


Decoding the κ Opioid Receptor (KOR): Advancements in Structural Understanding and Implications for Opioid Analgesic Development

Abstract
1. Introduction
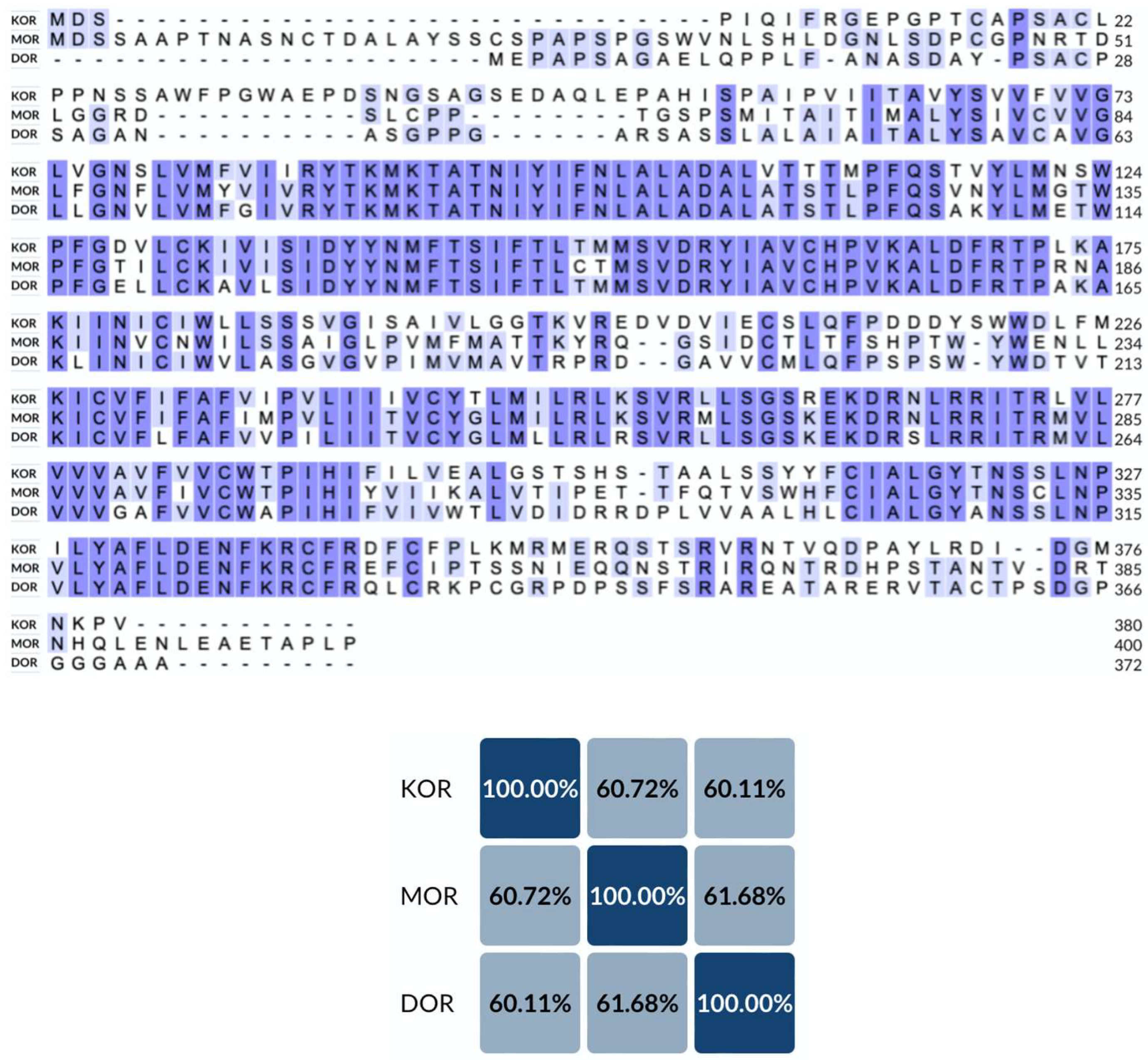
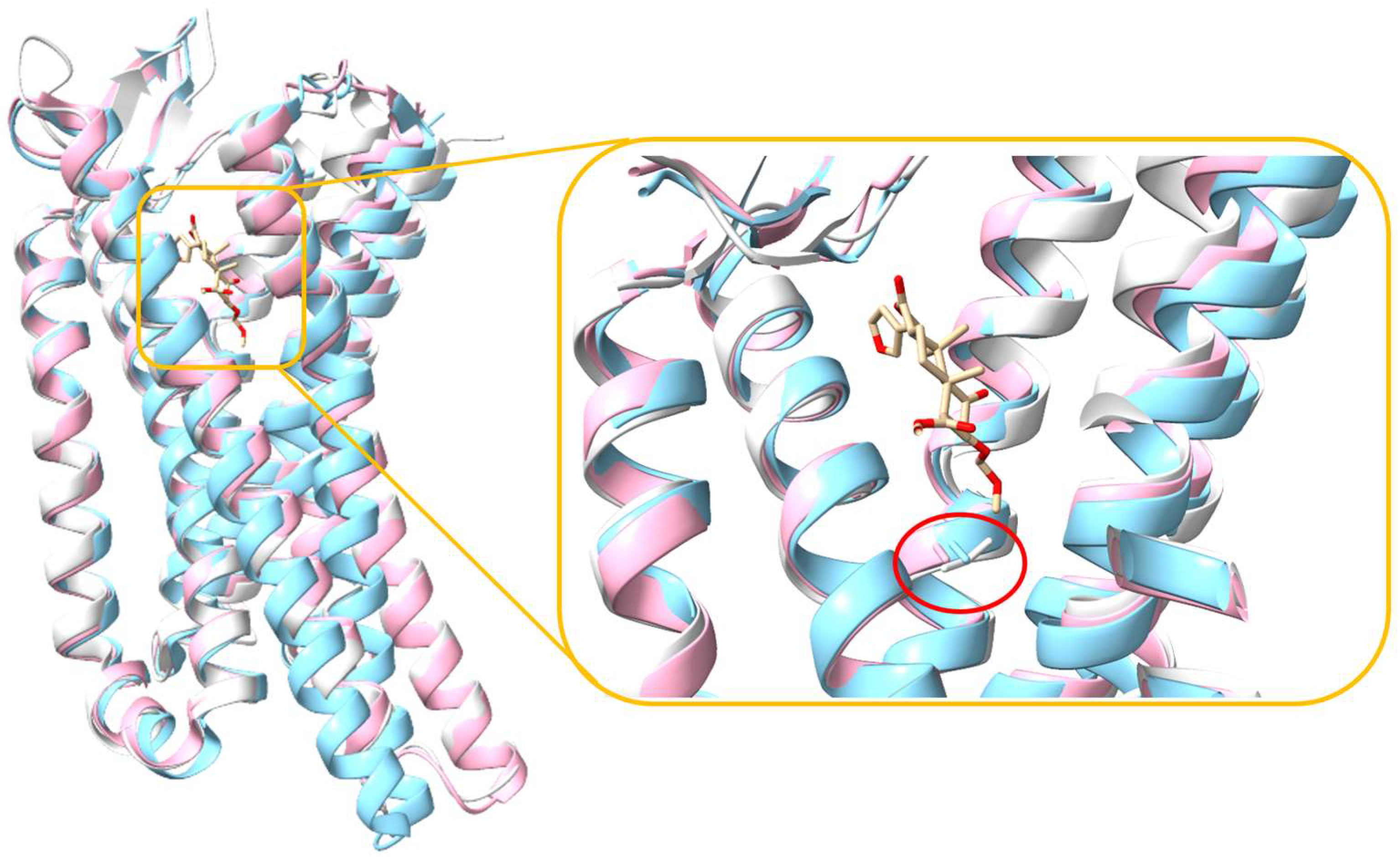
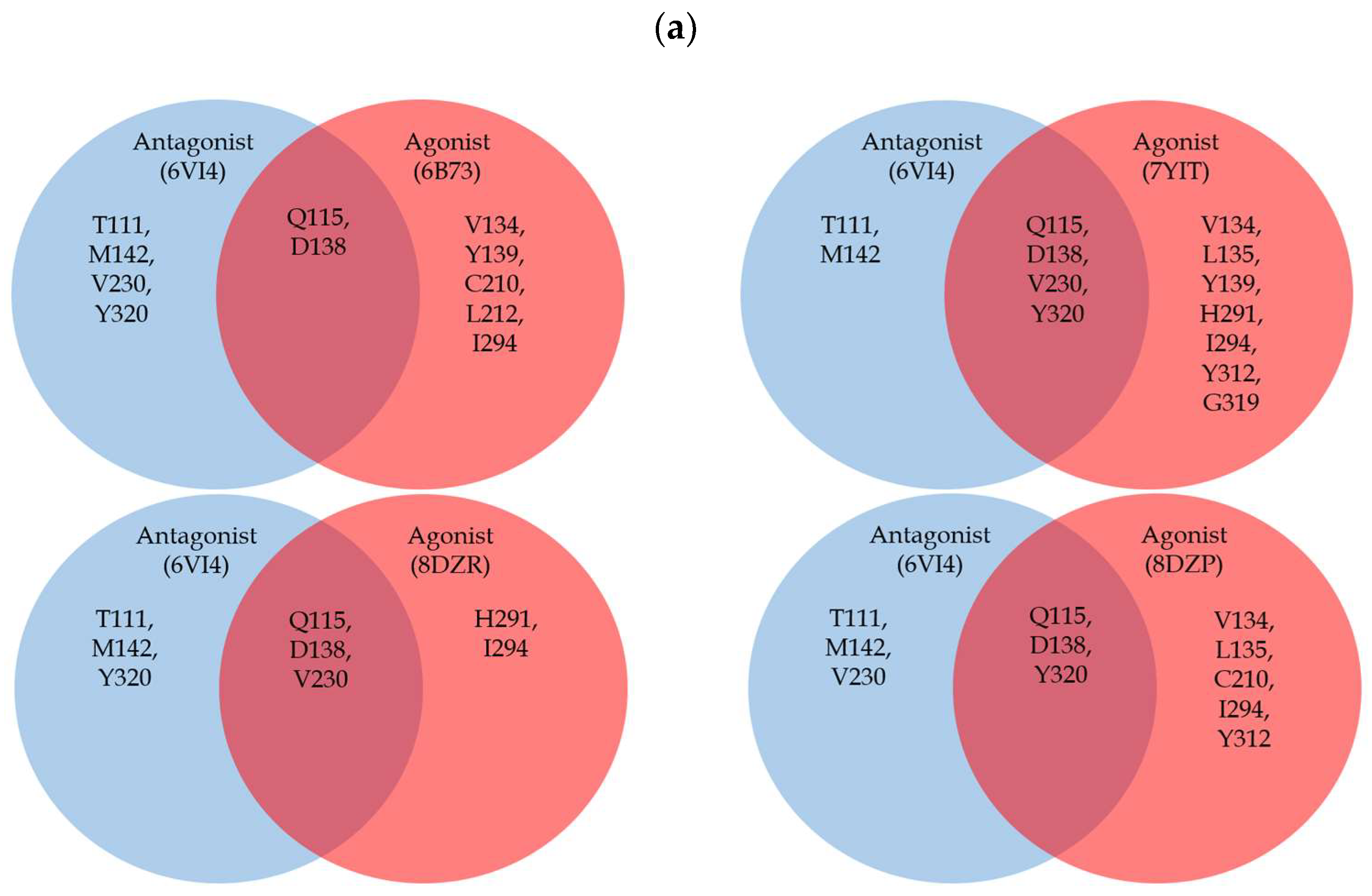
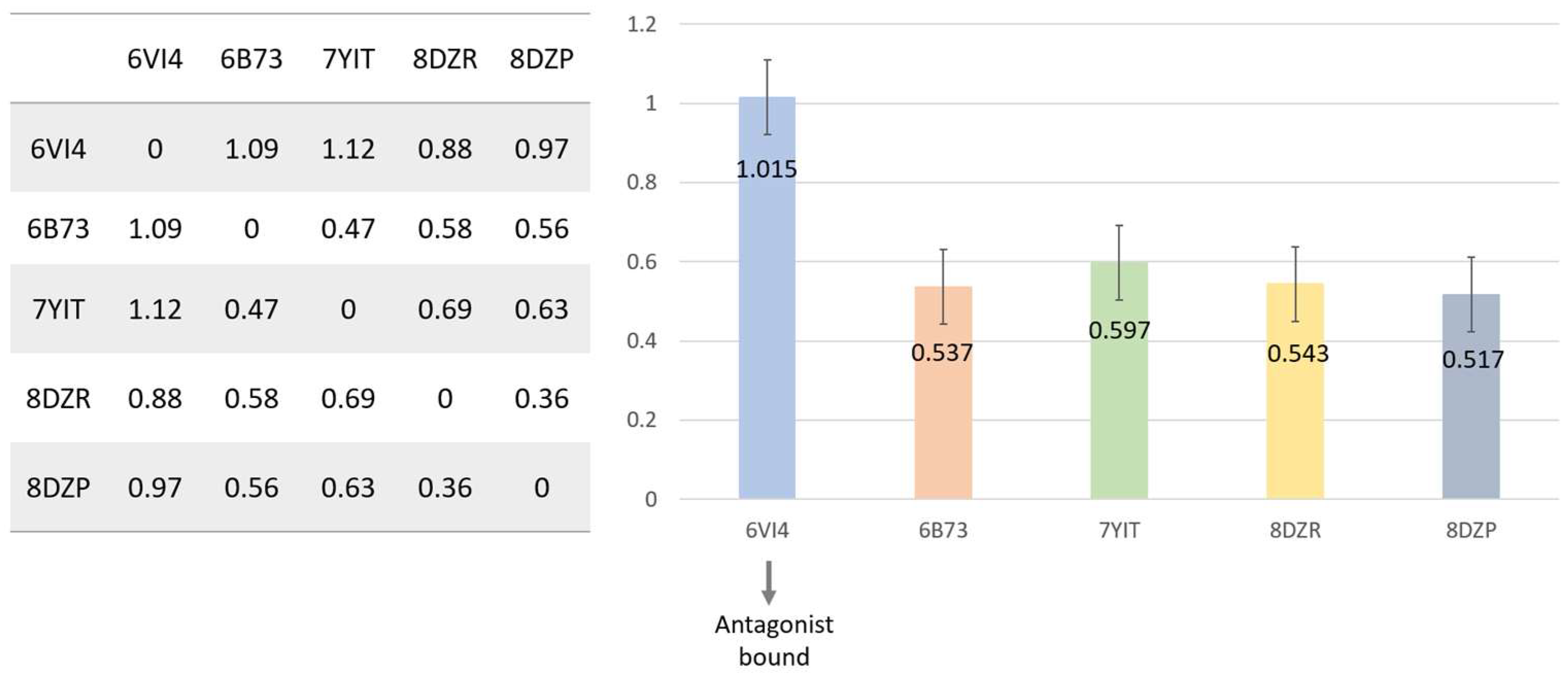
2. Structural Overview of KOR
3. Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rudd, R.A.; Seth, P.; David, F.; Scholl, L. Increases in Drug and Opioid-Involved Overdose Deaths—United States, 2010–2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1445–1452. [Google Scholar] [CrossRef]
- Rudd, R.A.; Aleshire, N.; Zibbell, J.E.; Gladden, R.M. Increases in Drug and Opioid Overdose Deaths--United States, 2000–2014. MMWR Morb. Mortal. Wkly. Rep. 2016, 64, 1378–1382. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.M.; Baldwin, G.T.; Manocchio, T.; White, J.O.; Mack, K.A. Trends in Methadone Distribution for Pain Treatment, Methadone Diversion, and Overdose Deaths—United States, 2002–2014. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 667–671. [Google Scholar] [CrossRef]
- Peterson, A.B.; Gladden, R.M.; Delcher, C.; Spies, E.; Garcia-Williams, A.; Wang, Y.; Halpin, J.; Zibbell, J.; McCarty, C.L.; DeFiore-Hyrmer, J.; et al. Increases in Fentanyl-Related Overdose Deaths—Florida and Ohio, 2013–2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 844–849. [Google Scholar] [CrossRef]
- Gladden, R.M.; Martinez, P.; Seth, P. Fentanyl Law Enforcement Submissions and Increases in Synthetic Opioid-Involved Overdose Deaths—27 States, 2013–2014. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 837–843. [Google Scholar] [CrossRef]
- Vadivelu, N.; Kai, A.M.; Kodumudi, V.; Sramcik, J.; Kaye, A.D. The Opioid Crisis: A Comprehensive Overview. Curr. Pain Headache Rep. 2018, 22, 16. [Google Scholar] [CrossRef] [PubMed]
- Darcq, E.; Kieffer, B.L. Opioid Receptors: Drivers to Addiction? Nat. Rev. Neurosci. 2018, 19, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W.; Pan, Y.-X. Mu Opioids and Their Receptors: Evolution of a Concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef]
- Portoghese, P.S. Relationships Between Stereostructure and Pharmacological Activities. Annu. Rev. Pharmacol. 1970, 10, 51–76. [Google Scholar] [CrossRef]
- Portoghese, P.S. A New Concept on the Mode of Interaction of Narcotic Analgesics with Receptors. J. Med. Chem. 1965, 8, 609–616. [Google Scholar] [CrossRef]
- Beckett, A.H.; Casy, A.F. 5 Analgesics and Their Antagonists: Biochemical Aspects and Structure-Activity Relationships. In Progress in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 1965; Volume 4, pp. 171–218. ISBN 978-0-444-53323-4. [Google Scholar]
- Pert, C.B.; Snyder, S.H. Opiate Receptor: Demonstration in Nervous Tissue. Science 1973, 179, 1011–1014. [Google Scholar] [CrossRef]
- Terenius, L. Stereospecific Interaction Between Narcotic Analgesics and a Synaptic Plasma Membrane Fraction of Rat Cerebral Cortex. Acta Pharmacol. Toxicol. 2009, 32, 317–320. [Google Scholar] [CrossRef]
- Simon, E.J.; Hiller, J.M.; Edelman, I. Stereospecific Binding of the Potent Narcotic Analgesic [3H]Etorphine to Rat-Brain Homogenate. Proc. Natl. Acad. Sci. USA 1973, 70, 1947–1949. [Google Scholar] [CrossRef] [PubMed]
- Miller’s Anesthesia, 9th ed.; Gropper, M.A., Miller, R.D., Cohen, N.H., Eds.; Elsevier: Philadelphia, PA, USA, 2020; ISBN 978-0-323-59604-6. [Google Scholar]
- Mollereau, C.; Parmentier, M.; Mailleux, P.; Butour, J.-L.; Moisand, C.; Chalon, P.; Caput, D.; Vassart, G.; Meunier, J.-C. ORL1, a Novel Member of the Opioid Receptor Family: Cloning, Functional Expression and Localization. FEBS Lett. 1994, 341, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Al-Hasani, R.; Bruchas, M.R. Molecular Mechanisms of Opioid Receptor-Dependent Signaling and Behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [PubMed]
- Le Merrer, J.; Becker, J.A.J.; Befort, K.; Kieffer, B.L. Reward Processing by the Opioid System in the Brain. Physiol. Rev. 2009, 89, 1379–1412. [Google Scholar] [CrossRef]
- Pathan, H.; Williams, J. Basic Opioid Pharmacology: An Update. Br. J. Pain 2012, 6, 11–16. [Google Scholar] [CrossRef]
- Snyder, S.H.; Pasternak, G.W. Historical Review: Opioid Receptors. Trends Pharmacol. Sci. 2003, 24, 198–205. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal Structure of the Μ-Opioid Receptor Bound to a Morphinan Antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef]
- Feng, Y.; He, X.; Yang, Y.; Chao, D.; Lazarus, L.H.; Xia, Y. Current Research on Opioid Receptor Function. Curr. Drug Targets 2012, 13, 230–246. [Google Scholar] [CrossRef]
- Tan, L.; Yan, W.; McCorvy, J.D.; Cheng, J. Biased Ligands of G Protein-Coupled Receptors (GPCRs): Structure–Functional Selectivity Relationships (SFSRs) and Therapeutic Potential. J. Med. Chem. 2018, 61, 9841–9878. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G Protein-Coupled Receptors: Structure- and Function-Based Drug Discovery. Signal Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Cong, X.; Maurel, D.; Déméné, H.; Vasiliauskaité-Brooks, I.; Hagelberger, J.; Peysson, F.; Saint-Paul, J.; Golebiowski, J.; Granier, S.; Sounier, R. Molecular Insights into the Biased Signaling Mechanism of the μ-Opioid Receptor. Mol. Cell 2021, 81, 4165–4175.e6. [Google Scholar] [CrossRef] [PubMed]
- Che, T.; Dwivedi-Agnihotri, H.; Shukla, A.K.; Roth, B.L. Biased Ligands at Opioid Receptors: Current Status and Future Directions. Sci. Signal. 2021, 14, eaav0320. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; Hollingsworth, S.A.; Blakemore, D.C.; Owen, R.M.; Storer, R.I.; Swain, N.A.; Aydin, D.; Torella, R.; Warmus, J.S.; Dror, R.O. Delineating the Ligand–Receptor Interactions That Lead to Biased Signaling at the μ-Opioid Receptor. J. Chem. Inf. Model. 2021, 61, 3696–3707. [Google Scholar] [CrossRef] [PubMed]
- Nagi, K.; Pineyro, G. Kir3 Channel Signaling Complexes: Focus on Opioid Receptor Signaling. Front. Cell. Neurosci. 2014, 8, 186. [Google Scholar] [CrossRef] [PubMed]
- Mafi, A.; Kim, S.-K.; Goddard, W.A. Mechanism of β-Arrestin Recruitment by the μ-Opioid G Protein-Coupled Receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 16346–16355. [Google Scholar] [CrossRef] [PubMed]
- Noor, N.; Patel, C.B.; Rockman, H.A. β-Arrestin: A Signaling Molecule and Potential Therapeutic Target for Heart Failure. J. Mol. Cell. Cardiol. 2011, 51, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Macé, G.; Miaczynska, M.; Zerial, M.; Nebreda, A.R. Phosphorylation of EEA1 by P38 MAP Kinase Regulates μ Opioid Receptor Endocytosis. EMBO J. 2005, 24, 3235–3246. [Google Scholar] [CrossRef]
- Belcheva, M.M.; Clark, A.L.; Haas, P.D.; Serna, J.S.; Hahn, J.W.; Kiss, A.; Coscia, C.J. μ and κ Opioid Receptors Activate ERK/MAPK via Different Protein Kinase C Isoforms and Secondary Messengers in Astrocytes. J. Biol. Chem. 2005, 280, 27662–27669. [Google Scholar] [CrossRef]
- Baker Rogers, J.; Higa, G.M. Spoken and Unspoken Matters Regarding the Use of Opioids in Cancer. J. Pain Res. 2022, 15, 909–924. [Google Scholar] [CrossRef] [PubMed]
- Lutz, P.-E.; Kieffer, B.L. Opioid Receptors: Distinct Roles in Mood Disorders. Trends Neurosci. 2013, 36, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Valentino, R.J.; Volkow, N.D. Untangling the Complexity of Opioid Receptor Function. Neuropsychopharmacol 2018, 43, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Barnett, M.E.; Knapp, B.I.; Bidlack, J.M. Unique Pharmacological Properties of the Kappa Opioid Receptor Signaling Through Gαz as Shown with Bioluminescence Resonance Energy Tranfer. Mol. Pharmacol. 2020, 98, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Sturaro, C.; Malfacini, D.; Argentieri, M.; Djeujo, F.M.; Marzola, E.; Albanese, V.; Ruzza, C.; Guerrini, R.; Calo’, G.; Molinari, P. Pharmacology of Kappa Opioid Receptors: Novel Assays and Ligands. Front. Pharmacol. 2022, 13, 873082. [Google Scholar] [CrossRef] [PubMed]
- Daibani, A.E. Molecular Mechanism of Biased Signaling at the Kappa Opioid Receptor. Nat. Commun. 2023, 11, 1338. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Huang, P.; Chiu, Y.-T.; Chen, C.; Wang, H.; Li, M.; Zheng, Y.; Ehlert, F.J.; Zhang, Y.; Liu-Chen, L.-Y. Comparison of Pharmacological Properties between the Kappa Opioid Receptor Agonist Nalfurafine and 42B, Its 3-Dehydroxy Analogue: Disconnect between in Vitro Agonist Bias and in Vivo Pharmacological Effects. ACS Chem. Neurosci. 2020, 11, 3036–3050. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jiang, Q.; Zhu, H.; Zhou, Y.; Lu, D.; Liu, X.; Chen, X.; Chen, J.; Wang, Y.; Liu, J.; et al. Uncovering Kappa-Opioid Receptor Agonist-Induced PAK1/2 Phosphorylation by Quantitative Phosphoproteomics. Biochem. Biophys. Res. Commun. 2019, 516, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Reichard, K.L.; Newton, K.A.; Rivera, Z.M.G.; Sotero de Menezes, P.M.; Schattauer, S.S.; Land, B.B.; Chavkin, C. Regulation of Kappa Opioid Receptor Inactivation Depends on Sex and Cellular Site of Antagonist Action. Mol. Pharmacol. 2020, 98, 548–558. [Google Scholar] [CrossRef]
- Brust, T.F. Biased Ligands at the Kappa Opioid Receptor: Fine-Tuning Receptor Pharmacology. Kappa Opioid Recept. 2022, 271, 115–135. [Google Scholar] [CrossRef]
- James, A.; Williams, J. Basic Opioid Pharmacology—An Update. Br. J. Pain 2020, 14, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.H.; Sawyer, B.J.; Akins, N.S.; Le, H.V. A Systematic Review on the Kappa Opioid Receptor and Its Ligands: New Directions for the Treatment of Pain, Anxiety, Depression, and Drug Abuse. Eur. J. Med. Chem. 2022, 243, 114785. [Google Scholar] [CrossRef] [PubMed]
- Jullié, D.; Gondin, A.B.; von Zastrow, M.; Canals, M. Opioid Pharmacology under the Microscope. Mol. Pharmacol. 2020, 98, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Hassanien, S.H.; Bassman, J.R.; Perrien Naccarato, C.M.; Twarozynski, J.J.; Traynor, J.R.; Iula, D.M.; Anand, J.P. In Vitro Pharmacology of Fentanyl Analogs at the Human Mu Opioid Receptor and Their Spectroscopic Analysis. Drug Test. Anal. 2020, 12, 1212–1221. [Google Scholar] [CrossRef]
- Ji, J.; Lin, W.; Vrudhula, A.; Xi, J.; Yeliseev, A.; Grothusen, J.R.; Bu, W.; Liu, R. Molecular Interaction Between Butorphanol and κ-Opioid Receptor. Anesth. Analg. 2020, 131, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-Based Discovery of Opioid Analgesics with Reduced Side Effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Huang, X.-P.; Mangano, T.J.; Zou, R.; Chen, X.; Zaidi, S.A.; Roth, B.L.; Stevens, R.C.; Katritch, V. Structure-Based Discovery of New Antagonist and Biased Agonist Chemotypes for the Kappa Opioid Receptor. J. Med. Chem. 2017, 60, 3070–3081. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Dimmito, M.P.; Mollica, A.; Zengin, G.; Benyhe, S.; Zador, F.; Stefanucci, A. Discovery of Novel µ-Opioid Receptor Inverse Agonist from a Combinatorial Library of Tetrapeptides through Structure-Based Virtual Screening. Molecules 2019, 24, 3872. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, A.; Wang, H.; Zaidi, S.A.; DiBerto, J.F.; Che, T.; Qu, Q.; Robertson, M.J.; Madasu, M.K.; El Daibani, A.; Varga, B.R.; et al. Structure-Based Design of Bitopic Ligands for the µ-Opioid Receptor. Nature 2023, 613, 767–774. [Google Scholar] [CrossRef]
- Che, T.; English, J.; Krumm, B.E.; Kim, K.; Pardon, E.; Olsen, R.H.J.; Wang, S.; Zhang, S.; Diberto, J.F.; Sciaky, N.; et al. Nanobody-Enabled Monitoring of Kappa Opioid Receptor States. Nat. Commun. 2020, 11, 1145. [Google Scholar] [CrossRef]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.-P.; Carroll, F.I.; et al. Structure of the Human κ-Opioid Receptor in Complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 2018, 172, 55–67.e15. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhang, J.; Nazarova, A.L.; Bernhard, S.M.; Krumm, B.E.; Zhao, L.; Lam, J.H.; Rangari, V.A.; Majumdar, S.; Nichols, D.E.; et al. Ligand and G-Protein Selectivity in the κ-Opioid Receptor. Nature 2023, 617, 417–425. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, B.; Wu, T.; Zhou, F.; Xu, F. Cryo-EM Structure of Human κ-Opioid Receptor-Gi Complex Bound to an Endogenous Agonist Dynorphin A. Protein Cell 2023, 14, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhuang, Y.; DiBerto, J.F.; Zhou, X.E.; Schmitz, G.P.; Yuan, Q.; Jain, M.K.; Liu, W.; Melcher, K.; Jiang, Y.; et al. Structures of the Entire Human Opioid Receptor Family. Cell 2023, 186, 413–427.e17. [Google Scholar] [CrossRef]
- Muratspahić, E.; Deibler, K.; Han, J.; Tomašević, N.; Jadhav, K.B.; Olivé-Marti, A.-L.; Hochrainer, N.; Hellinger, R.; Koehbach, J.; Fay, J.F.; et al. Design and Structural Validation of Peptide-Drug Conjugate Ligands of the Kappa-Opioid Receptor. Nat. Commun. 2023, 14, 8064. [Google Scholar] [CrossRef]
- Li, Z.; Liu, J.; Dong, F.; Chang, N.; Huang, R.; Xia, M.; Patterson, T.A.; Hong, H. Three-Dimensional Structural Insights Have Revealed the Distinct Binding Interactions of Agonists, Partial Agonists, and Antagonists with the µ Opioid Receptor. Int. J. Mol. Sci. 2023, 24, 7042. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Wang, Y.; He, B.; He, X.; Zhou, X.E.; Guo, S.; Rao, Q.; Yang, J.; Liu, J.; Zhou, Q.; et al. Molecular Recognition of Morphine and Fentanyl by the Human μ-Opioid Receptor. Cell 2022, 185, 4361–4375.e19. [Google Scholar] [CrossRef] [PubMed]
- Claff, T.; Yu, J.; Blais, V.; Patel, N.; Martin, C.; Wu, L.; Han, G.W.; Holleran, B.J.; Van der Poorten, O.; White, K.L.; et al. Elucidating the Active δ-Opioid Receptor Crystal Structure with Peptide and Small-Molecule Agonists. Sci. Adv. 2019, 5, eaax9115. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Sakkiah, S.; Guo, W.; Pan, B.; Ji, Z.; Yavas, G.; Azevedo, M.; Hawes, J.; Patterson, T.A.; Hong, H. Elucidating Interactions Between SARS-CoV-2 Trimeric Spike Protein and ACE2 Using Homology Modeling and Molecular Dynamics Simulations. Front. Chem. 2021, 8, 622632. [Google Scholar] [CrossRef] [PubMed]
- Sakkiah, S.; Kusko, R.; Pan, B.; Guo, W.; Ge, W.; Tong, W.; Hong, H. Structural Changes Due to Antagonist Binding in Ligand Binding Pocket of Androgen Receptor Elucidated Through Molecular Dynamics Simulations. Front. Pharmacol. 2018, 9, 492. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, C.; Sakkiah, S.; Tong, W.; Hong, H. Molecular Dynamics Simulations and Applications in Computational Toxicology and Nanotoxicology. Food Chem. Toxicol. 2018, 112, 495–506. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, W.; Fang, H.; Perkins, R.; Tong, W.; Hong, H. Homology Modeling, Molecular Docking, and Molecular Dynamics Simulations Elucidated α-Fetoprotein Binding Modes. BMC Bioinform. 2013, 14, S6. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.H. Deciphering the Molecular Basis of the Kappa Opioid Receptor Selectivity: A Molecular Dynamics Study. J. Mol. Graph. Model. 2021, 106, 107940. [Google Scholar] [CrossRef]
- An, X.; Bai, Q.; Bing, Z.; Zhou, S.; Shi, D.; Liu, H.; Yao, X. How Does Agonist and Antagonist Binding Lead to Different Conformational Ensemble Equilibria of the κ-Opioid Receptor: Insight from Long-Time Gaussian Accelerated Molecular Dynamics Simulation. ACS Chem. Neurosci. 2019, 10, 1575–1584. [Google Scholar] [CrossRef]


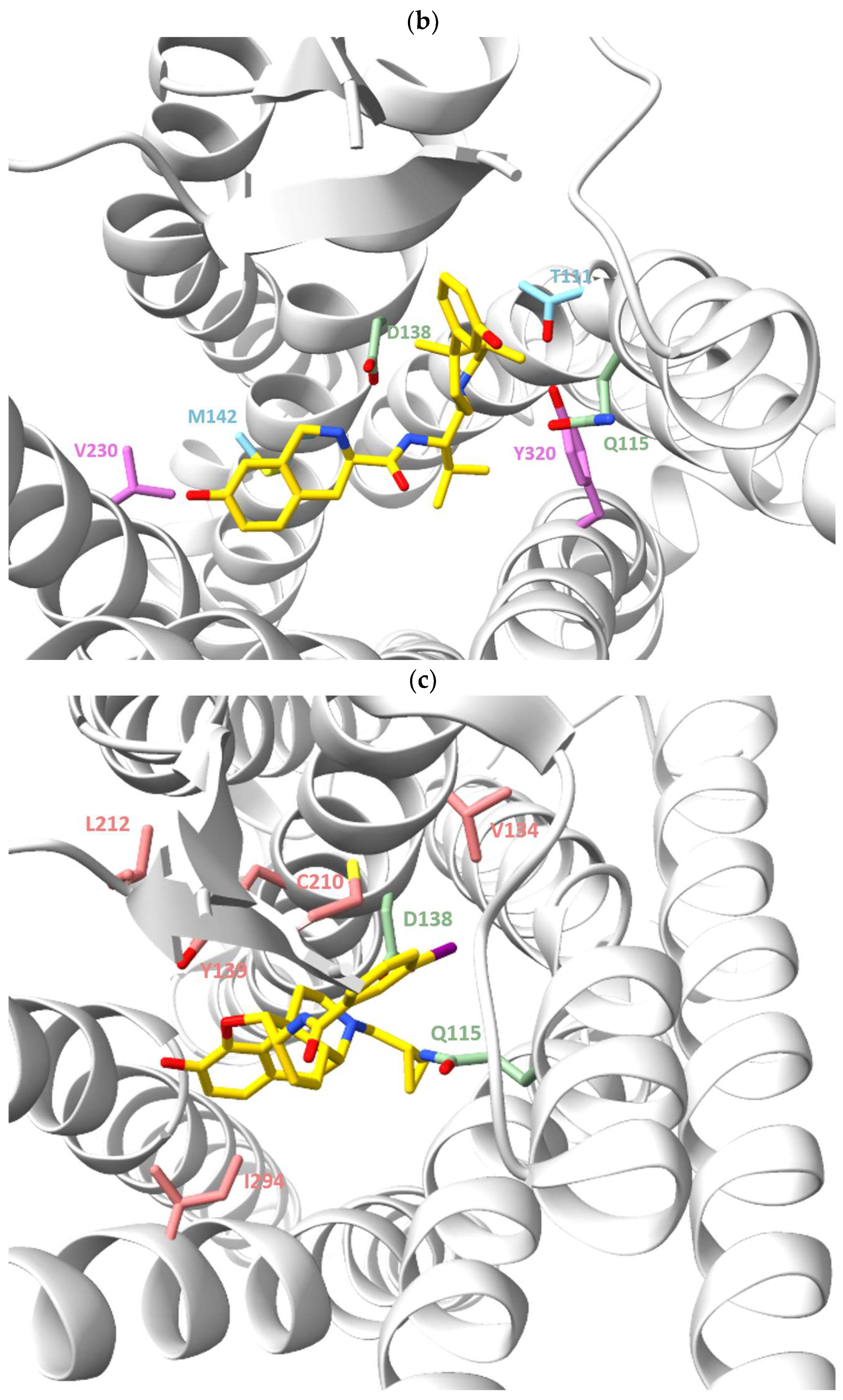
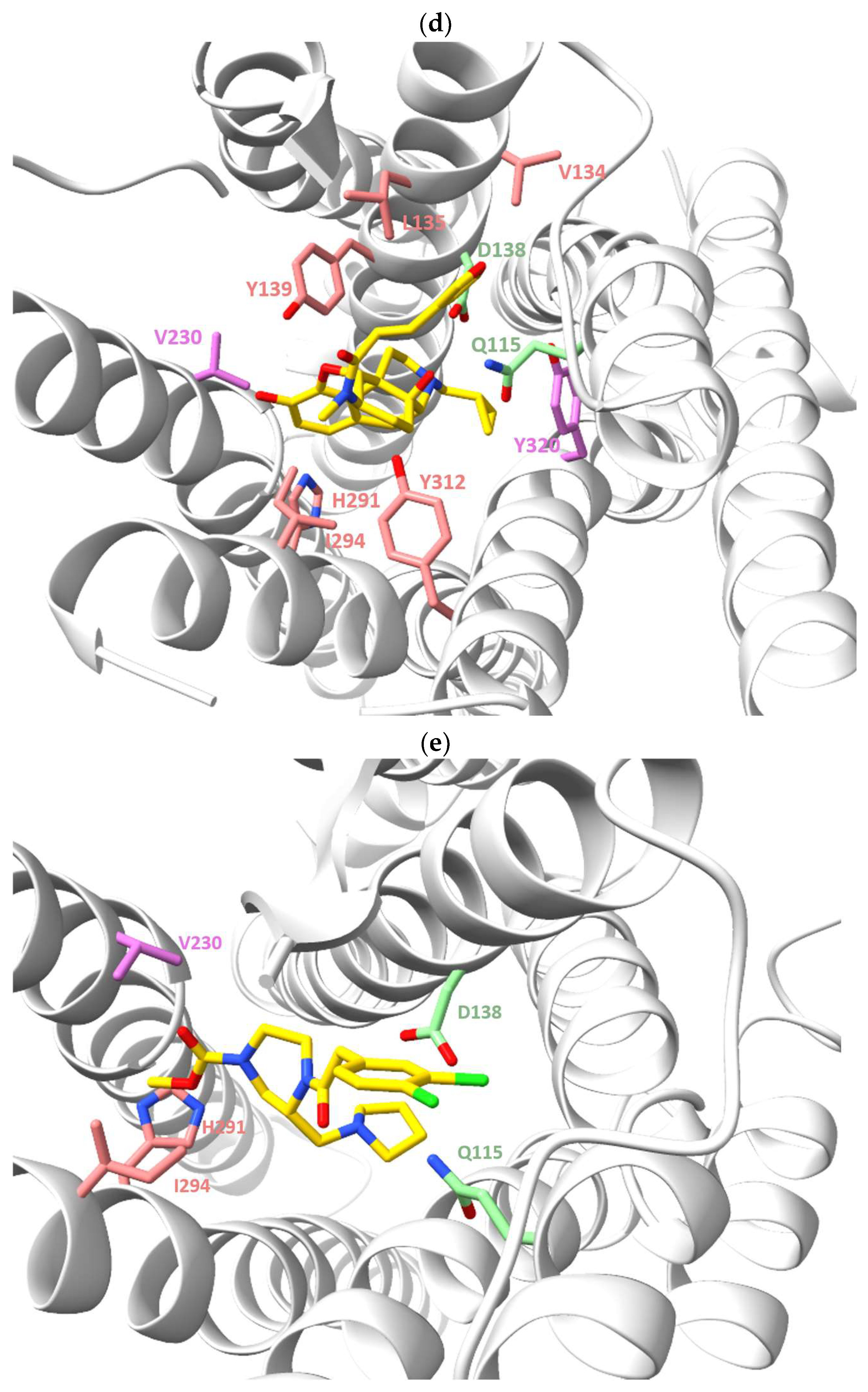
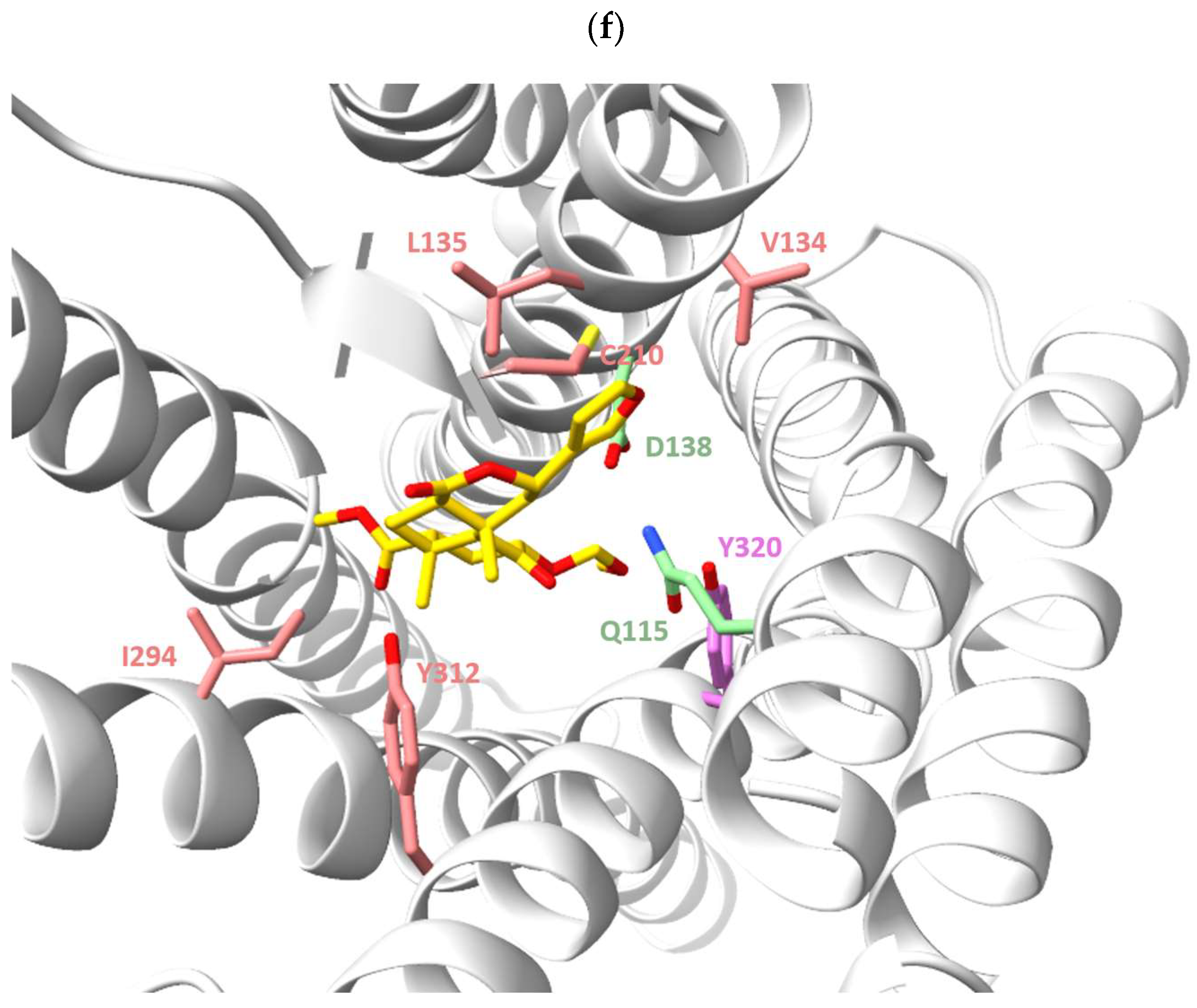
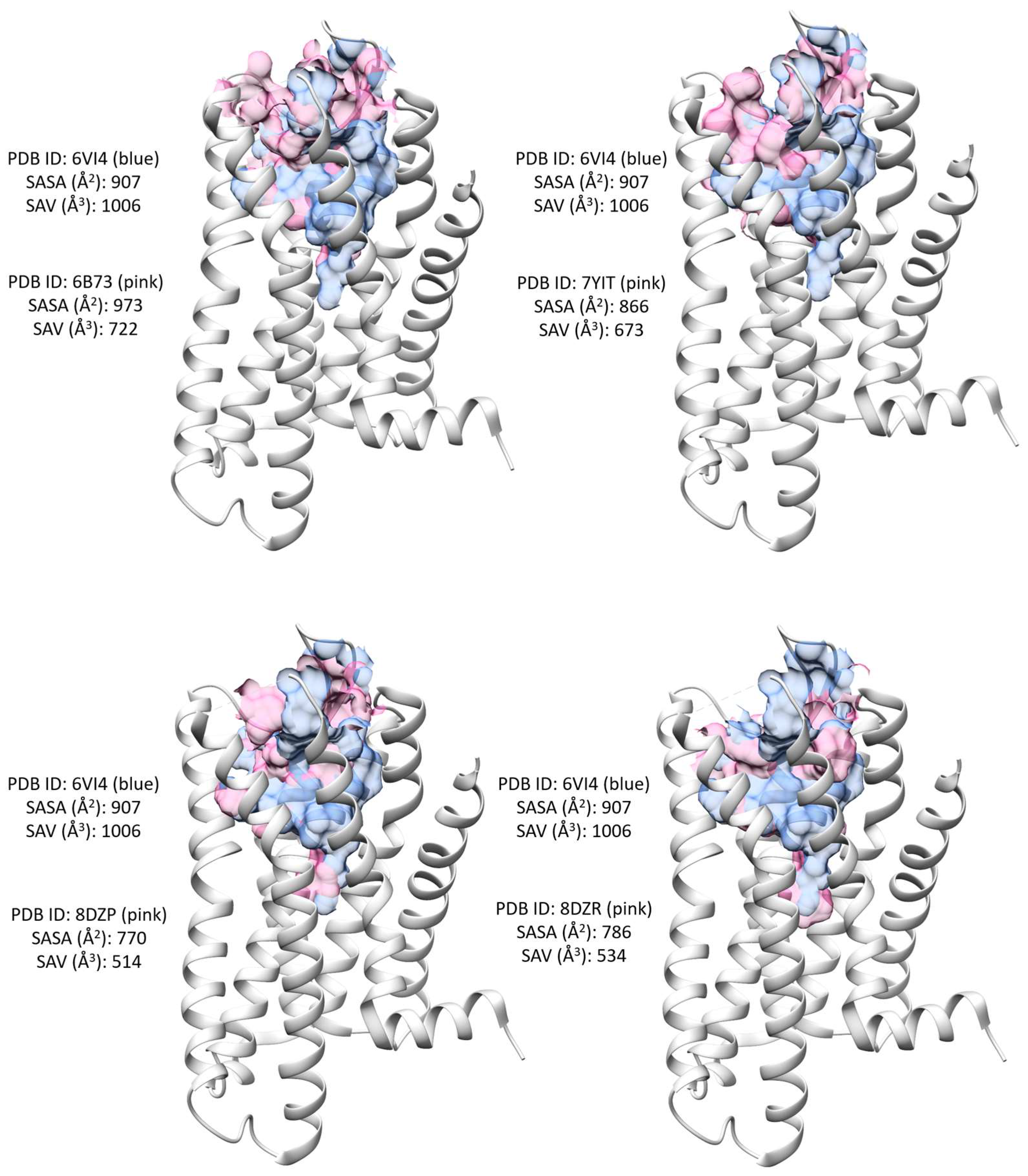

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
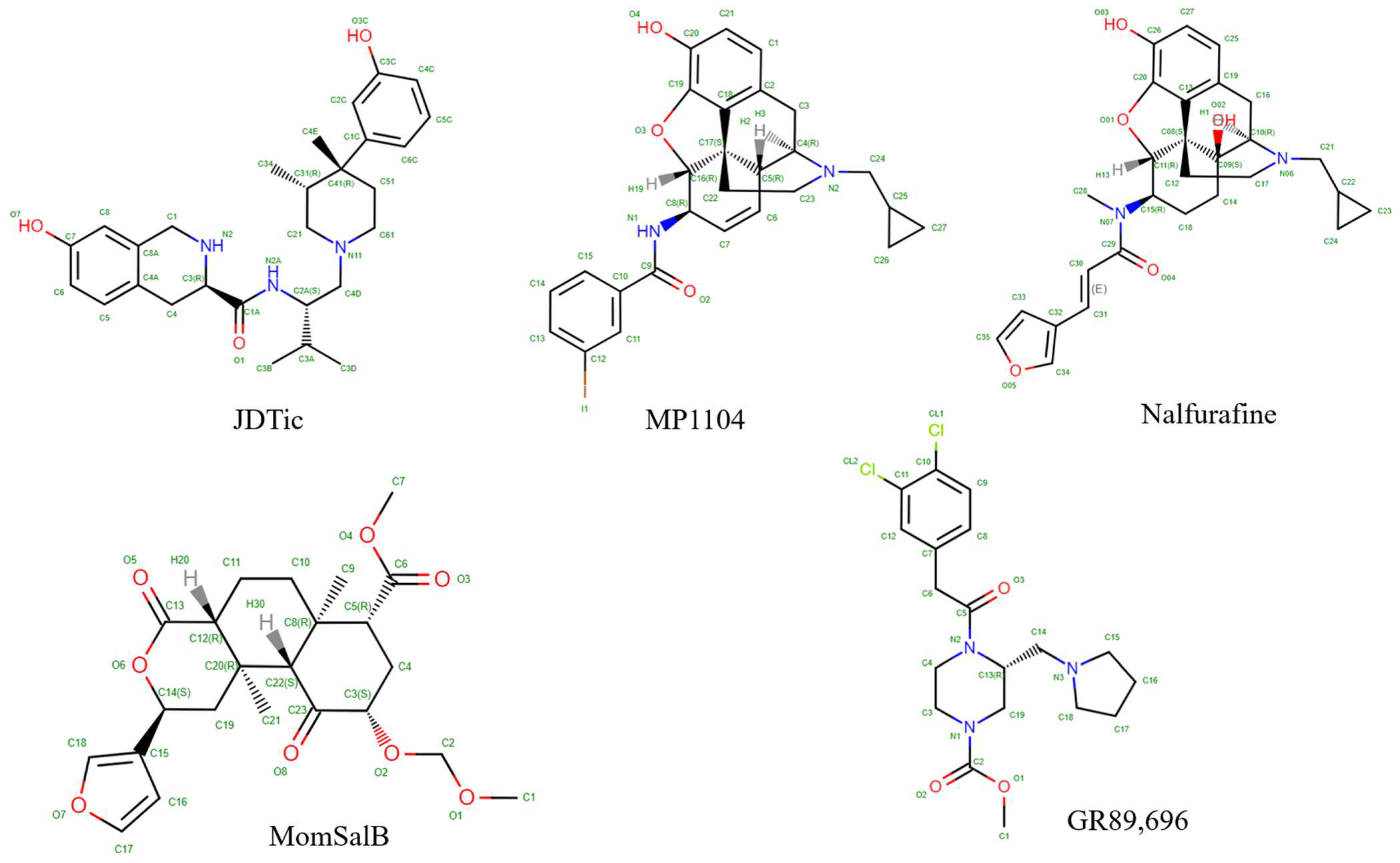
| PDB ID | Ligand | Ligand Type | Resolution | Complex with | Method | Reference |
|---|---|---|---|---|---|---|
| 6VI4 | JDTic | antagonist | 3.3 | Nanobody 6 | X-ray | [52] |
| 4DJH | JDTic | antagonist | 2.9 | Lysozyme | X-ray | [53] |
| 6B73 | MP1104 | agonist | 3.1 | Nanobody | X-ray | [54] |
| 7YIT | nalfurafine | agonist | 3.3 | Nanobody 39 | X-ray | [38] |
| 8DZR | GR89696 | agonist | 2.6 | G alpha gustducin protein, Gβ-1, Gγ-2, scFv16 | Cryo-EM | [55] |
| 8DZS | GR89696 | agonist | 2.7 | Guanine nucleotide-binding protein G(z) subunit alpha, Gβ-1, Gγ-2, scFv16 | Cryo-EM | [55] |
| 8DZP | momSalB | agonist | 2.7 | Gαi-1, Gβ-1, Gγ-2, scFv16 | Cryo-EM | [55] |
| 8DZQ | momSalB | agonist | 2.8 | Guanine nucleotide-binding protein G(o) subunit alpha, Gβ-1, Gγ-2, scFv16 | Cryo-EM | [55] |
| 7Y1F | dynorphin | agonist | 3.3 | Gαi-1, Gβ-1, Gγ-2, scFv16 | Cryo-EM | [56] |
| 8F7W | dynorphin | agonist | 3.2 | Gαi-1, Gβ-1, Gγ-2, scFv16 | Cryo-EM | [57] |
| 8FEG | De Novo Cyclic Peptide (DNCP)-β-naloxamine (NalA) | agonist | 2.5 | Gαi-1, Gβ-1, Gγ-2, scFv16 | Cryo-EM | [58] |
| PDB ID | 6VI4 | 6B73 | 7YIT | 8ZDR | 8DZP |
|---|---|---|---|---|---|
| Ligand | JDTic | MP1104 | nalfurafine | GR89,696 | momSalB |
| Ligand type | antagonist | agonist | agonist | agonist | agonist |
| Charged residues | 1 | 1 | 2 | 2 | 1 |
| Polar residues | 2 | 2 | 4 | 1 | 3 |
| Hydrophobic residues | 3 | 4 | 5 | 2 | 4 |
| Binding site SASA (Å2) | 907 | 973 | 866 | 770 | 786 |
| Binding site SA volume (Å3) | 1006 | 722 | 673 | 514 | 534 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Huang, R.; Xia, M.; Chang, N.; Guo, W.; Liu, J.; Dong, F.; Liu, B.; Varghese, A.; Aslam, A.; et al. Decoding the κ Opioid Receptor (KOR): Advancements in Structural Understanding and Implications for Opioid Analgesic Development. Molecules 2024, 29, 2635. https://doi.org/10.3390/molecules29112635
Li Z, Huang R, Xia M, Chang N, Guo W, Liu J, Dong F, Liu B, Varghese A, Aslam A, et al. Decoding the κ Opioid Receptor (KOR): Advancements in Structural Understanding and Implications for Opioid Analgesic Development. Molecules. 2024; 29(11):2635. https://doi.org/10.3390/molecules29112635
Chicago/Turabian StyleLi, Zoe, Ruili Huang, Menghang Xia, Nancy Chang, Wenjing Guo, Jie Liu, Fan Dong, Bailang Liu, Ann Varghese, Aasma Aslam, and et al. 2024. "Decoding the κ Opioid Receptor (KOR): Advancements in Structural Understanding and Implications for Opioid Analgesic Development" Molecules 29, no. 11: 2635. https://doi.org/10.3390/molecules29112635
APA StyleLi, Z., Huang, R., Xia, M., Chang, N., Guo, W., Liu, J., Dong, F., Liu, B., Varghese, A., Aslam, A., Patterson, T. A., & Hong, H. (2024). Decoding the κ Opioid Receptor (KOR): Advancements in Structural Understanding and Implications for Opioid Analgesic Development. Molecules, 29(11), 2635. https://doi.org/10.3390/molecules29112635