
Latest Developments of the Julia–Kocienski Olefination Reaction: Mechanistic Considerations



Abstract
:1. Introduction
2. Origins and Mechanism of the Julia–Kocienski Olefination Reaction
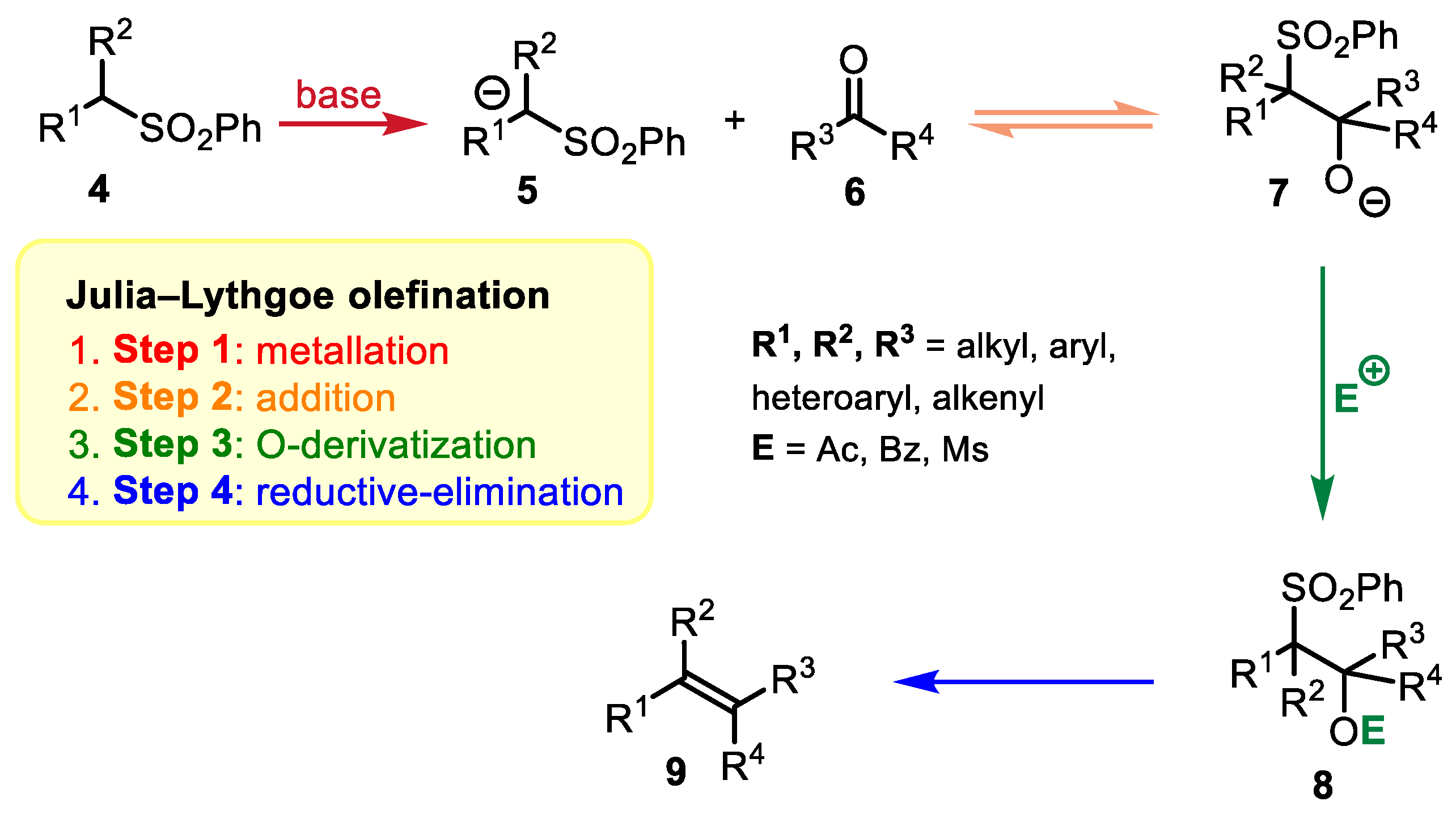
2.1. Julia–Lythgoe Olefination vs. Julia–Kocienski Olefination: A Comparison
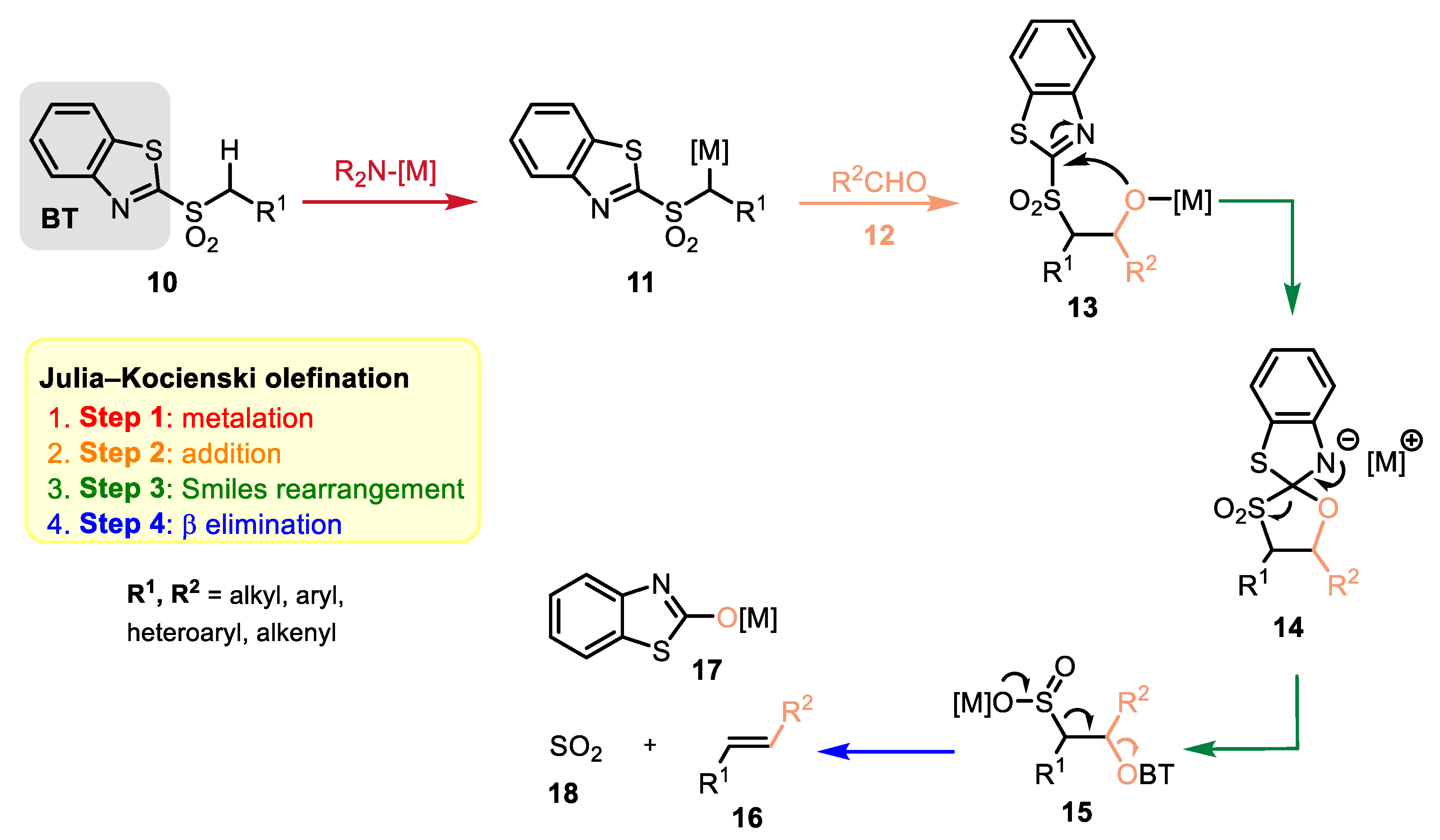
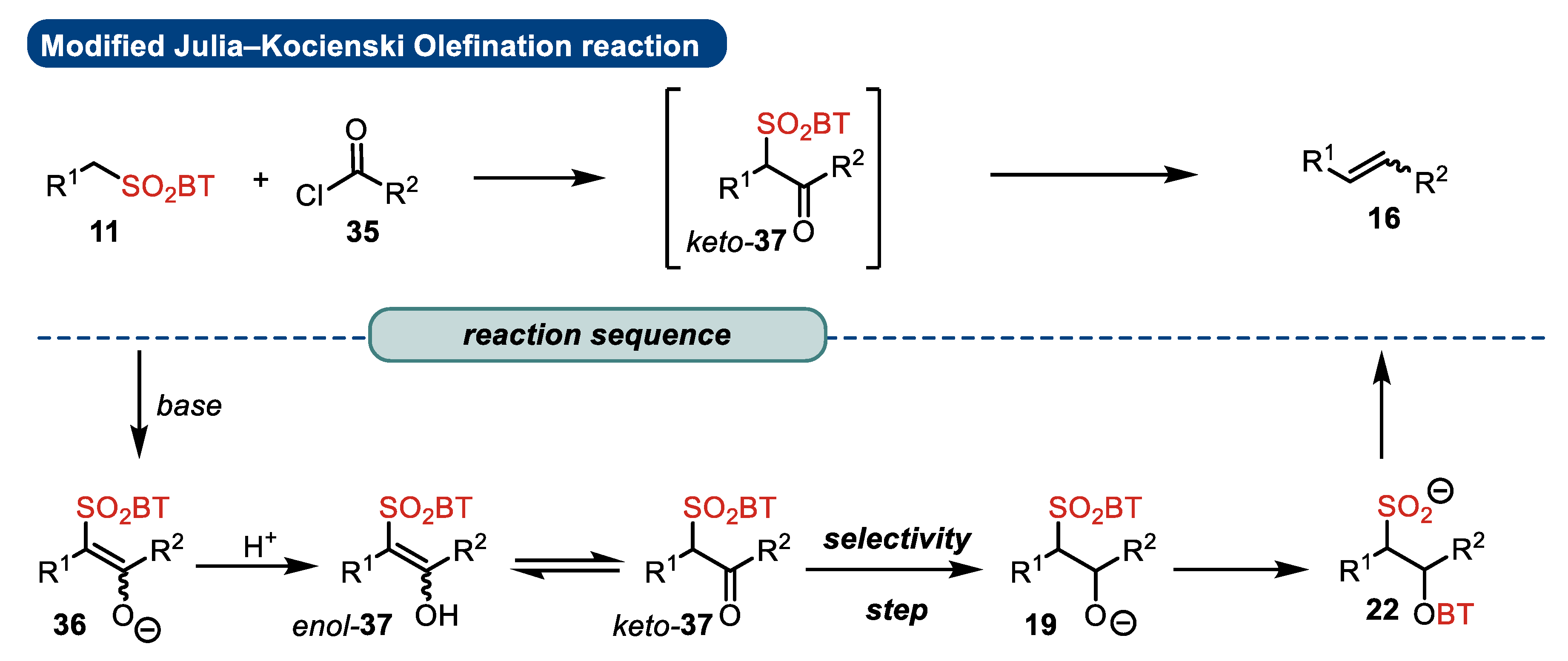
2.2. Reaction Mechanism and Its Impact on the Selectivity of Julia–Kocienski Olefination
- (1)
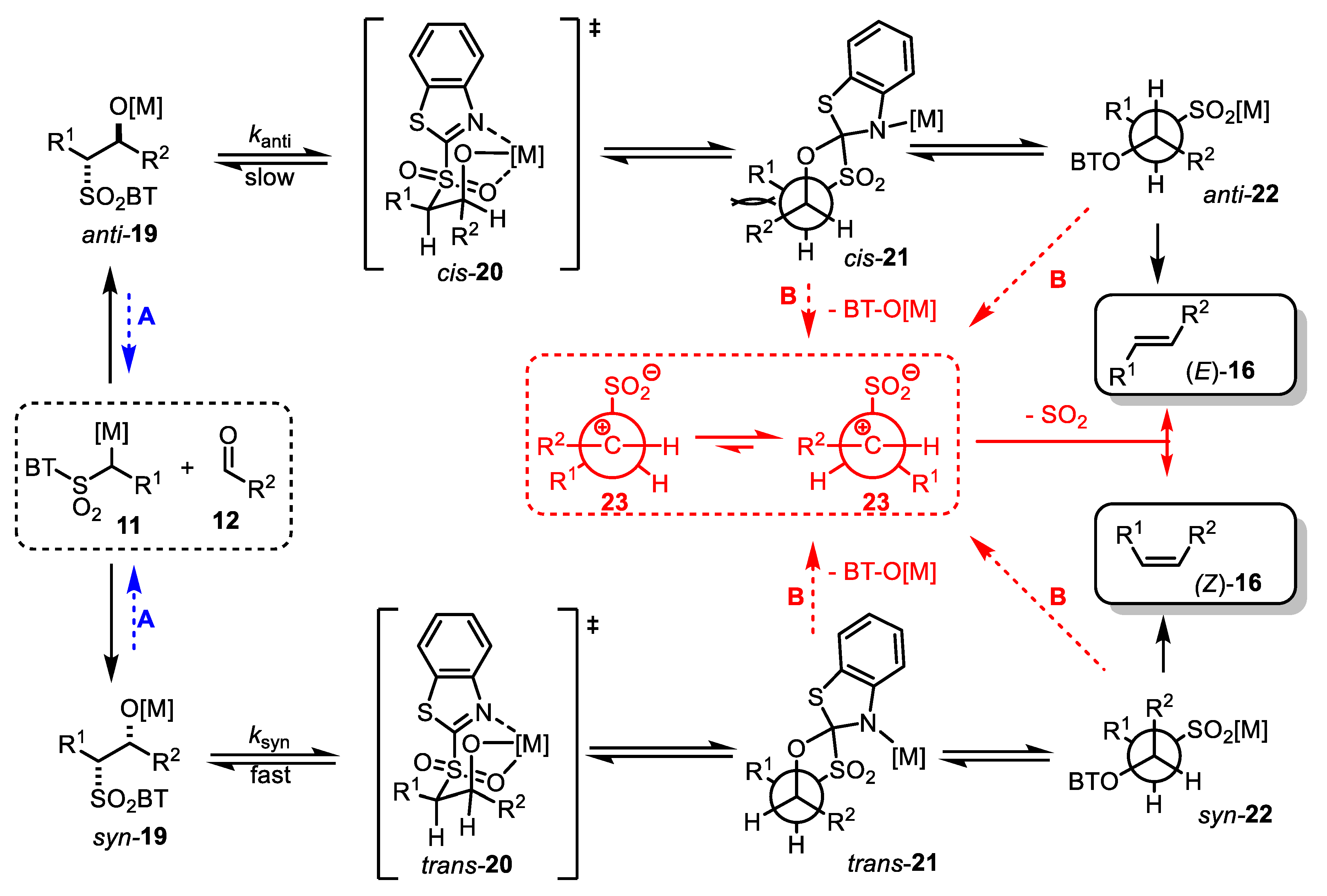
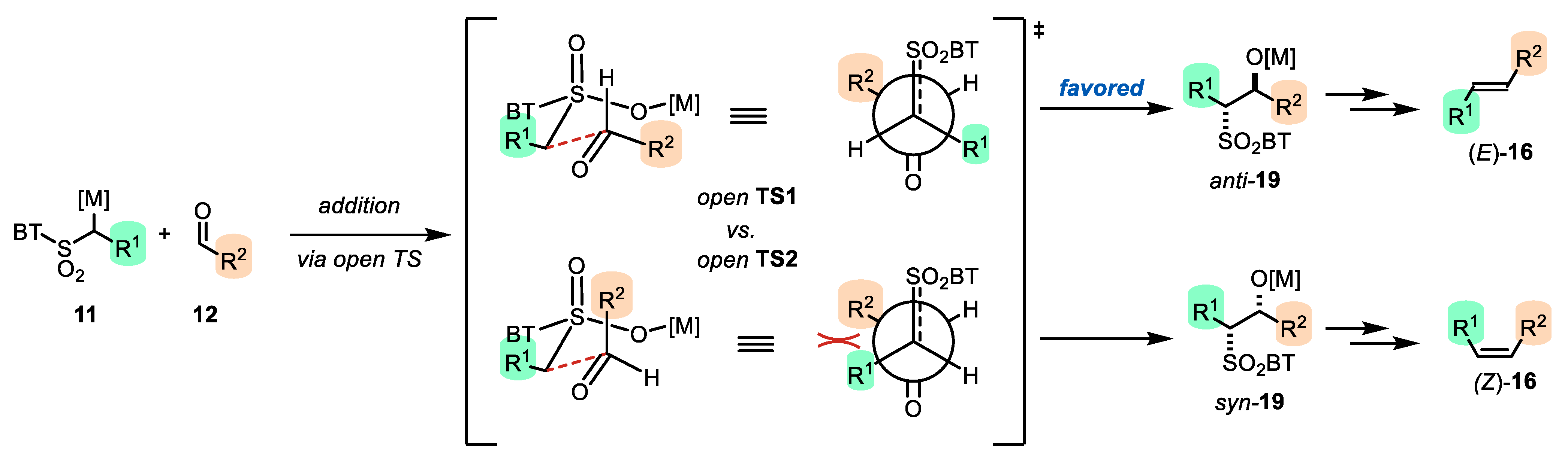
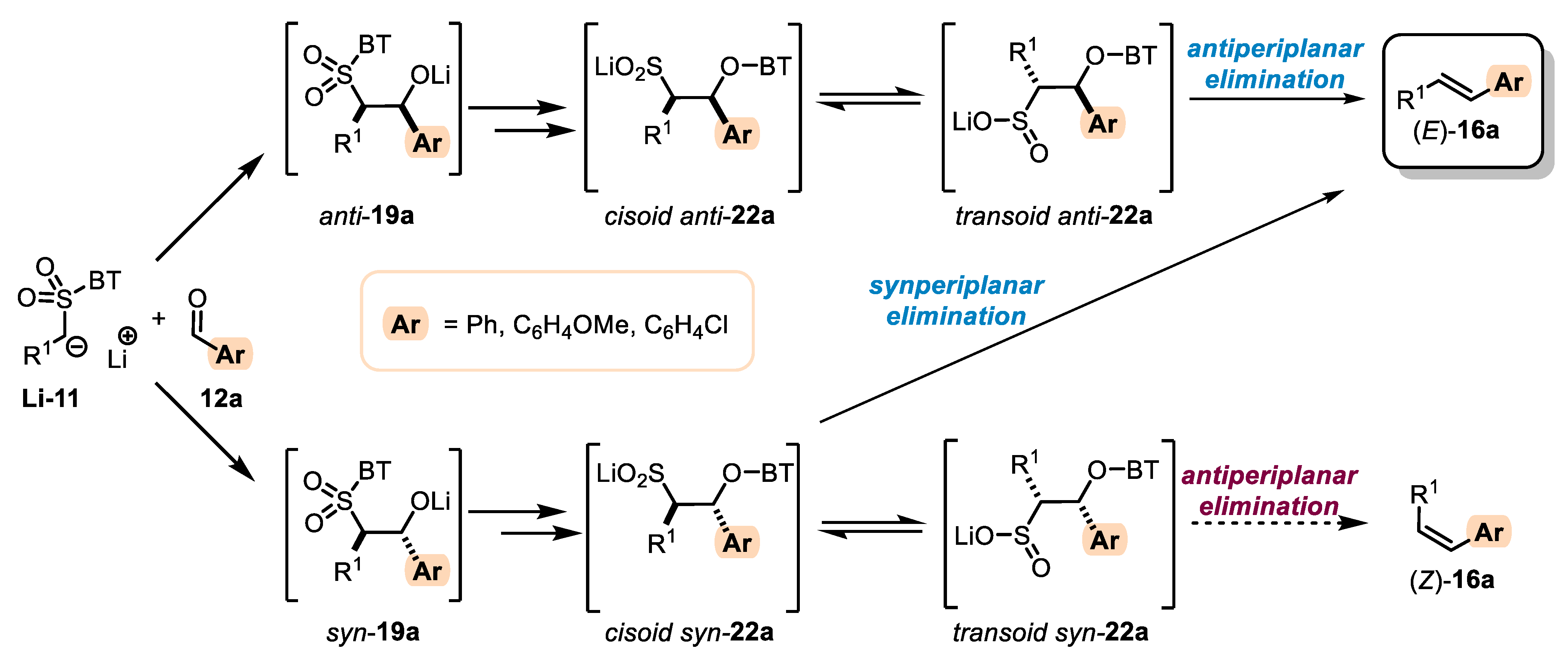
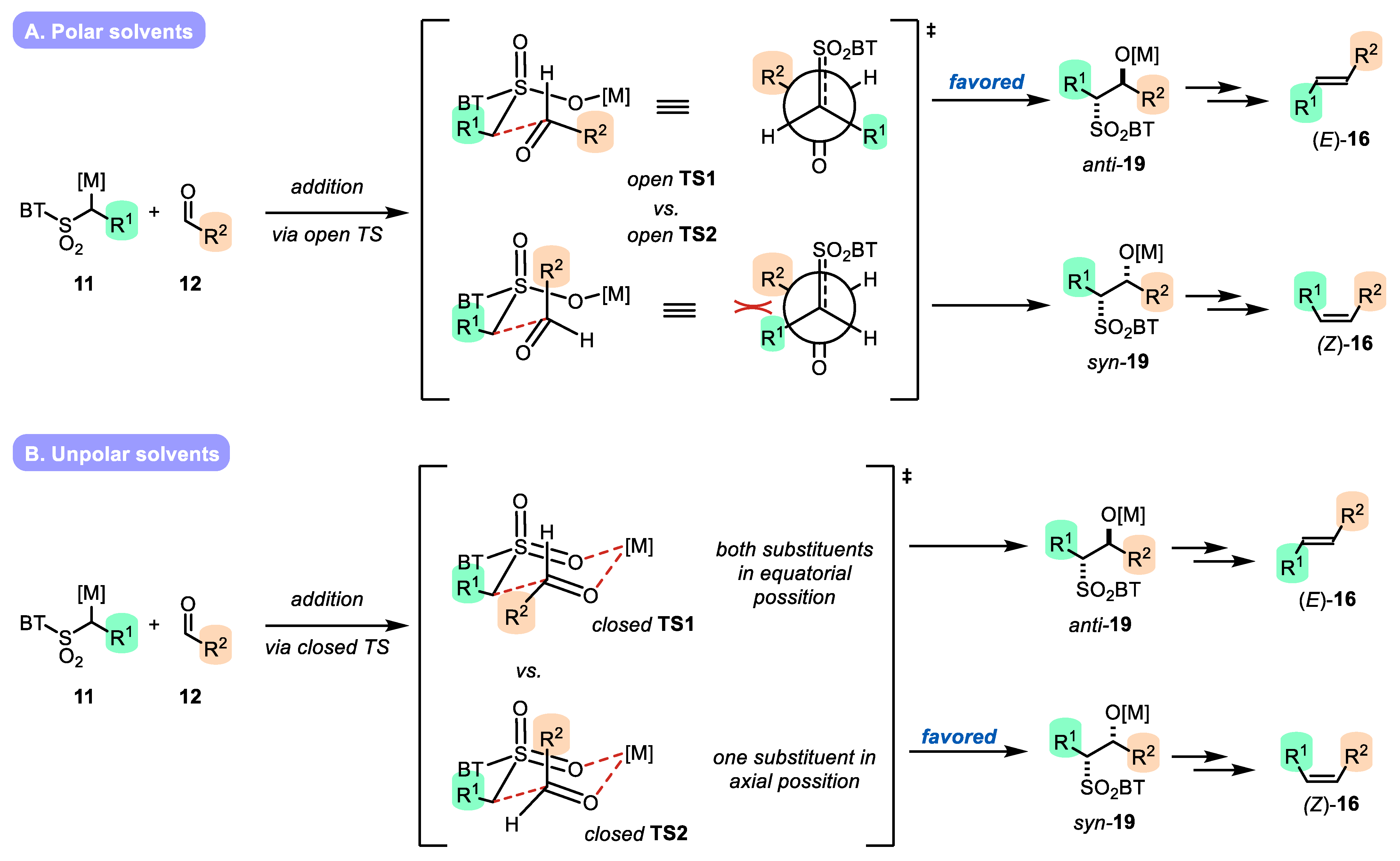
- The addition of metalated sulfone 11 to aldehyde 12 can provide anti-adduct anti-19 via TS1 or the syn-adduct syn-19 via TS2 (Figure 2). The selectivity in this step is extremely important, since all subsequent transformations of intermediate 19, the Smiles rearrangement, and the β-elimination process are stereospecific. Thus, the syn/anti-selectivity of the addition step determines the final (E/Z) olefin ratio. Therefore, in theory, the (E/Z) selectivity of the reaction can be swapped from (E) to (Z) if proper reaction conditions are applied.
- (2)
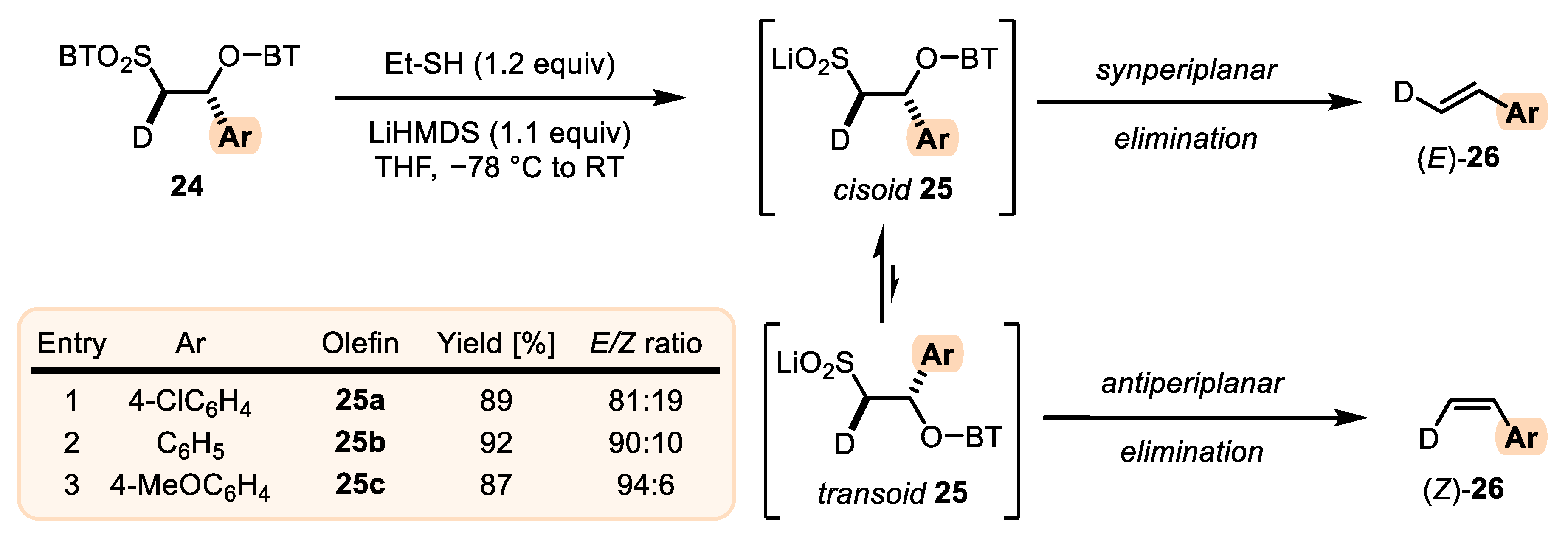
- When stabilized metalated sulfonyl anions 11 (R1 = Ph, alkenyl, etc.) are used, the addition of 11 to 12 becomes reversible (Scheme 3, path A). In this case, the original kinetically driven syn/anti-ratio of adduct 19 becomes less important in comparison with the Smiles rearrangement reaction rates (transformation of 19 to 22). In such cases, the rearrangement of anti-19 adduct leading to (E) olefin 16 is slower compared to the rearrangement of syn-19 to olefin (Z)-16 due to repulsive 1,2-interactions in the transition state (see cis-20).
- (3)
- For the elimination step, two borderline mechanisms are generally accepted. In the first, which is the most common, the rearranged intermediate 22 undergoes β-elimination. The elimination is stereospecific, and the syn-19 adduct-rearranged intermediate syn-22 furnishes the (Z) olefin and the anti-19 adduct-rearranged intermediate, the compound trans-22 (trans refers to the arrangement of R1 and R2 within the intermediate cycle), yields the (E) olefin. Alternatively, when (hetero)aryl aldehydes 12 (R2 = (hetero)aryl) are used, an alternative elimination pathway (path B) is postulated to occur. In this case, the elimination pathway should proceed through the formation of an intermediate carbocation 23. The steric requirements of R1 and R2 then play a crucial role in the final (E/Z) selectivity of the reaction. Path B was used to explain the unexpected (E) selectivity of the coupling reactions carried out using (hetero)aryl aldehydes 12 as substrates.
2.3. Recent Reaction Selectivity Improvements
2.3.1. Solvent Effect
- Metal cation
- Cosolvents
2.3.2. Additives
- Crown ethers
- Ammonium salts
- Chelating salts
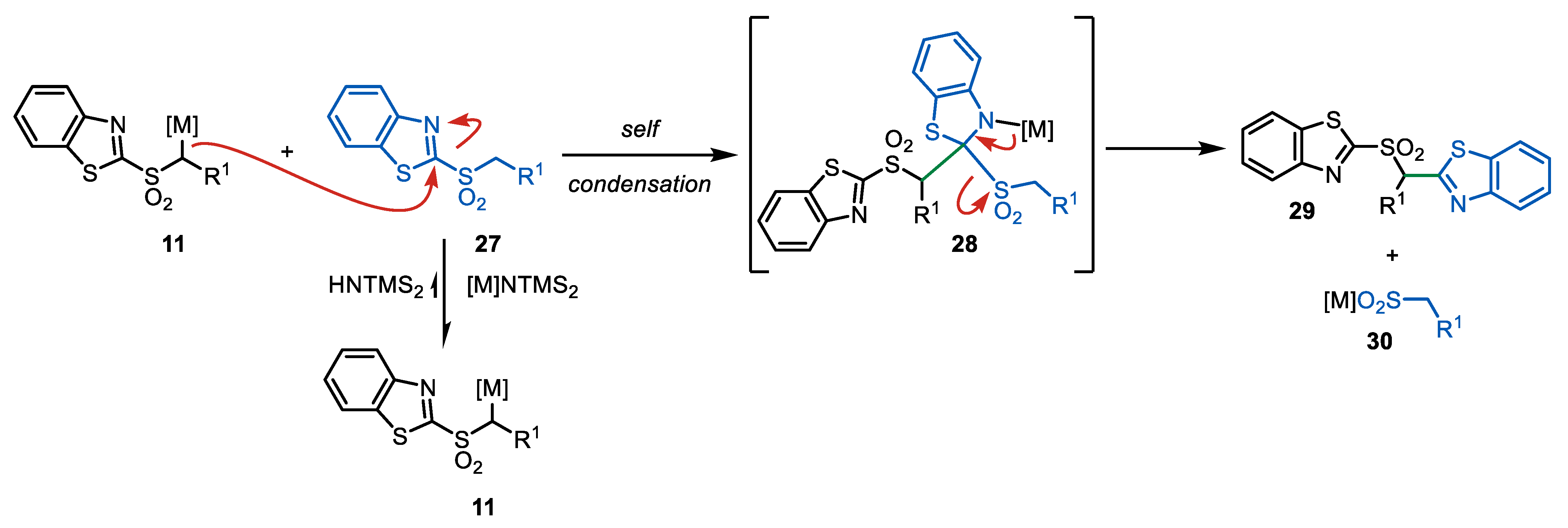
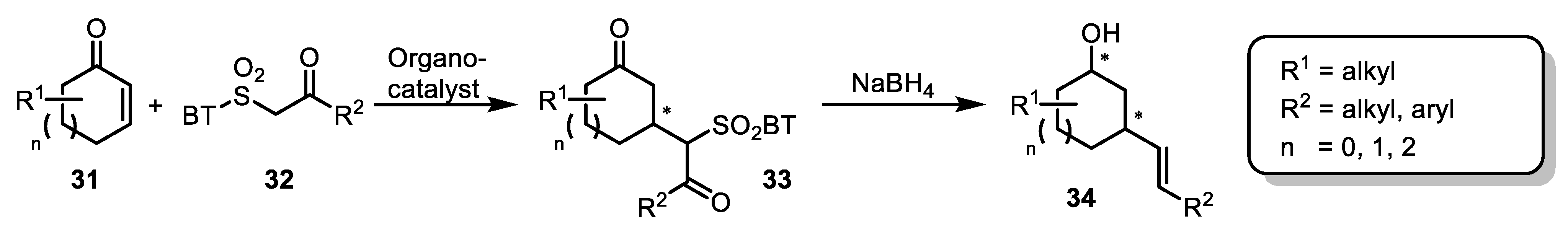
3. Julia–Kocienski Olefination—Extension to Carboxylic Acid Derivatives
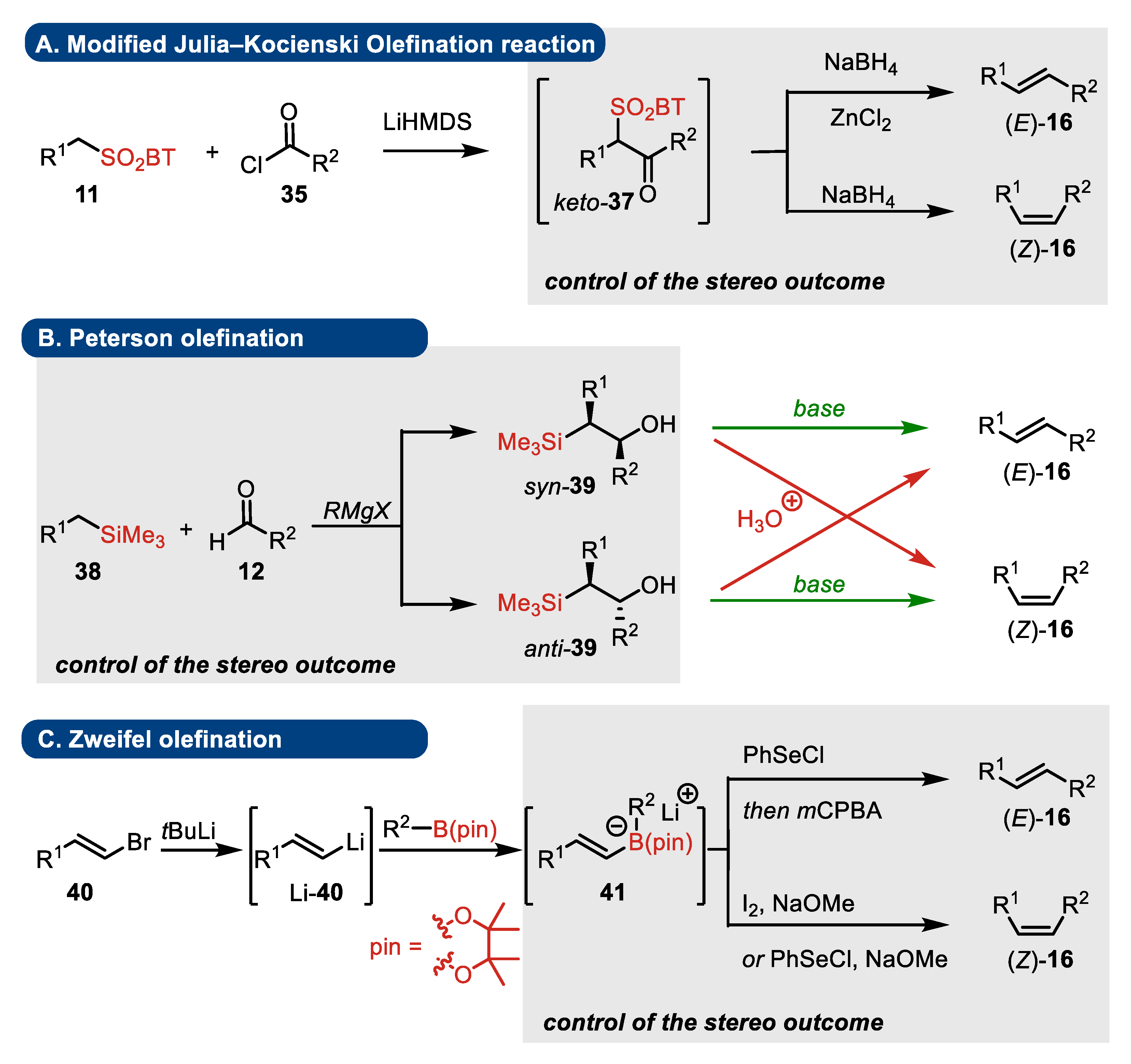
4. Julia–Kocienski, Peterson, and Zweifel Olefination Reactions: A Brief Comparison
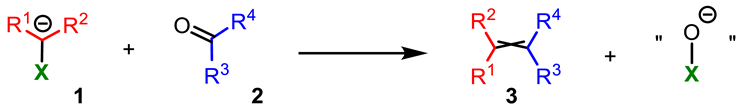
4.1. Modified Julia–Kocienski Reaction
- Substrates
- Elimination step
- Presence of stereogenic centers
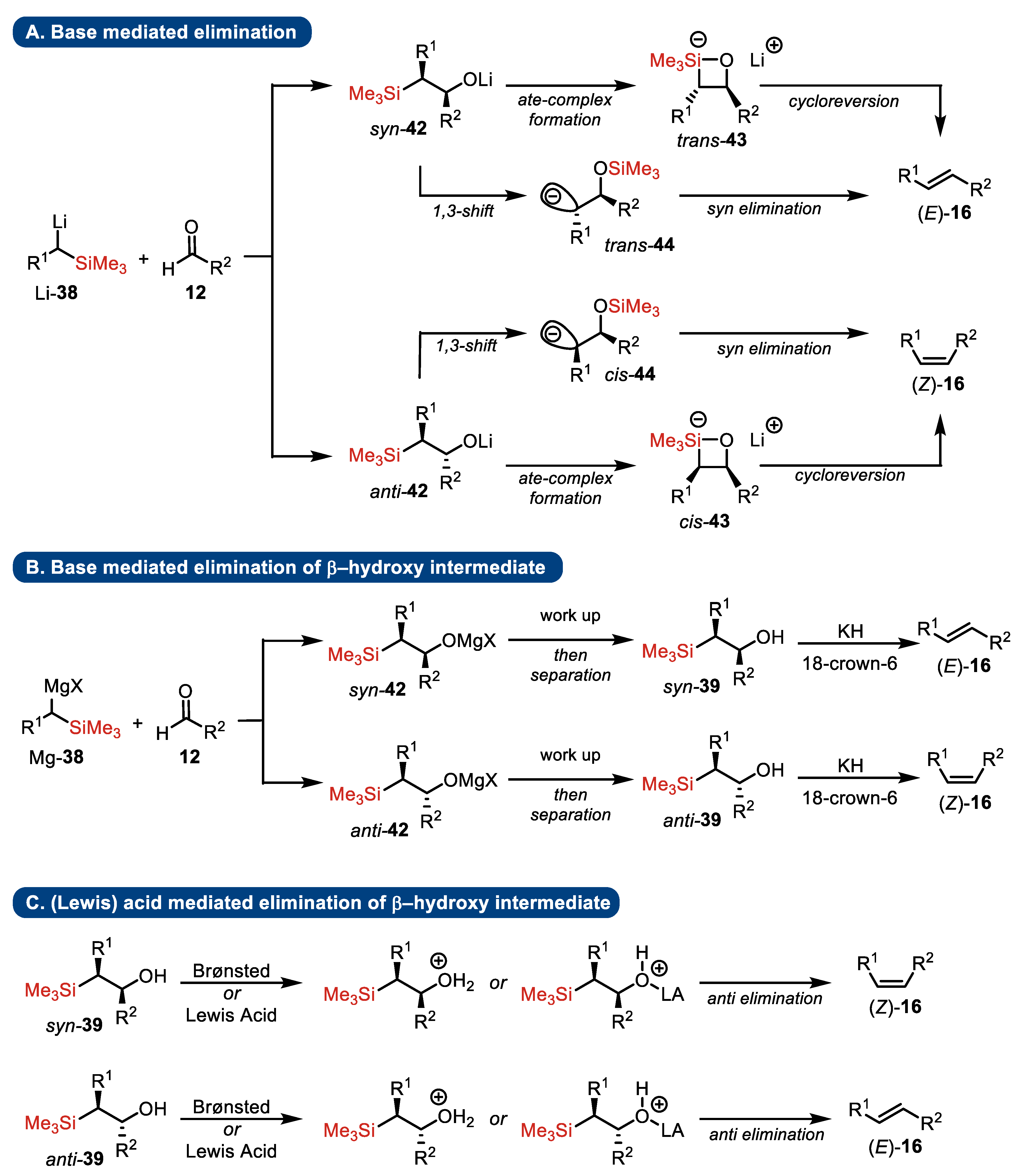
4.2. Peterson Olefination
- Substrates
- Elimination step
- Presence of stereogenic centers
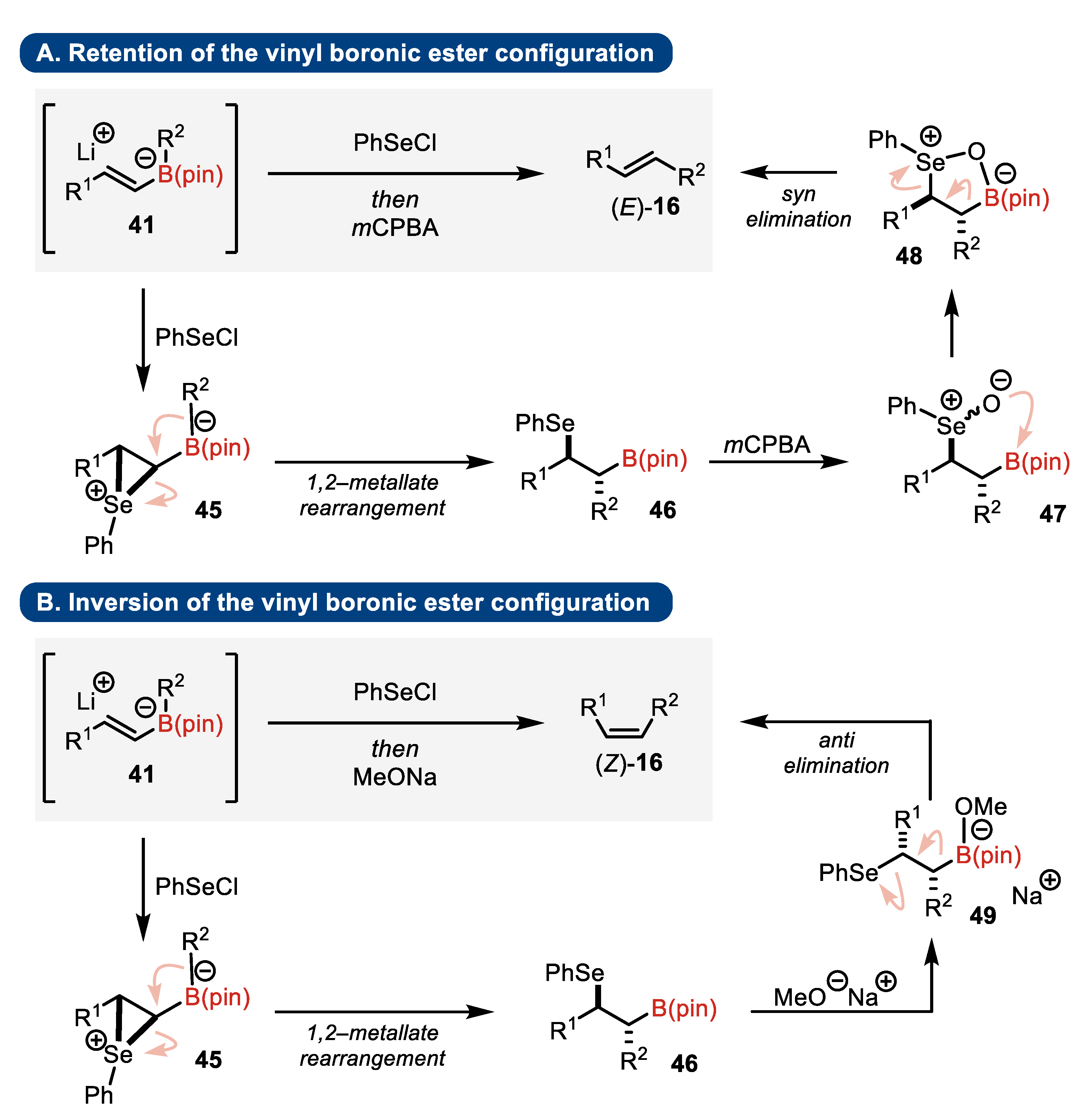
4.3. Zweifel Olefination
- Substrates
- Elimination step
- Presence of stereogenic centers
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Markó, I.E.; Pospíšil, J. Julia, Julia–Kocienski, and Related Sulfur-Based Alkenations. In Science of Synthesis; de Meijere, A., Ed.; Georg Thieme Verlag: Stuttgart, Germany, 2010; Volume 47, pp. 105–160. [Google Scholar]
- Johnson, C.R.; Shanklin, J.R.; Kirchhoff, R.A. Olefin Synthesis by Reductive Elimination of b-Hydroxysulfoximines. Methylenation of Carbonyl Compounds. J. Am. Chem. Soc. 1973, 95, 6462–6463. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Reitz, A.B. The Wittig Olefination Reaction and Modifications Involving Phosphoryl-Stabilized Carbanions. Stereochemistry, Mechanism, and Selected Synthetic Aspects. Chem. Rev. 1989, 89, 863–927. [Google Scholar] [CrossRef]
- Bisceglia, J.A.; Orelli, L.R. Recent Progress in the Horner-Wadsworth-Emmons Reaction. Curr. Org. Chem. 2015, 19, 744–775. [Google Scholar] [CrossRef]
- Van Staden, L.F.; Gravestock, D.; Ager, D.J. New Developments in the Peterson Olefination Reaction. Chem. Soc. Rev. 2002, 31, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Coombs, J.R.; Zhang, L.; Morken, J.P. Synthesis of Vinyl Boronates from Aldehydes by a Practical Boron-Wittig Reaction. Org. Lett. 2015, 17, 1708–1711. [Google Scholar] [CrossRef] [PubMed]
- Wittig, G.; Geissler, G. Zur Reaktionsweise Des Pentaphenyl-phosphors Und Einiger Derivate. Justus Liebigs Ann. Chem. 1953, 580, 44–57. [Google Scholar] [CrossRef]
- Wittig, G.; Schöllkopf, U. Über Triphenyl-phosphin-methylene Als Olefinbildende Reagenzien. Chem. Berichte 1954, 87, 1318–1330. [Google Scholar] [CrossRef]
- Chatterjee, B.; Bera, S.; Mondal, D. Julia-Kocienski Olefination: A Key Reaction for the Synthesis of Macrolides. Tetrahedron Asymmetry 2014, 25, 1–55. [Google Scholar] [CrossRef]
- Legnani, L.; Porta, A.; Caramella, P.; Toma, L.; Zanoni, G.; Vidari, G. Computational Mechanistic Study of the Julia-Kocieński Reaction. J. Org. Chem. 2015, 80, 3092–3100. [Google Scholar] [CrossRef]
- Aïssa, C. Mechanistic Manifold and New Developments of the Julia-Kocienski Reaction. Eur. J. Org. Chem. 2009, 2009, 1831–1844. [Google Scholar] [CrossRef]
- Blakemore, P.R. The Modified Julia Olefination: Alkene Synthesis via the Condensation of Metallated Heteroarylalkylsulfones with Carbonyl Compounds. J. Chem. Soc. Perkin 1 2002, 2, 2563–2585. [Google Scholar] [CrossRef]
- Blakemore, P.R.; Cole, W.J.; Kocieński, P.J.; Morley, A. A Stereoselective Synthesis of Trans-1,2-Disubstituted Alkenes Based on the Condensation of Aldehydes with Metallated 1-Phenyl-1 H -Tetrazol-5-Yl Sulfones. Synlett 1998, 1998, 26–28. [Google Scholar] [CrossRef]
- Robiette, R.; Pospíšil, J. On the Origin of E/Z Selectivity in the Modified Julia Olefination—Importance of the Elimination Step. Eur. J. Org. Chem. 2013, 836–840. [Google Scholar] [CrossRef]
- Baudin, J.B.; Hareau, G.; Julia, S.A.; Ruel, O. A Direct Synthesis of Olefins by Reaction of Carbonyl Compounds with Lithio Derivatives of 2-[Alkyl- or (2′-Alkenyl)- or Benzyl-Sulfonyl]-Benzothiazoles. Tetrahedron Lett. 1991, 32, 1175–1178. [Google Scholar] [CrossRef]
- Gueyrard, D. Extension of the Modified Julia Olefination on Carboxylic Acid Derivatives: Scope and Applications. Synlett 2018, 29, 34–45. [Google Scholar] [CrossRef]
- Julia, M.; Paris, J.M. Syntheses a l’aide de Sulfones v(+)- Methode de Synthese Generale de Doubles Liaisons. Tetrahedron Lett. 1973, 14, 4833–4836. [Google Scholar] [CrossRef]
- Kocienski, P.J.; Lythgoe, B.; Ruston, S. Scope and Stereochemistry of an Olefin Synthesis from β-Hydroxysulphones. J. Chem. Soc. Perkin 1 1978, 829–834. [Google Scholar] [CrossRef]
- Keck, G.E.; Savin, K.A.; Weglarz, M.A. Use of Samarium Diiodide as an Alternative to Sodium/Mercury Amalgam in the Julia-Lythgoe Olefination. J. Org. Chem. 1995, 60, 3194–3204. [Google Scholar] [CrossRef]
- Baudin, J.B.; Hareau, G.; Julia, S.A.; Lorne, R.; Ruel, O. Stereochemistry of Direct Olefin Formation from Carbonyl Compounds and Lithiated Heterocyclic Sulfones. Bull. Soc. Chim. Fr. 1993, 130, 856–878. [Google Scholar]
- Baudin, J.B.; Hareau, G.; Julia, S.A.; Ruel, O. Stereochemistry of the Olefin Formation from Anti and Syn Heterocyclic β-Hydroxy-Sulfones. Bull. Soc. Chim. Fr. 1993, 130, 336–357. [Google Scholar]
- Sakaine, G.; Leitis, Z.; Ločmele, R.; Smits, G. Julia-Kocienski Olefination: A Tutorial Review. Eur. J. Org. Chem. 2023, 26, e202201217. [Google Scholar] [CrossRef]
- Ouzounthanasis, K.A.; Rizos, S.R.; Koumbis, A.E. Julia-Kocienski Olefination in the Synthesis of Trisubstituted Alkenes: Recent Progress. Eur. J. Org. Chem. 2023, 26, e202300626. [Google Scholar] [CrossRef]
- Rinu, P.X.T.; Radhika, S.; Anilkumar, G. Recent Applications and Trends in the Julia-Kocienski Olefination. ChemistrySelect 2022, 7, e202200760. [Google Scholar] [CrossRef]
- Charette, A.B.; Berthelette, C.; St-Martin, D. An Expedient Approach to E, Z-Dienes Using the Julia Olefination. Tetrahedron Lett. 2001, 42, 5149–5153. [Google Scholar] [CrossRef]
- Kocienski, P.J.; Bell, A.; Blakemore, P.R. 1- Tert -Butyl-1 H -Tetrazol-5-Yl Sulfones in the Modified Julia Olefination. Synlett 2000, 2000, 365–366. [Google Scholar] [CrossRef]
- Alonso, D.A.; Fuensanta, M.; Nájera, C.; Varea, M. 3,5-Bis(Trifluoromethyl)Phenyl Sulfones in the Direct Julia−Kocienski Olefination. J. Org. Chem. 2005, 70, 6404–6416. [Google Scholar] [CrossRef] [PubMed]
- Mąkosza, M.; Bujok, R. Synthesis of Benzylidenecyclopropanes from γ-Halopropyl Pentachlorophenyl Sulfones Using a Julia-Kocienski Olefination. Synlett 2008, 2008, 586–588. [Google Scholar] [CrossRef]
- Pospíšil, J. Simple Protocol for Enhanced (E)-Selectivity in Julia–Kocienski Reaction. Tetrahedron Lett. 2011, 52, 2348–2352. [Google Scholar] [CrossRef]
- Jana, N.; Nanda, S. Asymmetric Total Syntheses of Cochliomycin A and Zeaenol. Eur. J. Org. Chem. 2012, 2012, 4313–4320. [Google Scholar] [CrossRef]
- Mohapatra, D.K.; Reddy, D.S.; Mallampudi, N.A.; Yadav, J.S. Stereoselective Total Syntheses of Paecilomycins e and F through a Protecting Group Directed Diastereoselective Intermolecular Nozaki-Hiyama-Kishi (NHK) Reaction. Eur. J. Org. Chem. 2014, 2014, 5023–5032. [Google Scholar] [CrossRef]
- Sánchez, D.; Andreou, T.; Costa, A.M.; Meyer, K.G.; Williams, D.R.; Barasoain, I.; Díaz, J.F.; Lucena-Agell, D.; Vilarrasa, J. Total Synthesis of Amphidinolide K, a Macrolide That Stabilizes F-Actin. J. Org. Chem. 2016, 80, 8511–8519. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M.; Britton, R. Enantioselective Total Synthesis of the Marine Macrolides Salarins A and C. J. Am. Chem. Soc. 2024, 146, 8456–8463. [Google Scholar] [CrossRef] [PubMed]
- Billard, F.; Robiette, R.; Pospíšil, J. Julia-Kocienski Reaction-Based 1,3-Diene Synthesis: Aldehyde-Dependent (E, E/E, Z)-Selectivity. J. Org. Chem. 2012, 77, 6358–6364. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.; Surendran, S.; Siddavatam, N.; Rajendar, G. The Influence of α-Coordinating Groups of Aldehydes on E/Z-Selectivity and the Use of Quaternary Ammonium Counter Ions for Enhanced E-Selectivity in the Julia–Kocienski Reaction. Org. Biomol. Chem. 2022, 20, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Rajendar, G.; Corey, E.J. A Systematic Study of Functionalized Oxiranes as Initiating Groups for Cationic Polycyclization Reactions. J. Am. Chem. Soc. 2015, 137, 5837–5844. [Google Scholar] [CrossRef]
- Tsubone, K.; Hashizume, K.; Fuwa, H.; Sasaki, M. Studies toward the Total Synthesis of Gambieric Acids: Convergent Synthesis of the GHIJ-Ring Fragment Having a Side Chain. Tetrahedron Lett. 2011, 52, 548–551. [Google Scholar] [CrossRef]
- Tsubone, K.; Hashizume, K.; Fuwa, H.; Sasaki, M. Studies toward the Total Synthesis of Gambieric Acids, Potent Antifungal Polycyclic Ethers: Convergent Synthesis of a Fully Elaborated GHIJ-Ring Fragment. Tetrahedron 2011, 67, 6600–6615. [Google Scholar] [CrossRef]
- Rej, R.K.; Kumar, R.; Nanda, S. Asymmetric Synthesis of Cytospolides C and D through Successful Exploration of Stereoselective Julia-Kocienski Olefination and Suzuki Reaction Followed by Macrolactonization. Tetrahedron 2015, 71, 3185–3194. [Google Scholar] [CrossRef]
- Eliel, E.L.; Frye, S.V.; Hortelano, E.R.; Chen, X.; Bai, X. Asymmetric Synthesis and Cram’s (Chelate) Rule. Pure Appl. Chem. 1991, 63, 1591–1598. [Google Scholar] [CrossRef]
- Bon, D.J.-Y.D.; Chrenko, D.; Kováč, O.; Ferugová, V.; Lasák, P.; Fuksová, M.; Zálešák, F.; Pospíšil, J. Julia-Kocienski-Like Connective C−C and C=C Bond-Forming Reaction. Adv. Synth. Catal. 2024, 366, 480–487. [Google Scholar] [CrossRef]
- Nielsen, M.; Jacobsen, C.B.; Paixão, M.W.; Holub, N.; Jørgensen, K.A. Asymmetric Organocatalytic Formal Alkynylation and Alkenylation of α,β-Unsaturated Aldehydes. J. Am. Chem. Soc. 2009, 131, 10581–10586. [Google Scholar] [CrossRef]
- Jacobsen, C.B.; Nielsen, M.; Worgull, D.; Zweifel, T.; Fisker, E.; Jørgensen, K.A. Asymmetric Organocatalytic Monofluorovinylations. J. Am. Chem. Soc. 2011, 133, 7398–7404. [Google Scholar] [CrossRef] [PubMed]
- Pospíšil, J.; Sato, H. Practical Synthesis of β-Acyl and β-Alkoxycarbonyl Heterocyclic Sulfones. J. Org. Chem. 2011, 76, 2269–2272. [Google Scholar] [CrossRef]
- Pospíšil, J.; Robiette, R.; Sato, H.; Debrus, K. Practical Synthesis of β-Oxo Benzo[d]Thiazolyl Sulfones: Scope and Limitations. Org. Biomol. Chem. 2012, 10, 1225–1234. [Google Scholar] [CrossRef]
- Bettens, T.; Alonso, M.; Geerlings, P.; De Proft, F. Mechanochemical Felkin–Anh Model: Achieving Forbidden Reaction Outcomes with Mechanical Force. J. Org. Chem. 2023, 88, 2046–2056. [Google Scholar] [CrossRef] [PubMed]
- Ager, D.J. Peterson Alkenation. In Science of Synthesis: Houben-Weyl Methods of Molecular Transformations Vol. 47a: Alkenes; Georg Thieme Verlag: Stuttgart, Germany, 2014; p. 85. [Google Scholar]
- Armstrong, R.; Aggarwal, V. 50 Years of Zweifel Olefination: A Transition-Metal-Free Coupling. Synthesis 2017, 49, 3323–3336. [Google Scholar] [CrossRef]
- Li, X.; Song, Q. Recent Progress on the Zweifel Olefination: An Update. Synthesis 2023. [Google Scholar] [CrossRef]
- Fletcher, S. The Mitsunobu Reaction in the 21st Century. Org. Chem. Front. 2015, 2, 739–752. [Google Scholar] [CrossRef]
- Dickman, M.H.; Pope, M.T. Peroxo and Superoxo Complexes of Chromium, Molybdenum, and Tungsten. Chem. Rev. 1994, 94, 569–584. [Google Scholar] [CrossRef]
- Adam, W.; Ortega-Schulte, C.M. An Effective Synthesis of α-Cyanoenamines by Peterson Olefination. Synlett 2003, 2003, 414–416. [Google Scholar] [CrossRef]
- Fürstner, A.; Brehm, C.; Cancho-Grande, Y. Stereoselective Synthesis of Enamides by a Peterson Reaction Manifold. Org. Lett. 2001, 3, 3955–3957. [Google Scholar] [CrossRef]
- Ando, K.; Wada, T.; Okumura, M.; Sumida, H. Stereoselective Synthesis of Z-α,β-Unsaturated Sulfones Using Peterson Reagents. Org. Lett. 2015, 17, 6026–6029. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, T.A.; Kelly, C.B.; Cywar, R.M.; Leadbeater, N.E. Methylenation of Perfluoroalkyl Ketones Using a Peterson Olefination Approach. J. Org. Chem. 2014, 79, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, B. Hydrosilylation of Carbon—Carbon Multiple Bonds in Organic Synthesis. In Hydrosilylation: A Comprehensive Review on Recent Advances; Marciniec, B., Ed.; Springer: Dordrecht, The Netherlands, 2009; pp. 87–123. ISBN 978-1-4020-8172-9. [Google Scholar]
- Marciniec, B. Hydrosilylation of Alkenes and Their Derivatives. In Hydrosilylation: A Comprehensive Review on Recent Advances; Marciniec, B., Ed.; Springer: Dordrecht, The Netherlands, 2009; pp. 3–51. ISBN 978-1-4020-8172-9. [Google Scholar]
- Armstrong, R.J.; García-Ruiz, C.; Myers, E.L.; Aggarwal, V.K. Stereodivergent Olefination of Enantioenriched Boronic Esters. Angew. Chem. Int. Ed. 2017, 129, 804–808. [Google Scholar] [CrossRef]
- Linne, Y.; Lohrberg, D.; Struwe, H.; Linne, E.; Stohwasser, A.; Kalesse, M. 1,2-Metallate Rearrangement as a Toolbox for the Synthesis of Allylic Alcohols. J. Org. Chem. 2023, 88, 12623–12629. [Google Scholar] [CrossRef]
- Yeung, K.; Mykura, R.C.; Aggarwal, V.K. Lithiation–Borylation Methodology in the Total Synthesis of Natural Products. Nat. Synth. 2022, 1, 117–126. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activating Unit X | Olefination Method | Litt. Reference |
| PhSO2 | Julia–Lythgoe | Ref. [1] |
| ActSO2 | Julia–Kocienski | Ref. [1] |
| PhSO(NMe) | Johnson | Ref. [2] |
| R3P+ | Wittig | Ref. [3] |
| R2P(=O) | Wittig–Horner | Ref. [3] |
| (RO)2P(=O) | Horner–Wadsworth–Emmons (HWE) | Ref. [4] |
| R3Si | Peterson | Ref. [5] |
| R2B | Boron–Wittig | Ref. [6] |
| Key Features | Julia–Lythgoe | Julia–Kocienski |
|---|---|---|
| Practical Difference | Two-pot protocol | One-pot protocol |
| Origin of Stereoselectivity | Reductive elimination Step | Addition step |
| Scope of olefin formation | ||
| Terminal | ✔ | ✔ |
| 1,2-disubstituted | ✔ | ✔ |
| Trisubstituted | ✔ | ≈ |
| Tetrasubstituted | ≈ | X |
| Scope of (E)-Stereoselectivity | ||
| 1,2-disubstituted | ✔ | ✔ |
| Trisubstituted | ≈ | X |
| Tetrasubstituted | ≈ | X |
| Scope of (Z)-Stereoselectivity | ||
| 1,2-disubstituted | X | ✔ if the TBT-activating group is used; |
| Trisubstituted | X | X |
| Tetrasubstituted | X | X |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrenko, D.; Pospíšil, J. Latest Developments of the Julia–Kocienski Olefination Reaction: Mechanistic Considerations. Molecules 2024, 29, 2719. https://doi.org/10.3390/molecules29122719
Chrenko D, Pospíšil J. Latest Developments of the Julia–Kocienski Olefination Reaction: Mechanistic Considerations. Molecules. 2024; 29(12):2719. https://doi.org/10.3390/molecules29122719
Chicago/Turabian StyleChrenko, Daniel, and Jiří Pospíšil. 2024. "Latest Developments of the Julia–Kocienski Olefination Reaction: Mechanistic Considerations" Molecules 29, no. 12: 2719. https://doi.org/10.3390/molecules29122719
APA StyleChrenko, D., & Pospíšil, J. (2024). Latest Developments of the Julia–Kocienski Olefination Reaction: Mechanistic Considerations. Molecules, 29(12), 2719. https://doi.org/10.3390/molecules29122719