
Total Syntheses and Antibacterial Studies of Natural Isoflavones: Scandenone, Osajin, and 6,8-Diprenylgenistein
 , ,
, ,
Abstract
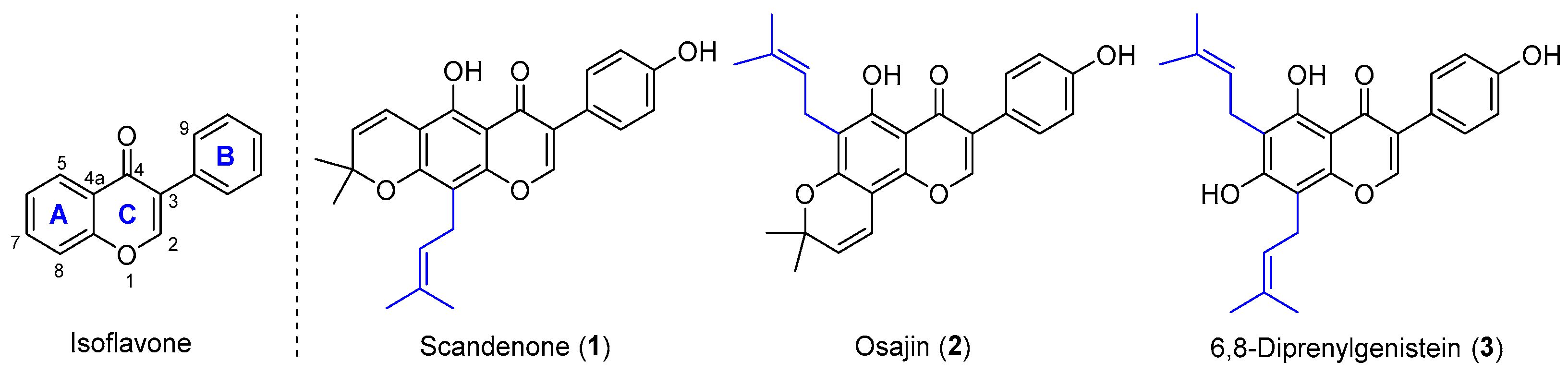
:1. Introduction
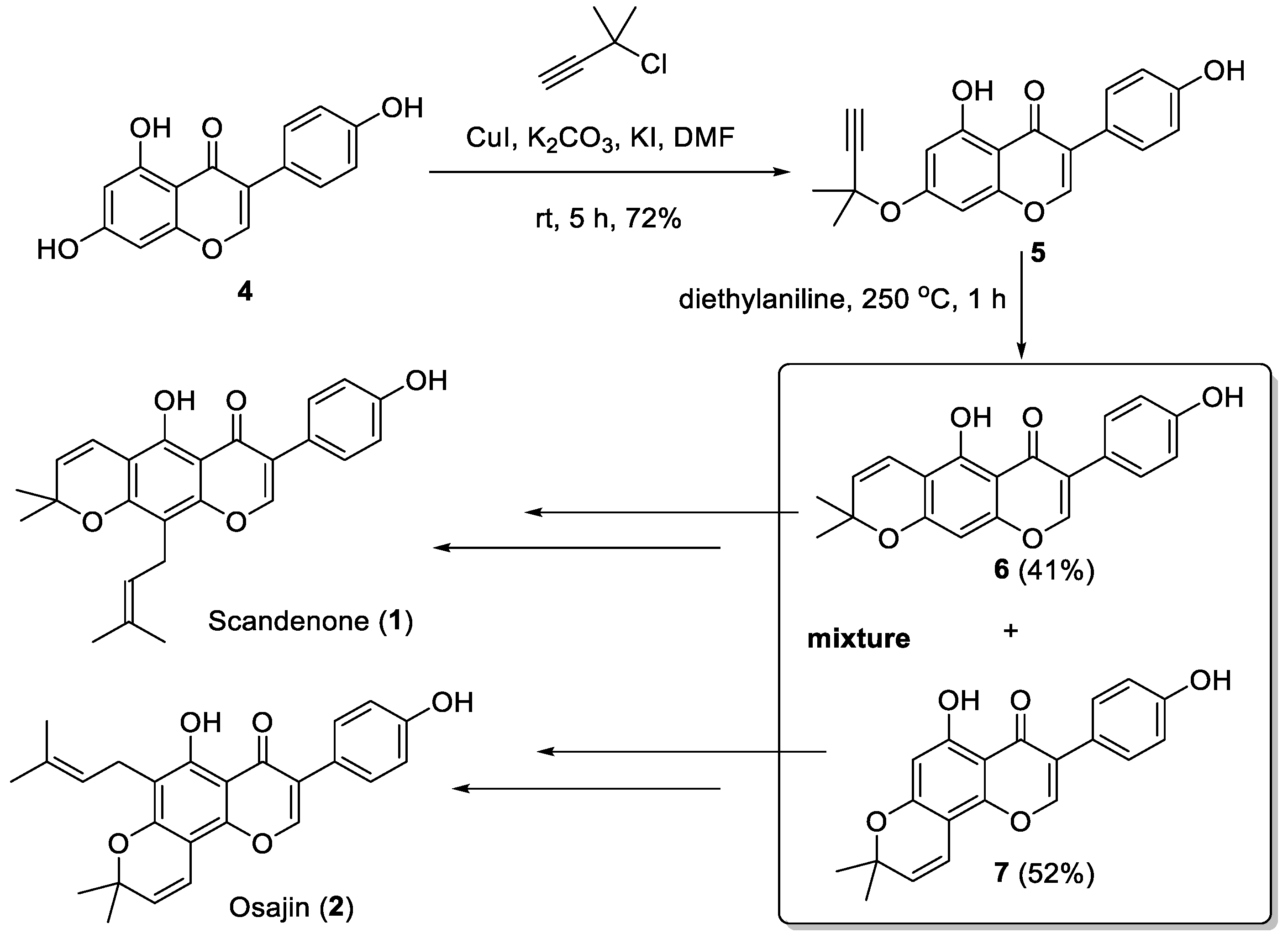
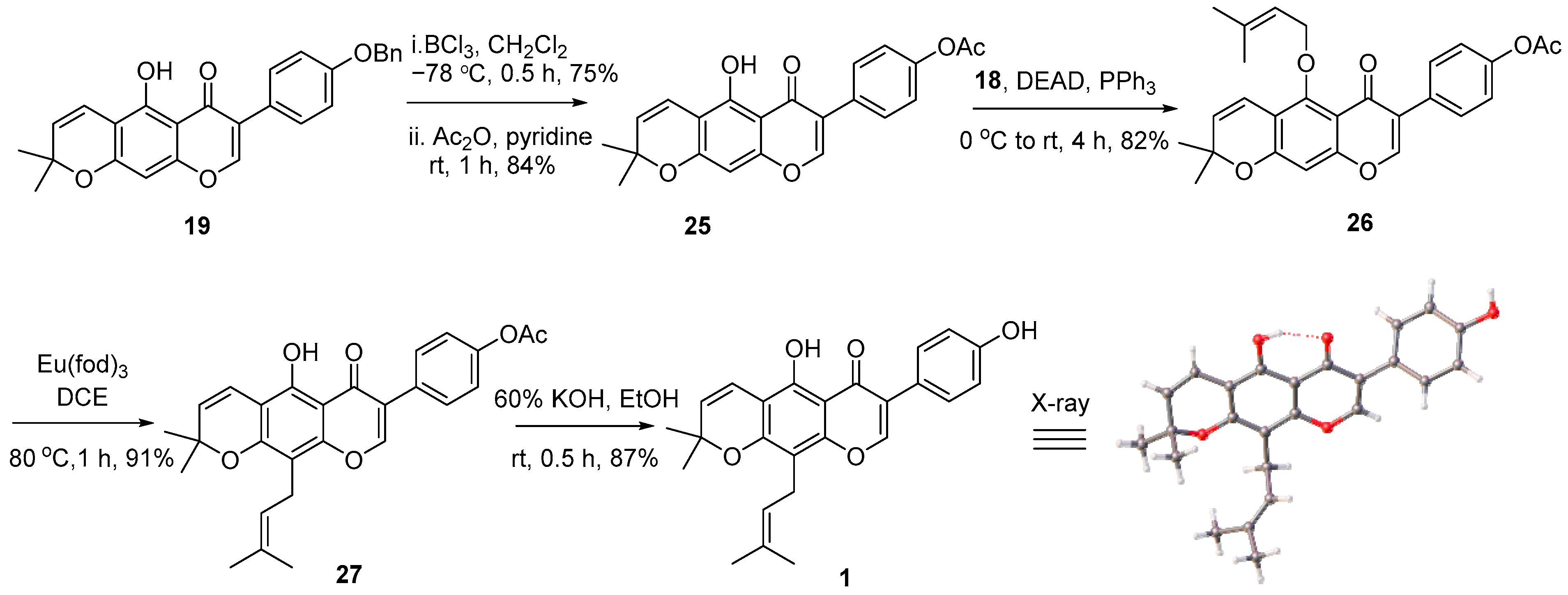
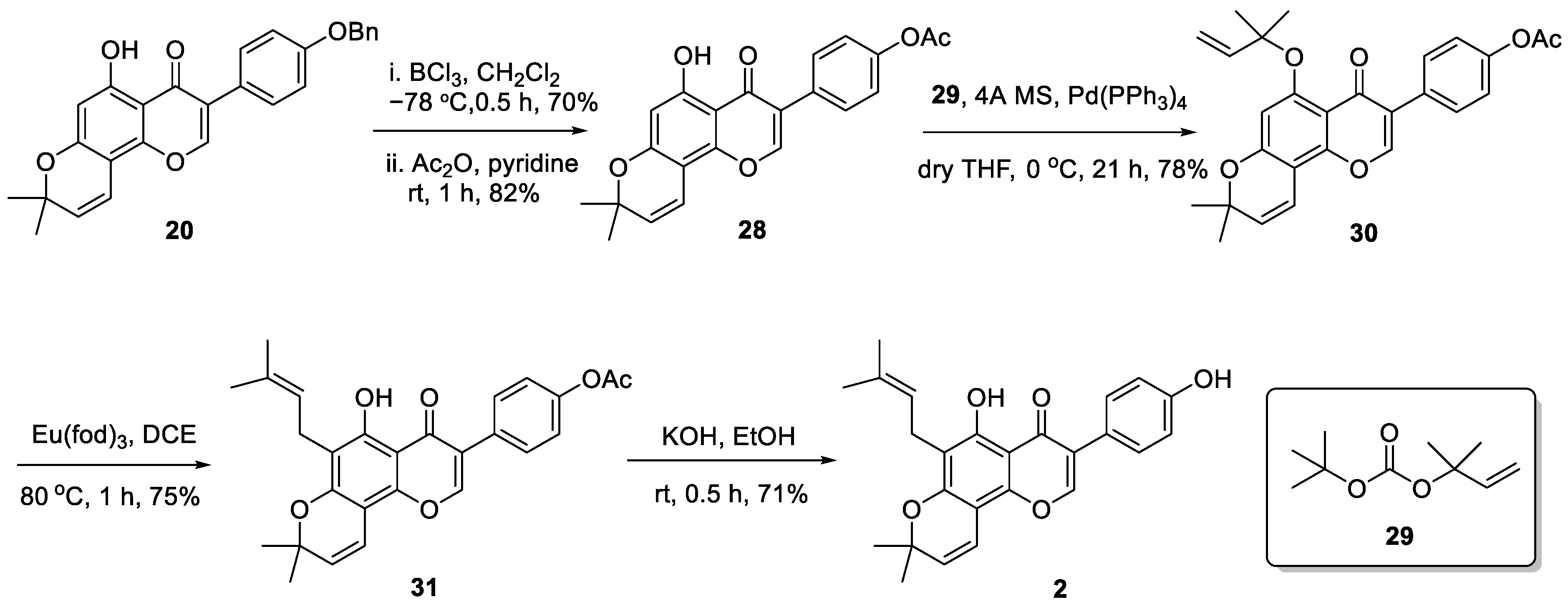
2. Results and Discussions
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Synthesis and Characterization of the Compounds
3.2.1. 1-(2-Hydroxy-4,6-bis(methoxymethoxy)phenyl)ethan-1-one (9)
3.2.2. (E)-3-(Dimethylamino)-1-(2-hydroxy-4,6-bis(methoxymethoxy)phenyl)prop-2-en-1-one (11)
3.2.3. 3-Iodo-5,7-bis(methoxymethoxy)-4H-chromen-4-one (12)
3.2.4. 3-(4-Hydroxyphenyl)-5,7-bis(methoxymethoxy)-4H-chromen-4-one (14)
3.2.5. 3-(4-(Benzyloxy)phenyl)-5,7-dihydroxy-4H-chromen-4-one (15)
3.2.6. 3-(4-(Benzyloxy)phenyl)-5,7-dihydroxy-6,8-bis(3-methylbut-2-en-1-yl)-4H-chromen-4-one (16)
3.2.7. 5,7-Dihydroxy-3-(4-hydroxyphenyl)-6,8-bis(3-methylbut-2-en-1-yl)-4H-chromen-4-one (3)
3.2.8. 3-(4-(Benzyloxy)phenyl)-5-hydroxy-7-((2-methylbut-3-yn-2-yl)oxy)-4H-chromen-4-one (24)
3.2.9. 3-(4-(Benzyloxy)phenyl)-5-hydroxy-8,8-dimethyl-4H,8H-pyrano[2,3-f]chromen-4-one (19)
3.2.10. (4-(Benzyloxy)phenyl)-5-hydroxy-8,8-dimethyl-4H,8H-pyrano[2,3-f]chromen-4-one (20)
3.2.11. 4-Hydroxy-7-(4-hydroxyphenyl)-2,2-dimethyl-2H,6H-pyrano[3,2-g]chromen-6-one (6)
3.2.12. 4-(5-Hydroxy-2,2-dimethyl-6-oxo-2H,6H-pyrano[3,2-g]chromen-7-yl)phenyl acetate (25)
3.2.13. 4-(8,8-Dimethyl-5-((3-methylbut-2-en-1-yl)oxy)-4-oxo-4H,8H-pyrano[2,3-f]chromen-3-yl)phenyl acetate (26)
3.2.14. 4-(5-Hydroxy-2,2-dimethyl-10-(3-methylbut-2-en-1-yl)-6-oxo-2H,6H-pyrano[3,2-g]chromen-7-yl)phenyl acetate (27)
3.2.15. 5-Hydroxy-7-(4-hydroxyphenyl)-2,2-dimethyl-10-(3-methylbut-2-en-1-yl)-2H,6H-pyrano[3,2-g]chromen-6-one (1)
3.2.16. 5-Hydroxy-3-((4-hydroxyphenyl)-8,8dimethyl-4H,8H-pyrano[2,3-f]chromen-4-one (7)
3.2.17. 3-(4-(Benzyloxy)phenyl)-5-hydroxy-7-((2-methylbut-3-yn-2-yl)oxy)-4H-chromen-4-one (28)
3.2.18. 4-(8,8-Dimethyl-5-((2-methylbut-3-en-2-yl)oxy)-4-oxo-4H,8H-pyrano[2,3-f]chromen-3-yl)phenyl acetate (30)
3.2.19. 4-(5-Hydroxy-8,8-dimethyl-6-(3-methylbut-2-en-1-yl)-4-oxo-4H,8H-pyrano[2,3-f]chromen-3-yl)phenyl acetate (31)
3.2.20. 5-Hydroxy-3-(4-hydroxyphenyl)-8,8-dimethyl-6-(3-methylbut-2-en-1-yl)-4H,8H-pyrano[2,3-f]chromen-4-one (2)
3.2.21. MICs Tests
3.2.22. Scanning Electron Microscopy (SEM) Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Al-Maharik, N. Isolation of naturally occurring novel isoflavonoids: An update. Nat. Prod. Rep. 2019, 36, 1156–1195. [Google Scholar] [CrossRef]
- Křížová, L.; Dadáková, K.; Kašparovská, J.; Kašparovský, T. Isoflavones. Molecules 2019, 24, 1076. [Google Scholar] [CrossRef]
- Qi, Y.; Xie, L.; Deng, Z.; Zhang, B.; Li, H. Stability and antioxidant activity of 10 isoflavones and anthocyanidins during in vitro digestion. Food Biosci. 2023, 56, 103189. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, H.; Cheng, M.; Cao, S.; Qiao, M.; Zhang, B.; Ding, L.; Qiu, F. New hepatoprotective isoflavone glucosides from Pueraria lobata (Willd.) Ohwi. Nat. Prod. Res. 2019, 33, 24. [Google Scholar] [CrossRef] [PubMed]
- Çiçek, S.S.; Pérez, M.G.; Wenzel-Storjohann, A.; Bezerra, R.B.; Segovia, J.F.O.; Girreser, U.; Kanzaki, I.; Tasdemir, D. Antimicrobial prenylated isoflavones from the leaves of the Amazonian medicinal plant Vatairea guianensis Aubl. J. Nat. Prod. 2022, 85, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Jeon, J.H.; Jeong, M.; Ryu, S.M.; Jeon, W.K.; Jang, D.S.; Shim, S.H. Chemical Constituents of Apios americana tubers and their inhibitory activities on nitric oxide production in lipopolysaccharide-stimulated RAW 264.7 macrophages. J. Nat. Prod. 2018, 81, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Ito, C.; Itoigawa, M.; Tan, H.T.; Tokuda, H.; Mou, X.Y.; Mukainaka, T.; Ishikawa, T.; Nishino, H.; Furukawa, H. Anti-tumor-promoting effects of isoflavonoids on Epstein-Barr virus activation and two-stage mouse skin carcinogenesis. Cancer Lett. 2000, 152, 187–192. [Google Scholar] [CrossRef]
- Ito, C.; Murata, T.; Itoigawa, M.; Itoigawa, M.; Nakao, K.; Kumagai, M.; Kaneda, N.; Furukawa, H. Induction of apoptosis by isoflavonoids from the leaves of Millettia taiwaniana in human leukemia HL-60 Cells. Planta Med. 2006, 72, 424–429. [Google Scholar] [CrossRef]
- Talla, E.; Yankep, E.; Mbafor, J.T. Chemical constituents from root barks of Erythrina mildbraedii and stem barks of Erythrina addisoniae. Bull. Chem. Soc. Ethiop. 2014, 28, 155–159. [Google Scholar] [CrossRef]
- Ozçelik, B.; Orhan, I.; Toker, G. Antiviral and antimicrobial assessment of some selected flavonoids. Z. Naturforschung C 2006, 61, 623–638. [Google Scholar] [CrossRef]
- Wang, B.; Ternai, B.; Polya, G. Specific inhibition of cyclic AMP-dependent protein kinase by warangalone and robustic acid. Phytochemistry 1997, 44, 787–796. [Google Scholar] [CrossRef]
- Sreelatha, T.; Hymavathi, A.; Rao, V.R.S.; Devanand, P.; Rani, P.U.; Rao, J.M.; Babu, K.S. A new benzil derivative from Derris scandens: Structure-insecticidal activity study. Bioorgan. Med. Chem. 2010, 20, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Raksat, A.; Maneerat, W.; Andersen, R.J.; Pyne, S.G.; Laphookhieo, S. Antibacterial prenylated isoflavonoids from the stems of Millettia extensa. J. Nat. Prod. 2018, 81, 1835–1840. [Google Scholar] [CrossRef] [PubMed]
- Peleyeju, G.B.; Emmanuel, T.; Tata, C.M.; Fotsing, M.C.D.; Niemann, N.; Rhyman, L.; Arderne, C.; Ndinteh, D.T.; Ramasami, P. Crystal structure and antibacterial activity of scandenone (warangalone) from Erythrina plants. J. Mol. Struct. 2019, 1191, 43–51. [Google Scholar] [CrossRef]
- Yang, R.; Hanwell, H.; Zhang, J.; Tsao, R.; Meckling, K.A. Antiproliferative activity of pomiferin in normal (MCF-10A) and transformed (MCF-7) breast epithelial cells. J. Agric. Food Chem. 2011, 59, 13328–13336. [Google Scholar] [CrossRef] [PubMed]
- Hošek, J.; Toniolo, A.; Neuwirth, O.; Bolego, C. Prenylated and geranylated flavonoids increase production of reactive oxygen species in mouse macrophages but inhibit the inflammatory response. J. Nat. Prod. 2013, 76, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Diopan, V.; Babula, P.; Shestivska, V.; Adam, V.; Zemlicka, M.; Dvorska, M.; Hubalek, J.; Trnkova, L.; Havel, L.; Kizek, R. Electrochemical and spectrometric study of antioxidant activity of pomiferin, isopomiferin, osajin and catalposide. J. Pharmaceut. Biomed. 2008, 48, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Nkengfack, A.E.; Kouam, J.; Vouffo, T.W.; Meyer, M.; Tempesta, M.S.; Fomum, Z.T. An isoflavanone and a coumestan from Erythrina sigmoidea. Phytochemistry 1994, 35, 521–526. [Google Scholar] [CrossRef]
- Dong, H.; Yu, P.; Long, B.; Peng, T.; He, Y.; Xu, B.; Liao, L.; Lu, L. Total synthesis of Kuwanons A and B and discovery of their antibacterial mechanism. J. Nat. Prod. 2023, 86, 2022–2030. [Google Scholar] [CrossRef]
- Dong, H.; Wu, M.; Li, Y.; Lu, L.; Qin, J.; He, Y.; Shi, Z. Total syntheses and anti-inflammatory evaluations of pongamosides A−C, natural furanoflavonoid glucosides from fruit of Pongamia pinnata (L.) Pierre. J. Nat. Prod. 2022, 85, 1118–1127. [Google Scholar] [CrossRef]
- Yu, P.; Long, B.; Feng, C.; Yang, T.; Jiang, X.; He, Y.; Dong, H. Total syntheses of pongaflavone and its natural analogues. J. Asian Nat. Prod. Res. 2023, 25, 1085–1096. [Google Scholar] [CrossRef]
- Wu, M.; Xu, L.; Li, Y.; Yu, P.; Lu, L.; Xie, S.; He, Y.; Dong, H. First total syntheses of Kanjone and its natural analogues. J. Asian Nat. Prod. Res. 2023, 25, 277–286. [Google Scholar] [CrossRef]
- Wang, R.; Ma, R.; Feng, K.; Lu, H.; Zhao, W.; Jin, H. Total synthesis and anti-inflammatory evaluation of osajin, scandenone and analogues. Pharmaceuticals 2024, 17, 86. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, X.; Chen, L.; Yin, Z.; Zuo, Z. Discovery and evaluation of cytisine N-isoflavones as novel EGFR/HER2 dual inhibitors. Bioorganic Chem. 2022, 127, 105868. [Google Scholar] [CrossRef]
- Dong, H.; Wu, M.; Xiang, S.; Song, T.; Li, Y.; Long, B.; Feng, C.; Shi, Z. Total syntheses and antibacterial evaluations of neocyclomorusin and related flavones. J. Nat. Prod. 2022, 85, 2217–2225. [Google Scholar] [CrossRef]
- Luan, N.N.T.; Okada, T.; Arata, R.; Prudhvi, L.; Miyaguchi, M.; Kodama, Y.; Awale, S.; Toyooka, N. Structure-activity relationship study of 4′-O-methylgrynullarin derivatives for the development of novel anticancer agents based on anti-austerity strategy. Tetrahedron 2022, 122, 132931. [Google Scholar] [CrossRef]
- Kwesiga, G.; Greese, J.; Kelling, A.; Sperlich, E.; Schmidt, B. The Suzuki-Miyaura cross-coupling-Claisen rearrangement-cross-metathesis approach to prenylated isoflavones. J. Org. Chem. 2023, 88, 1649–1664. [Google Scholar] [CrossRef]
- Xiao, L.; Tan, W.; Li, Y. First total synthesis of (±)-Kenusanone B. Synth. Commun. 1998, 28, 2861–2869. [Google Scholar] [CrossRef]
- Franov, L.J.; Hart, J.D.; Pullella, G.A.; Sumby, C.J.; George, J.H. Bioinspired total synthesis of erectones A and B, and the revised structure of hyperelodione D. Angew. Chem. Int. Ed. 2022, 61, e2022004. [Google Scholar] [CrossRef]
- Mzozoyana, V.; Heerden, F.R. Synthesis of fluorine-containing prenylated benzophenones. Synth. Commun. 2019, 50, 2226–2235. [Google Scholar] [CrossRef]
- Jentsch, N.G.; Zhang, X.; Magolan, J. Efficient synthesis of cannabigerol, grifolin, and piperogalin via alumina-promoted allylation. J. Nat. Prod. 2020, 83, 2587–2591. [Google Scholar] [CrossRef]
- Dong, H.; Liao, L.; Yu, P.; Long, B.; Che, Y.; Lu, L.; Xu, B. Total syntheses and antibacterial evaluations of cudraflavones A-C and related flavones. Bioorganic Chem. 2023, 40, 106764. [Google Scholar] [CrossRef]
- Dong, H.; Liao, L.; Long, B.; Che, Y.; Peng, T.; He, Y.; Mei, L.; Xu, B. Total synthesis and antibacterial evaluation of lupinifolin and its natural analogues. J. Nat. Prod. 2024, 87, 1044–1058. [Google Scholar] [CrossRef]
- Zheng, S.; Li, X.; Tan, H.; Yu, C.; Zhang, J.; Shen, Z. Studies on the total synthesis of Hirtellanine A: Regioselective synthesis of benzopyran. Eur. J. Org. Chem. 2013, 2013, 1356–1366. [Google Scholar] [CrossRef]
- Masao, T.; Yasuhiko, K.; Hideo, T. Synthesis of 4′,5- and 3′,4′,5-oxygenated pyranoisoflavones: Alpinum isoflavone and related compounds, and revised structure of derrone. Heterocycles 1992, 34, 505–516. [Google Scholar]
- Song, M.; Liu, Y.; Li, T.; Liu, X.; Hao, Z.; Ding, S.; Panichayupakaranant, P.; Zhu, K.; Shen, J. Plant natural flavonoids against multidrug resistant pathogens. Adv. Sci. 2021, 8, 2100749. [Google Scholar] [CrossRef]
- Sychrová, A.; Škovranová, G.; Čulenová, M.; Fialová, S. Prenylated flavonoids in topical infections and wound healing. Molecules 2022, 27, 4491. [Google Scholar] [CrossRef]
- Lin, Y.; Kuang, Y.; Li, K.; Wang, S.; Ji, S.; Chen, K.; Song, W.; Qiao, X.; Ye, M. Nrf2 activators from Glycyrrhiza inflata and their hepatoprotective activities against CCl4-induced liver injury in mice. Bioorganic Med. Chem. 2017, 25, 5522–5530. [Google Scholar] [CrossRef]
- Khalid, S.A.; Waterman, P.G. Thonningine-A and thonningine-B: Two 3-phenylcoumarins from the seeds of Millettia thonningii. Phytochemistry 1983, 22, 1001–1003. [Google Scholar]
- Peter, A.; Stainton, P. The extractives from Derris scandens. Part II. The isolation of osajin and two new isoflavones, scandenone and scandinone. J. Chem. Soc. C 1966, 701–704. [Google Scholar] [CrossRef]
- Chouna, H.; Dize, D.; Kagho, D.; Bankeu, J.; Fongang, Y.; Tali, M.; Ponou, B.; Bitchagno, G.; Awantu, A.; Tapondjou, L.; et al. Constituents from ripe figs of Ficus vallis-choudae Delile (Moraceae) with antiplasmodial activity. Parasitol. Res. 2022, 121, 2121–2127. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Entry | Conditions | Yields of Isolated 16 (%) |
| 1 | compound 17, KOH, H2O, 0 °C to rt | - |
| 2 | compound 17, K2CO3, acetone, rt | 7 |
| 3 | compound 17, DBU, THF, 50 °C | 20 |
| 4 | compound 18, p-toluenesulfonic acid, CH2Cl2, rt | 14 |
| 5 | compound 18, acidic alumina, DCE, 80 °C | 72 |
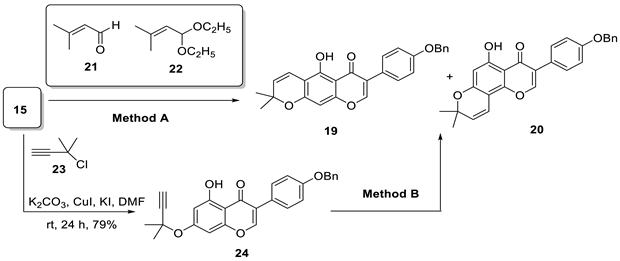 | ||||
|---|---|---|---|---|
| Entry | Method | Conditions | 19, Yield 1 (%) | 20, Yield 1 (%) |
| 1 | Method A | 21, Ca(OH)2, CH3OH, 0 °C to rt | 27 | 22 |
| 2 | Method A | 22, 3-picoline, toluene, 110 °C | 25 | 27 |
| 3 | Method B | DMF, 130 °C | 9 | 77 |
| 4 | Method B | xylene, 130 °C | 55 | 33 |
| 5 | Method B | xylene, KOH, 130 °C | 93 | - |
| 6 | Method B | diethylaniline, 250 °C | 41 | 40 |
| Compound | MIC (μg/mL) | |||
|---|---|---|---|---|
| S. aureus ATCC29213 | E. faecalis ATCC29212 | MRSA ATCC33591 | E. coli ATCC25922 | |
| 1 | 16 | 16 | 4 | >128 |
| 2 | 2 | 8 | 2 | >128 |
| 3 | 8 | 16 | 4 | >128 |
| Ampicillin | 32 | 1 | >128 | 4 |
| Vancomycin | 2 | 2 | 1 | 128 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, H.; Che, Y.; Zhu, X.; Zhong, Y.; Lin, J.; Wang, J.; Du, W.; Song, T. Total Syntheses and Antibacterial Studies of Natural Isoflavones: Scandenone, Osajin, and 6,8-Diprenylgenistein. Molecules 2024, 29, 2574. https://doi.org/10.3390/molecules29112574
Dong H, Che Y, Zhu X, Zhong Y, Lin J, Wang J, Du W, Song T. Total Syntheses and Antibacterial Studies of Natural Isoflavones: Scandenone, Osajin, and 6,8-Diprenylgenistein. Molecules. 2024; 29(11):2574. https://doi.org/10.3390/molecules29112574
Chicago/Turabian StyleDong, Hongbo, Yufei Che, Xingtong Zhu, Yi Zhong, Jiafu Lin, Jian Wang, Weihong Du, and Tao Song. 2024. "Total Syntheses and Antibacterial Studies of Natural Isoflavones: Scandenone, Osajin, and 6,8-Diprenylgenistein" Molecules 29, no. 11: 2574. https://doi.org/10.3390/molecules29112574