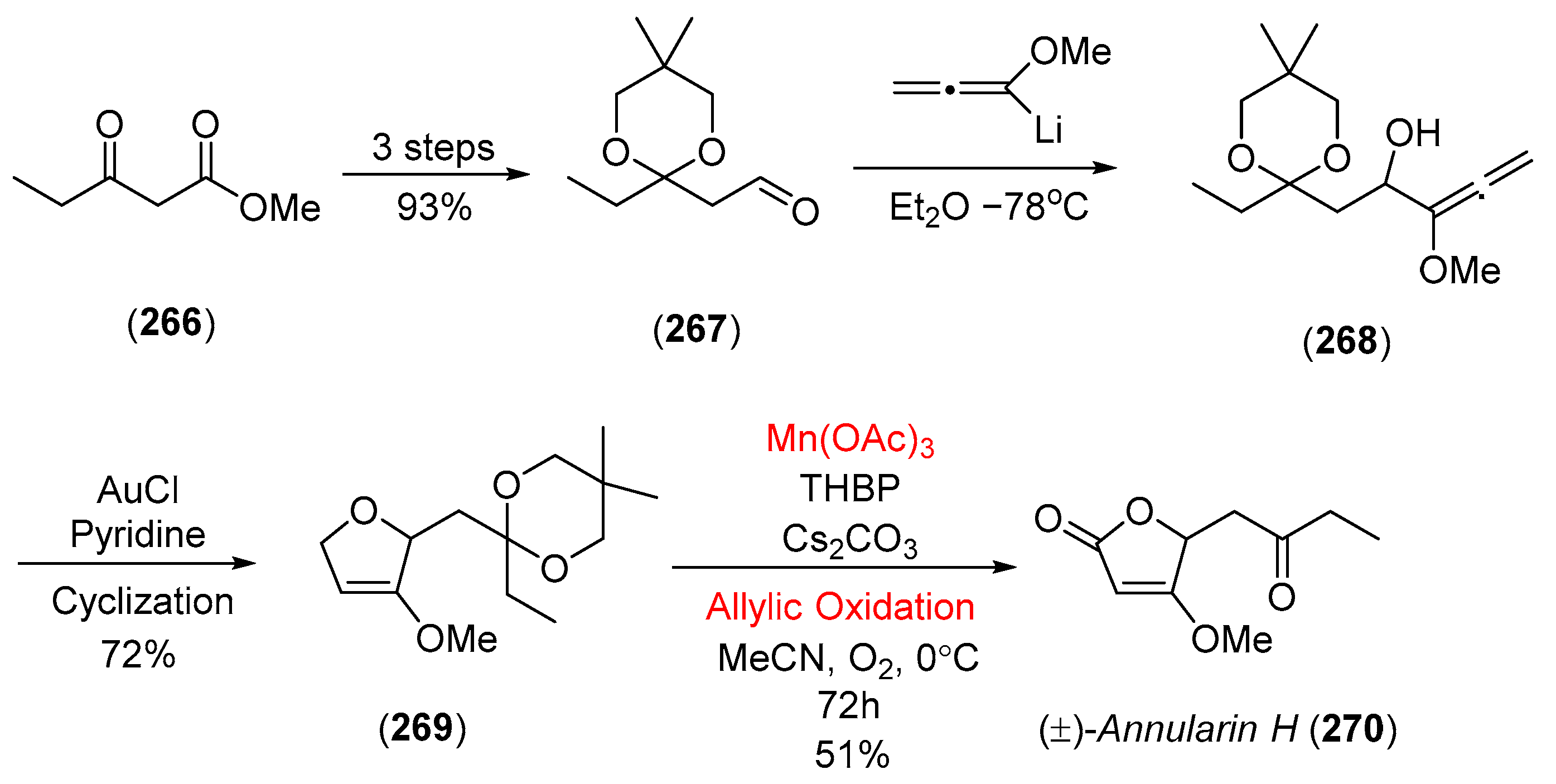
- Brasholz, M.; Reissig, H.U. Refined protocols for the preparation of 3-alkoxy-2,5-dihydrofurans, allylic oxidation to β-alkoxybutenolides and short synthesis of (±)-annularin H. Synlett 2007, 2007, 1294. [Google Scholar] [CrossRef]
- Singh, O.V.; Huang, W.J.; Chen, C.H.; Lee, S.S. Manganese(III) acetate mediated oxidation of aporphines: A convenient and useful synthesis of oxoaporphines. Tetr. Lett. 2007, 48, 8166. [Google Scholar] [CrossRef]
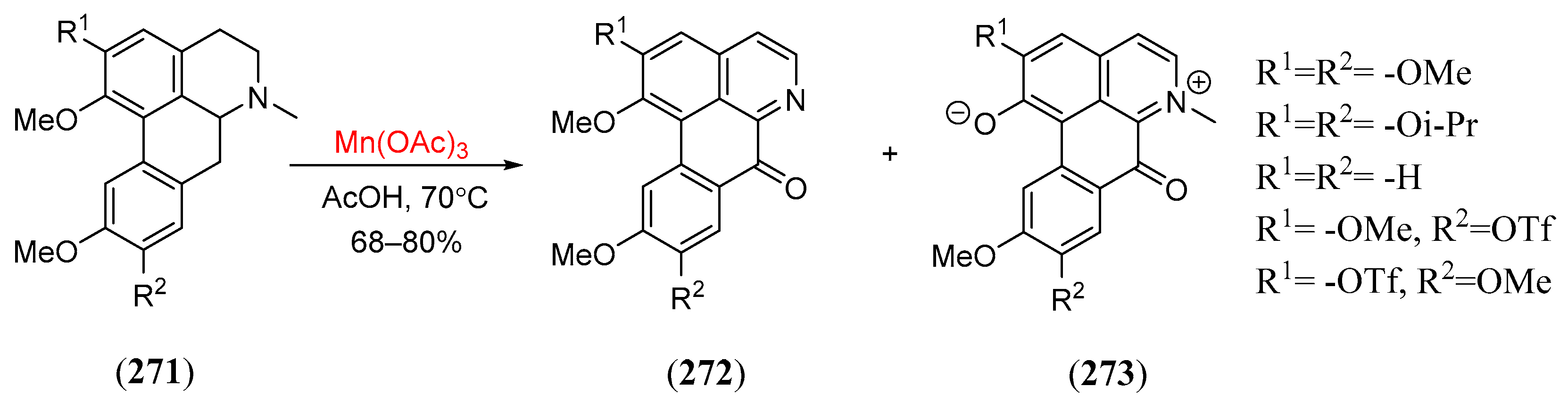
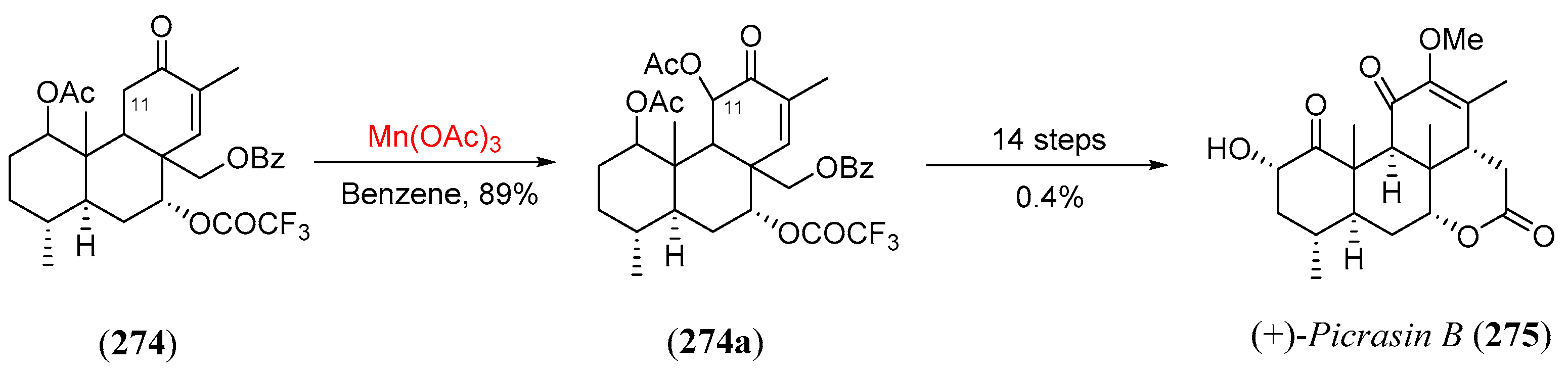
- Kawada, K.; Kim, M.; Watt, D.S. An enantioselective total synthesis of (+)-picrasin B. Tetr. Lett. 1989, 30, 5989. [Google Scholar] [CrossRef]
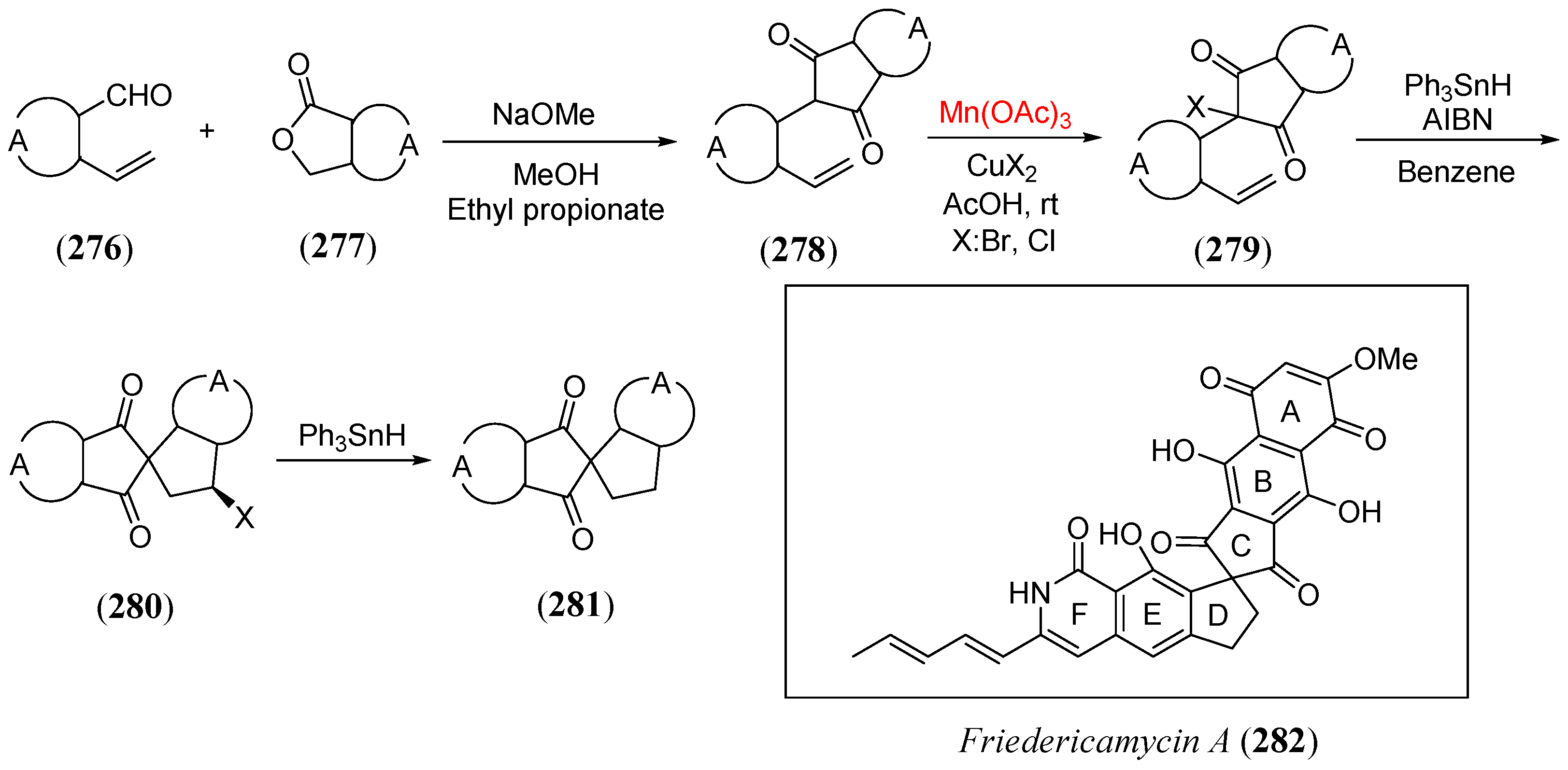
- Rama Rao, A.V.; Singh, A.K.; Reddy, K.M.; Ravikumar, K. Regioselective free radical cyclization: A general method for the synthesis of the spiro[4.4]nonane system of fredericamycin A. J. Chem. Soc. Perkin Trans. 1993, 1, 3171. [Google Scholar]




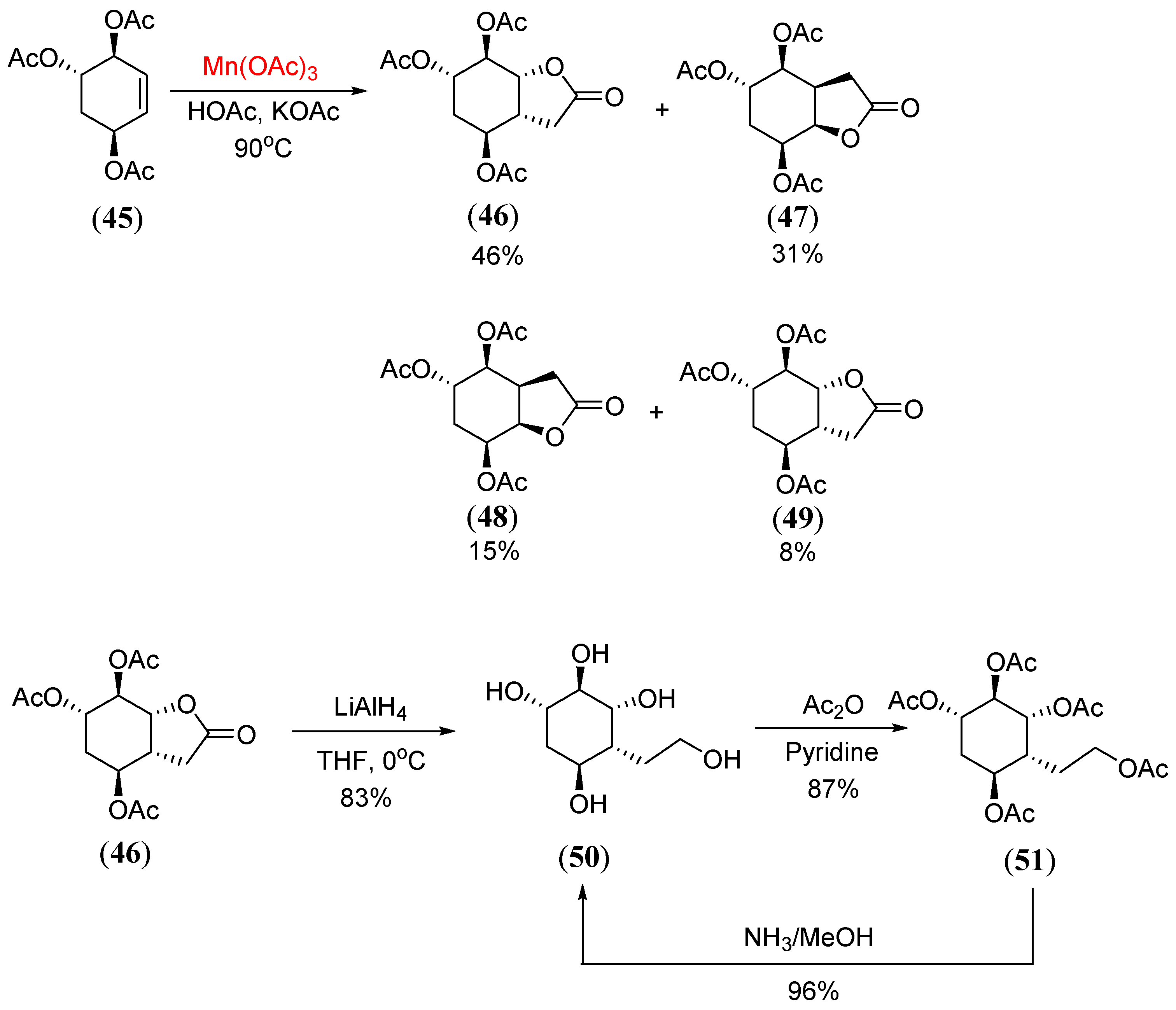
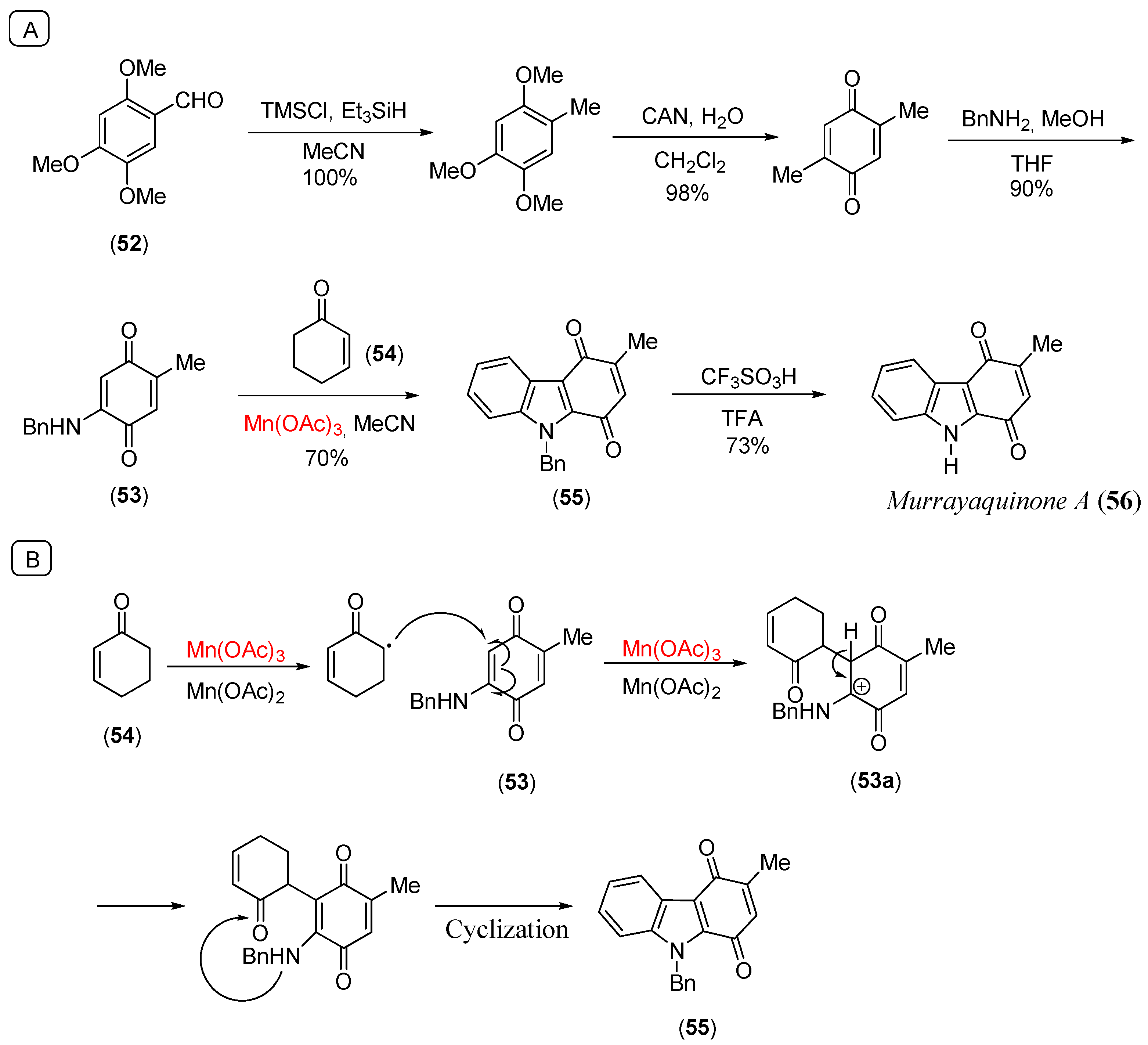
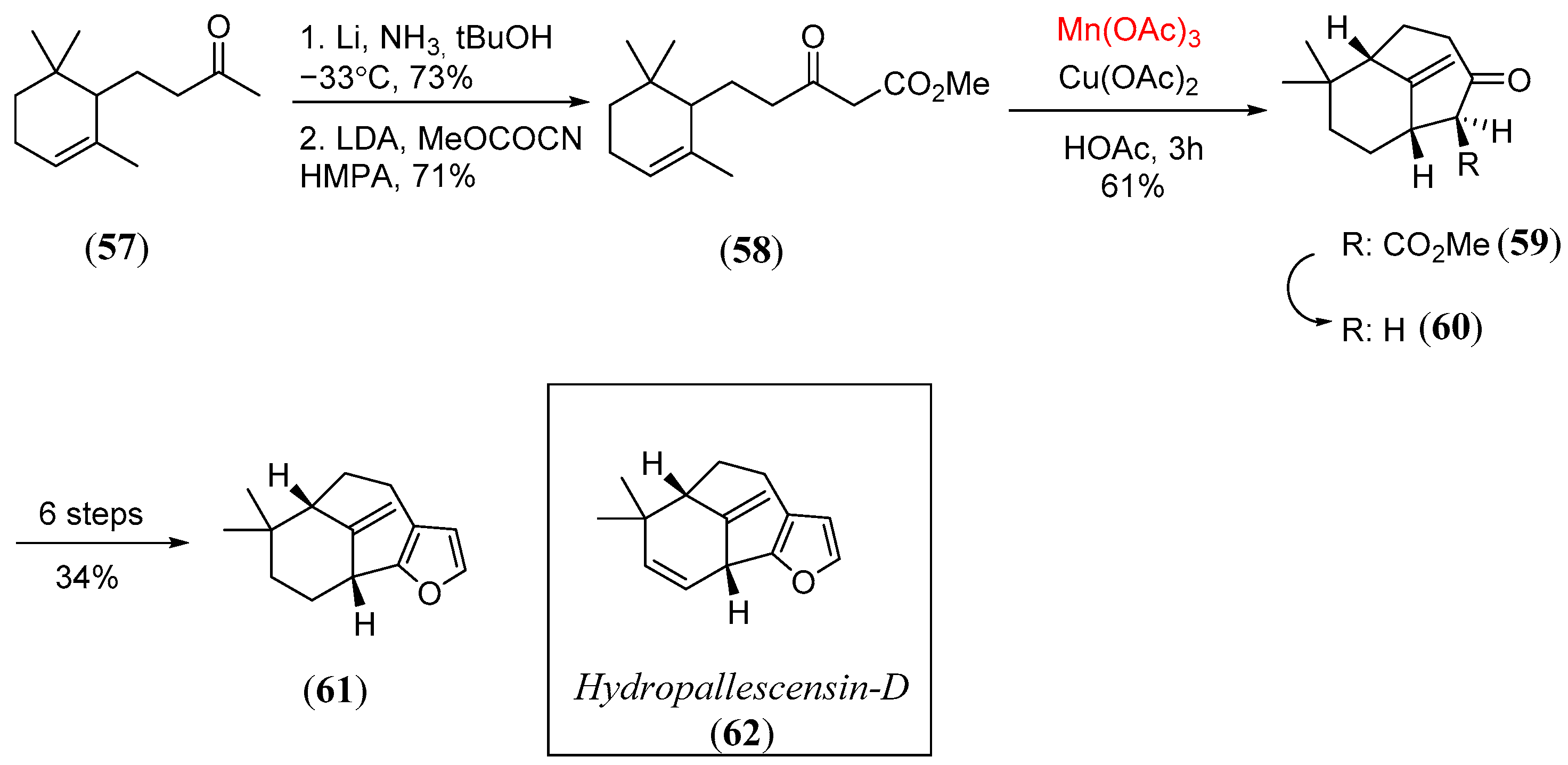
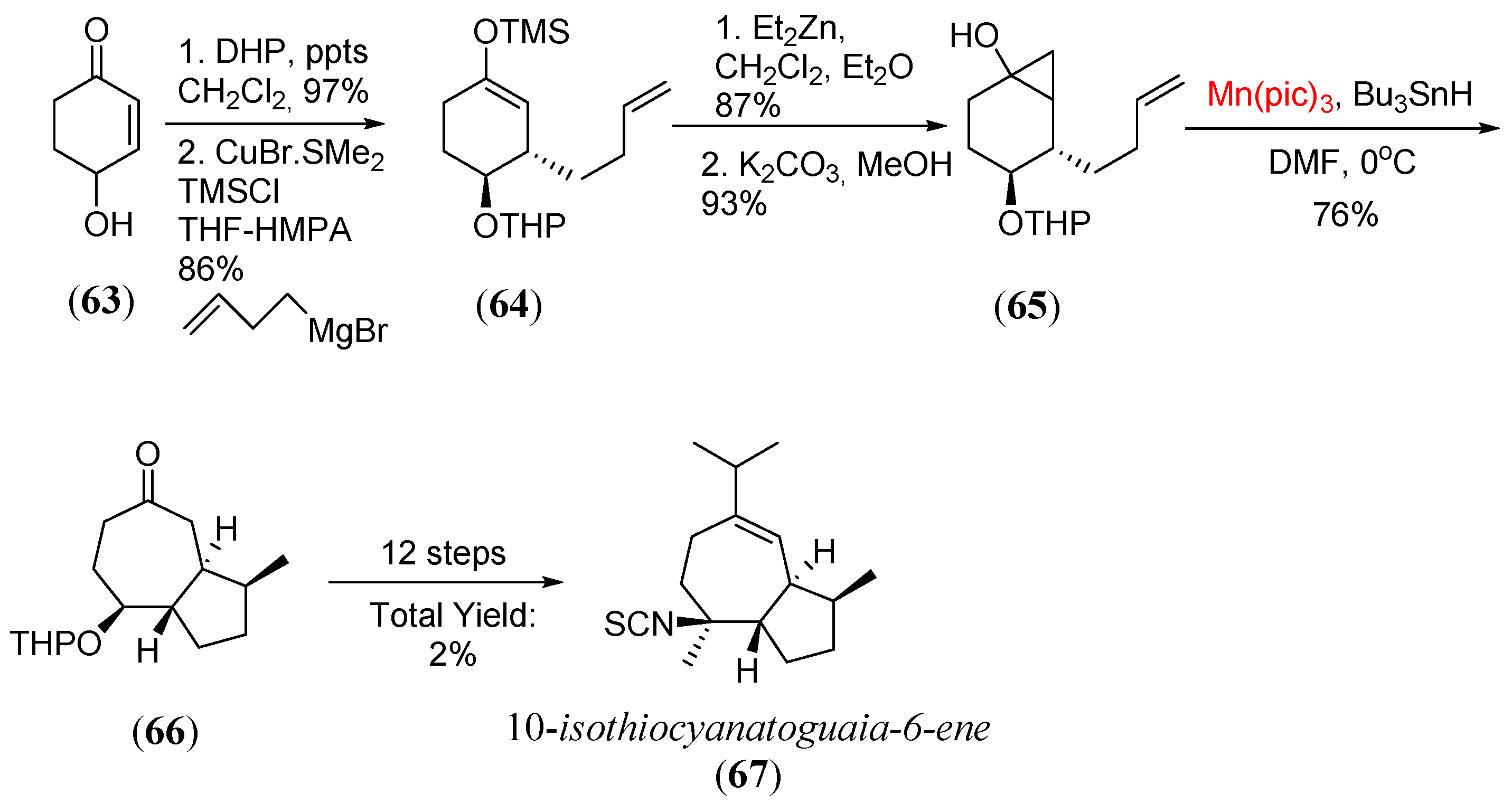



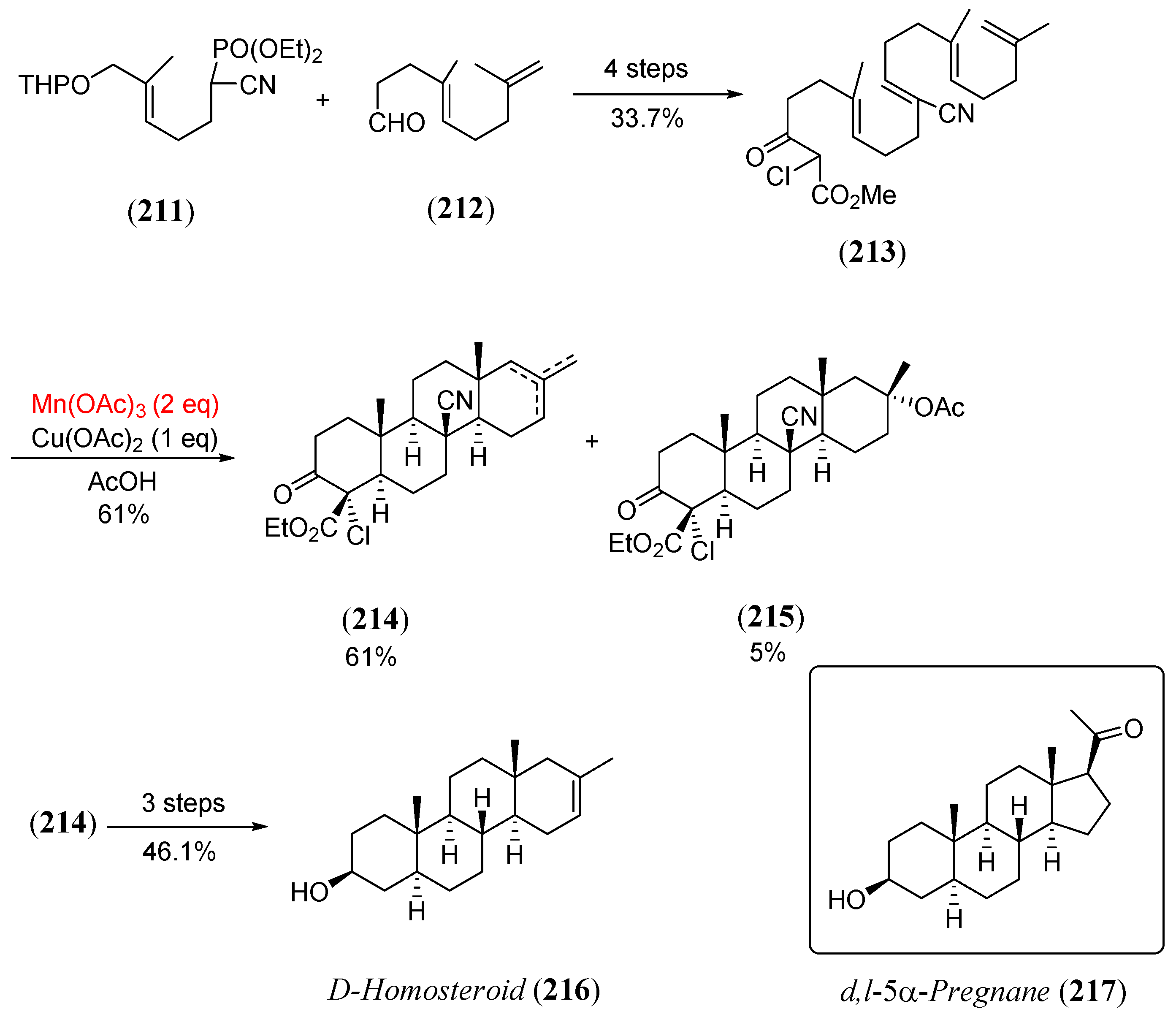
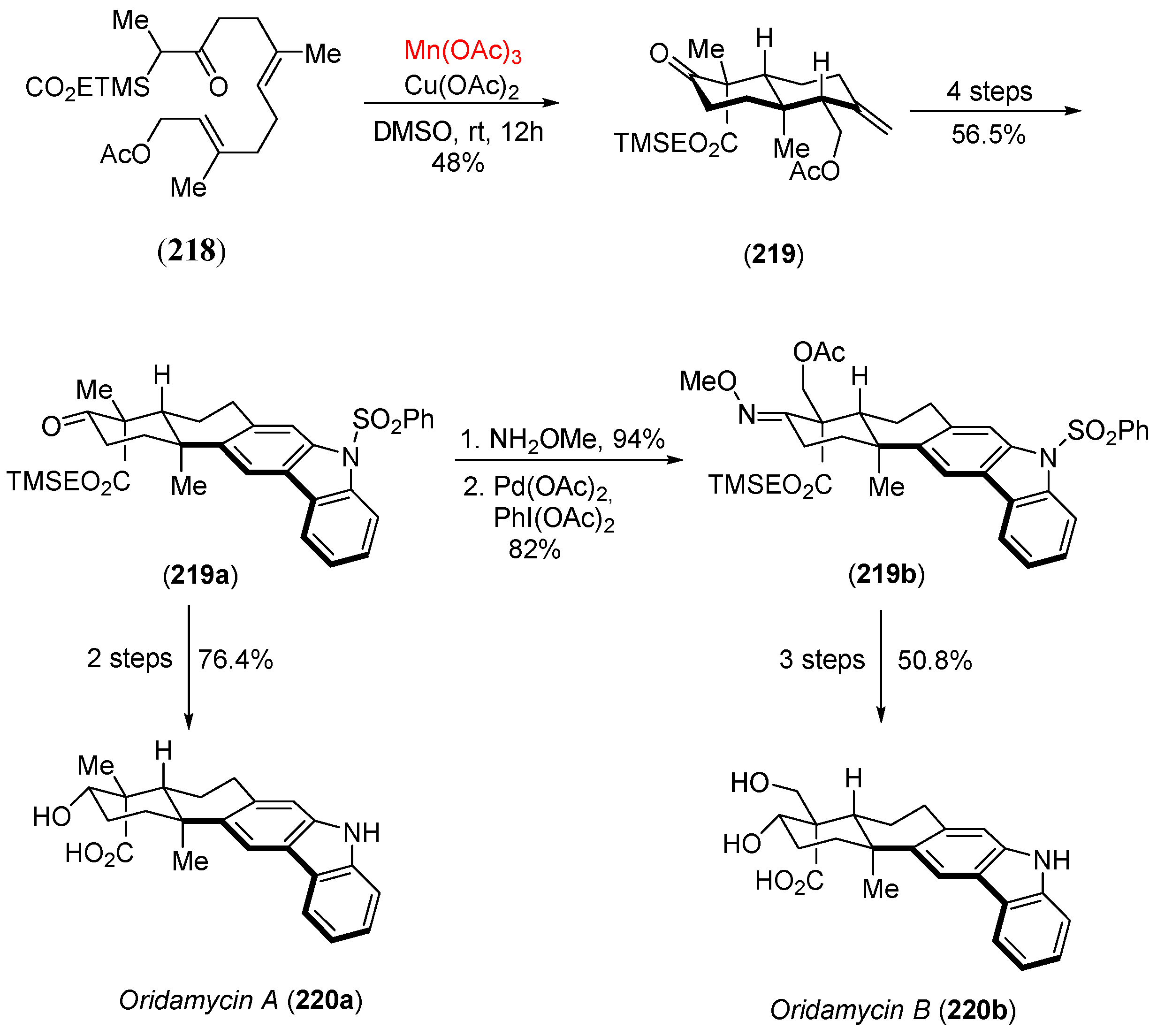
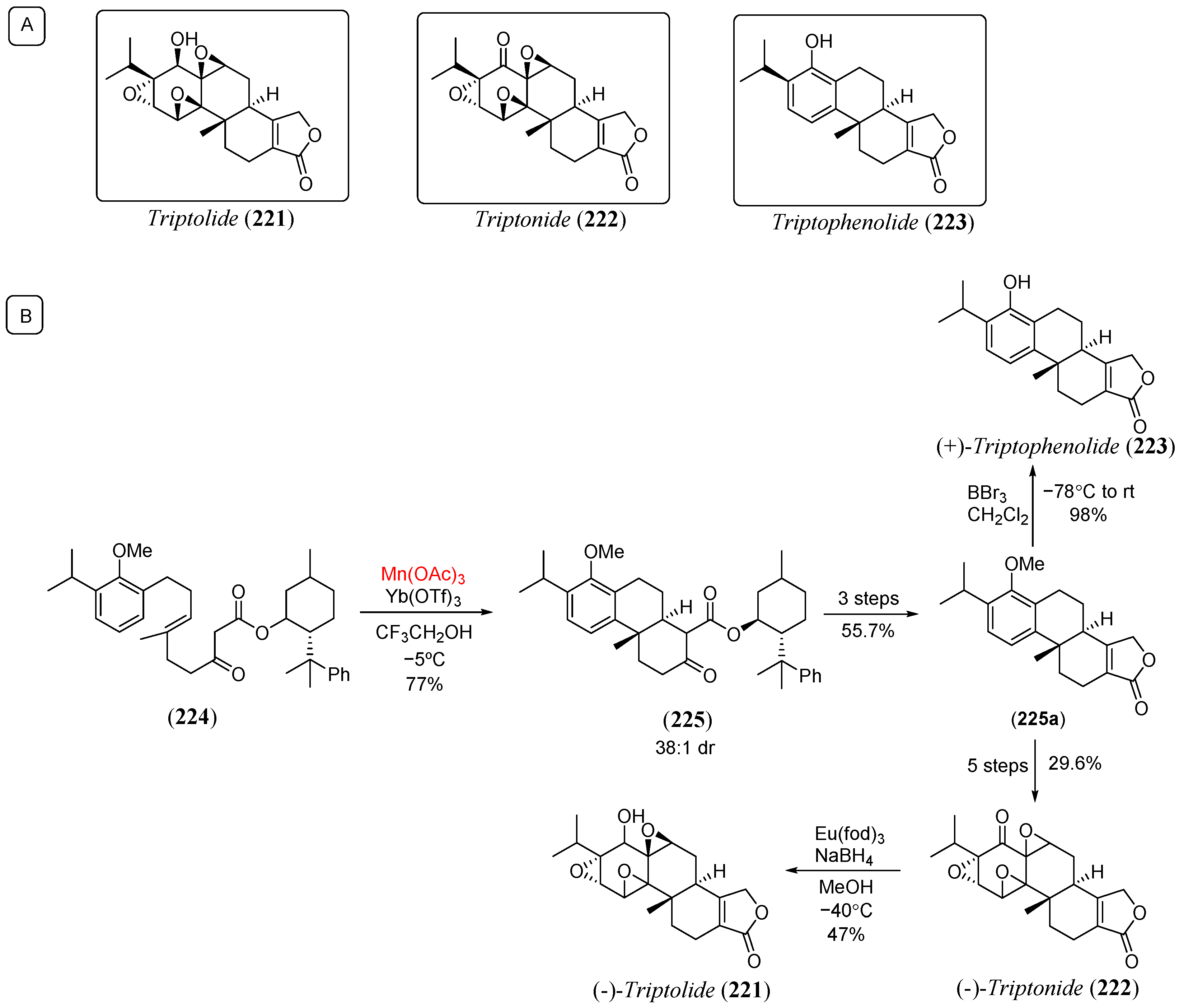
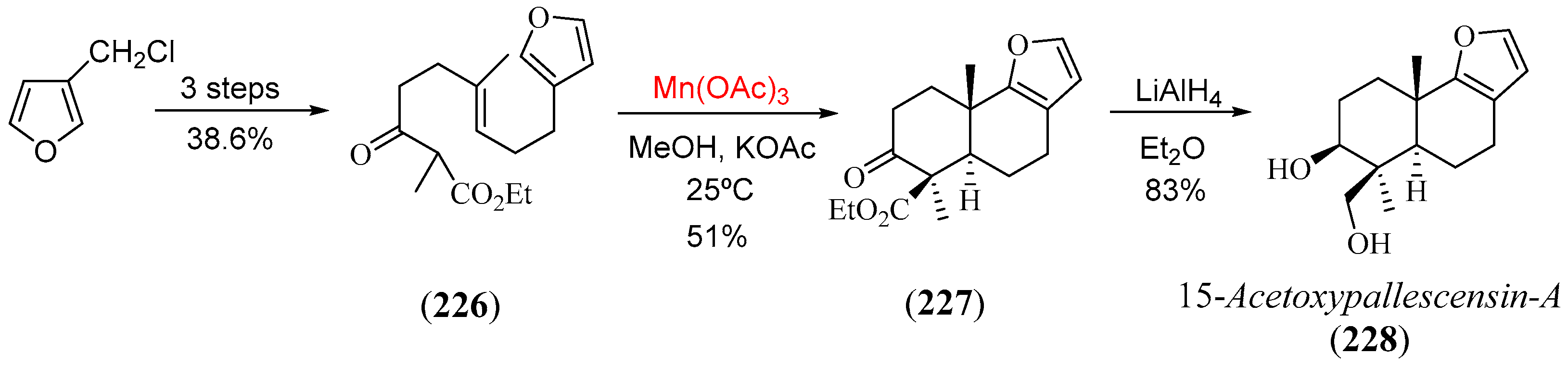
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biçer, E.; Yılmaz, M. Recent Advances in Manganese(III)-Assisted Radical Cyclization for the Synthesis of Natural Products: A Comprehensive Review. Molecules 2024, 29, 2264. https://doi.org/10.3390/molecules29102264
Biçer E, Yılmaz M. Recent Advances in Manganese(III)-Assisted Radical Cyclization for the Synthesis of Natural Products: A Comprehensive Review. Molecules. 2024; 29(10):2264. https://doi.org/10.3390/molecules29102264
Chicago/Turabian StyleBiçer, Emre, and Mehmet Yılmaz. 2024. "Recent Advances in Manganese(III)-Assisted Radical Cyclization for the Synthesis of Natural Products: A Comprehensive Review" Molecules 29, no. 10: 2264. https://doi.org/10.3390/molecules29102264
APA StyleBiçer, E., & Yılmaz, M. (2024). Recent Advances in Manganese(III)-Assisted Radical Cyclization for the Synthesis of Natural Products: A Comprehensive Review. Molecules, 29(10), 2264. https://doi.org/10.3390/molecules29102264