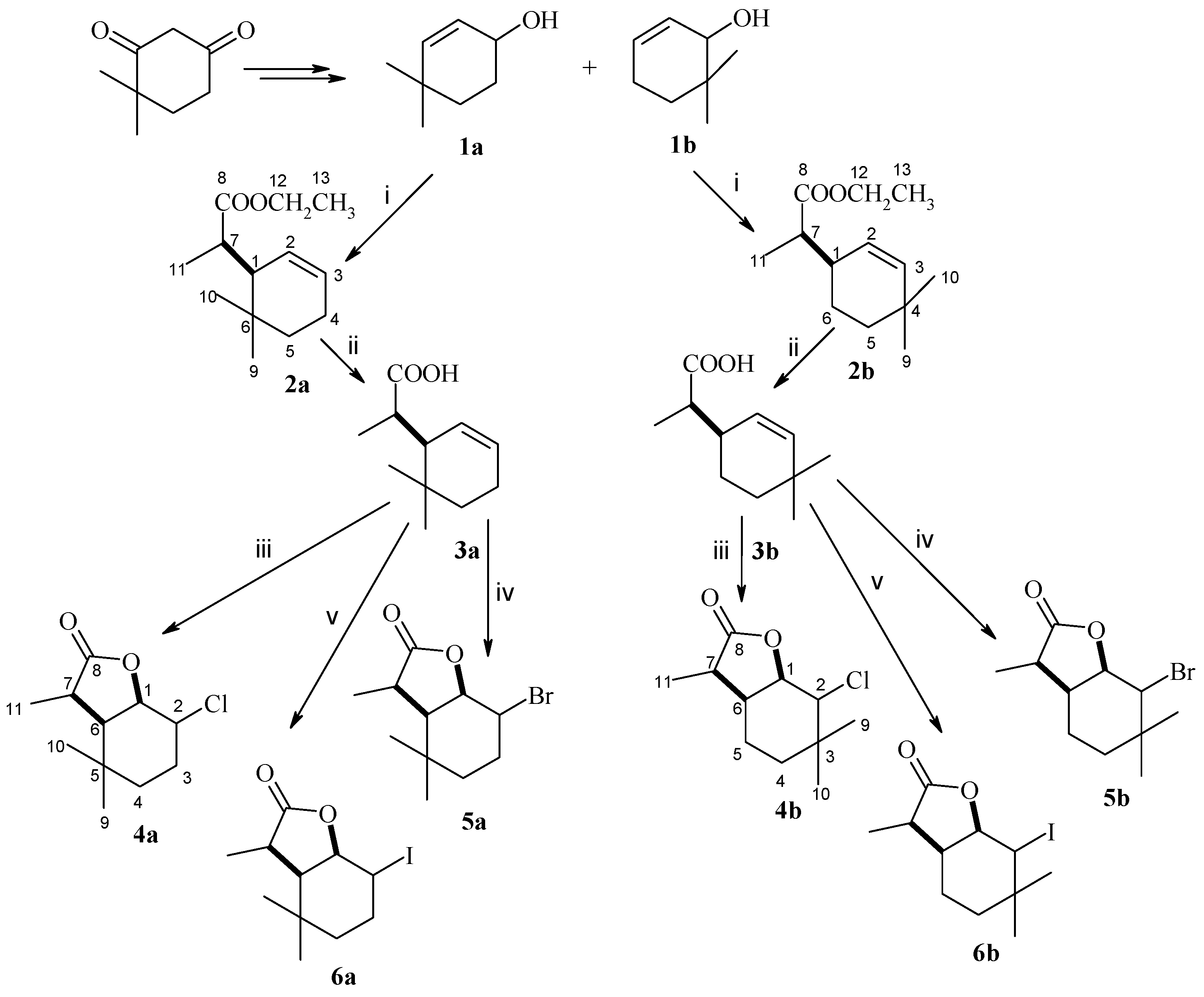
4.2. Organic Synthesis
(1R, 7S or 1S, 7R)-Ethyl ester of (4,4,7-trimethylcyclohex-2-en-1-yl)acetic acid (isomer A) and (1R, 7R or 1S, 7S)-Ethyl Ester of (4,4,7-trimethylcyclohex-2-en-1-yl)acetic acid (isomer B) (
2a), The known allylic alcohol
1a [
24] was subjected to Claisen rearrangement with orthopropionate modification. Briefly, a mixture of alcohol (3.5 g), triethyl orthopropionate (20 cm
3) and a catalytic amount of propionic acid was heated at 138 °C for 30 h. The progress of the reaction was monitored by TLC and GC. The obtained ester was purified by column chromatography using a 19:1 mixture of hexane and acetone as eluent. Ester
2a (3.9 g, 68%) was obtained as a pair of diastereoisomers A and B in the ratio 72%:28%. The spectral data of the obtained compound
2a are as follows:
1H NMR (600 MHz, CDCl
3): 0.86 (s, 3H, CH
3-9B), 0.87 (s, 3H, CH
3-9A), 0.94 (s, 3H, CH
3-10B), 0.96 (s, 3H, CH
3-10A), 1.10–1.14 (M, 2H, CH
2-5B), 1.09 (d,
J = 7.2 Hz, 3H, CH
3-11B), 1.20–1.26 (m, 9H, CH
3-13A, CH
3-13B, CH
3-11A), 1.33–1.42 (m, 2H, CH
2-4B), 1.58 (m, 2H, CH
2-5A), 1.96–2.00 (m, 2H, CH
2-4A), 2.27–2.37 (m, 2H, H-1A and H-1B), 2.58 (qd,
J = 6.8 and 4.8 Hz, 1H, H-7B), 2.71 (qd,
J = 7.2 and 3.6 Hz, 1H, H-7A), 4.06 (q,
J = 7.2 Hz,2H, CH
2-12A), 4.11 (q,
J = 6.8 Hz,2H, CH
2-12B), 5.49–5.54 (m, 2H, H-3A and H-3B), 5.69–5.74 (m, 2H, H-2A and H-3B),
13C NMR (151 MHz, CDCl
3): 9.26 (C-11B), 14.19 (C-11A), 14.61 (C-13B), 18.71 (C-13A), 22.78 (C-4A), 23.07 (C-9A), 24.74 (C-9B), 25.56 (C-4B), 28.79 (C-10B), 29.04 (C-10A), 29.18 (C-6B), 29.62 (C-6A), 34.82 (C-5B), 36.03 (C-5A), 39.86 (C-7B), 39.95 (C-7A), 45.90 (C-1B), 49.85 (C-1A), 60.02 (C-12A), 60.36 (C-12B), 126.34 (C-2A), 126.77 (C-2B), 139.02127.03 (C-3A), 127.55 (C-3B), 176.20 (C-8A), 177.55 (C-8B), ESIHRMS: calculated for C
13H
22O
2, 211.1698 (M + H)
+, found 211.1700.
(1R, 7S or 1S, 7R)-(4,4,7-trimethylcyclohex-2-en-1-yl)acetic acid (isomer A) and (1R, 7R or 1S, 7S)-(4,4,7-trimethylcyclohex-2-en-1-yl)acetic acid (isomer B) (
3a), Ester
2a (3.9 g) was given alkaline hydrolysis according to a known procedure [
32]. Briefly, the ester and 5% potassium hydroxide solution in ethanol were heated for 6 h under reflux conditions. After the reaction was completed, the ethanol was evaporated, the residue was dissolved in water, acidified with 1 mol hydrochloric acid, and then extracted with diethyl ether. As a result, 1.5 g (44%) of acid
3a was obtained, as a pair of isomers A and B in the ratio 74%:26%. The spectral data of compound
3a are as follows:
1H NMR (400 MHz, CDCl
3): 0.89 (s, 3H, CH
3-9A), 0.92 (s, 3H, CH
3-9B), 0.96 (s, 3H, CH
3-10A), 0.98 (s, 3H, CH
3-10B), 1.11 (d,
J = 7.2 Hz, 3H, CH
3-11A), 1.27 (d,
J = 7.6 Hz, 3H, CH
3-11B), 1.29–1.34 (m, 1H, CH
2-5B), 1.98–2.00 (m, 2H, CH
2-4A), 2.04–2.06 (m, 1H, H-1B), 2.38–2.40 (m, 1H, H-1A), 2.64–2.70 (qd,
J = 7.2 and 4.4 Hz, 1H, H-7a), 2.73–2.76 (qd,
J = 7.2 and 4.0 Hz, 1H, H-7B), 5.49–5.50 (m, 1H, H-3B), 5.51–5.53 (m, 1H, H-3A), 5.69–5.70 (m, 1H, H-2B), 5.73–5.77 (m, 1H, H-2A),
13C NMR (151 MHz, CDCl
3): 14.10 (C-11A), 17.82 (C-11B), 22.75 (C-9A), 23.32 (C-4A), 24.70 (C-9B), 29.12 (C-10A), 29.60 (C-10B), 32.31 (C-6A), 32.60 (C-6B), 34.93 (C-5A), 35.79 (C-5B), 39.45 (C-7A), 40.11 (C-7B), 45.80 (C-1B), 45.80 (C-4B), 49.50 (C-1A), 126.30 (C-2A), 126.72 (C-2B), 127.03 (C-3B), 127.90 (C-3B), 182.36 (C-8B), 183.870 (C-8A), ESIHRMS: calculated for C
11H
18O
2, m/z 183.1385 (M + H)
+, found 183.1378.
(1S, 2R, 6R, 7S or 1R, 2S, 6S, 7R)-2-Chloro-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (isomer A) and (1S, 2S, 6R, 7R or 1R, 2R, 6S, 7S)-2-Chloro-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (isomer B) (
4a), Chlorolactone was prepared according to a known procedure [
32]. Briefly, acid
3a (0.5 g) and N-chlorosuccinimide (0.9 g) dissolved in THF (30 mL) were stirred for 24 h at room temperature. The reaction mixture was diluted with water, and the product was extracted with diethyl ether. After purification of the reaction product by column chromatography, using a mixture of hexane/acetone 3:1 as eluent, 0.35 g (59%) of chlorolactone
4a was obtained as a pair of isomers A and B, occurring in the ratio 72%:28%. The spectral data of compound
4a are as follows: Isomer A:
1H NMR (600 MHz, CDCl
3): 1.09 (s, 3H, CH
3-9), 1.11 (s, 3H, CH
3-10), 1.41–1.44 (m, 1H, one of CH
2-4), 1.46 (d,
J = 7.2 Hz, 3H, CH
3-11), 1.59–1.62 (m, 1H, one of CH
2-4), 1.88-1.94 (m, 1H, one of CH
2-3), 2.15–2.20 (m, 1H, CH
2-3), 2.47 (dd,
J = 7.2 and 6.0 Hz, 1H, H-6), 2.76 (q,
J = 7.8 Hz, 1H, H-7), 4.23–4.28 (m, H-2), 4.49–4.52 (m, 1H, H-1),
13C NMR (151 MHz, CDCl
3): 14.07 (C-11), 24.67 (C-9), 27.83 (C-3), 30.80 (C-5), 32.38 (C-10), 34.22 (C-4), 39.64 (C-7), 47.58 (C-6), 58.11 (C-2), 82.26 (C-1), 178.93 (C-8). Isomer B:
1H NMR (400 MHz, CDCl
3): 1.04 (s, 3H, CH
3-10), 1.08 (s, 3H, CH
3-9), 1.33 (d,
J = 6.8 Hz, 3H, CH
3-11), 1.42–1.53 (m, 1H, CH
2-4), 1.84–1.92 (m, 1H, one of CH
2-3), 2.01–2.04 (m, 1H, H-6), 2.06–2.11 (m, 1H, one of CH
2-3), 2.36–2.44 (m, 1H, H-7), 3.62–3.69 (m, 1H, H-2), 4.97 (dd,
J = 9.2 and 7.2 Hz, 1H, H-1),
13C NMR (100 MHz, CDCl
3): 17.29 (C-11), 28.15 (C-9), 29.481 (C-10), 29.85 (C-3), 32.70(C-5), 34.29 (C-4), 35.90 (C-7), 54.62 (C-6), 60.75 (C-2), 81.62 (C-1), 178.85 (C-8). ESIHRMS: calculated for C
11H
17ClO
2, m/z 217.0995 and 219.0969 (M + H)
+, found 217.0999 and 219.0970, calculated for C
11H
17ClO
2Na, m/z 239.0815 and 241.0788 (M + H)
+, found 239.0823 and 241.0790.
(1S, 2R, 6R, 7S or 1R, 2S, 6S, 7R)-2-Bromo-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (isomer A) and (1S, 2S, 6R, 7R or 1R, 2R, 6S, 7S)-2-Bromo-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (isomer B) (
5a), Bromolactone was obtained according to a known procedure [
32]. Briefly, acid
3a (0.5 g) and N-bromosuccinimide (0.7 g) dissolved in THF (30 mL) were stirred for 24 h at room temperature. Water was added to the reaction mixture and the product was extracted with diethyl ether. After purification of the reaction product by column chromatography, using a mixture of hexane/acetone 3:1 as eluent, 0.54g (75%) of bromolactone
5a was obtained as a pair of isomers A and B, occurring in the ratio 76%:24%. The spectral data of compound
5a are as follows: Isomer A:
1H NMR (600 MHz, CDCl
3): 1.09 (s, 3H, CH
3-9), 1.12 (s, 3H, CH
3-10), 1.43–1.46 (m, 1H, one of CH
2-4), 1.47 (d,
J = 7.2 Hz, 3H, CH
3-11), 1.62 (m, 1H, one of CH
2-4), 2.00–2.06 (m, 3H, one of CH
2-3), 2.21–2.27 (m 1H, one of CH
2-3), 2.49 (dd,
J = 7.2 and 6.6 Hz, 1H, H-6), 2.77 (q,
J = 7.2 Hz, 1H, H-7), 4.36 (m, H-2), 4.61–4.65 (t,
J = 6.0 Hz, 1H, H-1),
13C NMR (151 MHz, CDCl3): 17.18 (C-11), 28.08 (C-3), 29.47 (C-5), 30.81 (C-9), 32.45 (C-10), 35.69 (C-4), 39.82 (C-7), 49.70 (C-6), 50.12 (C-2), 82.47 (C-1), 178.92 (C-8). Isomer B:
1H NMR (600 MHz, CDCl
3): 1.06 (s, 3H, CH
3-10), 1.12 (s, 3H, CH
3-9), 1.37 (d,
J = 6.6 Hz, 3H, CH
3-11), 1.42–1.44 (m, 1H, one of CH
2-4), 1.49–1.54 (m, 1H, one of CH
2-4), 2.02–2.10 (m, 2H, one of CH
2-3, H-6), 2.16–2.19 (m, 1H, one of CH
2-3), 2.45–2.51 (m, 1H, H-7), 3.74–3.79 (m, 1H, H-2), 4.66 (dd,
J = 9.6 and 7.2 Hz, 1H, H-1),
13C NMR (151 MHz, CDCl3): 17.14 (C-11), 28.08 (C-9), 29.047 (C-10), 30.72 (C-3), 32.64 (C-5), 35.22 (C-4), 35.62 (C-7), 51.72 (C-2), 54.84 (C-6), 81.70 (C-1), 178.65 (C-8). ESIHRMS: calculated for C
11H
17BrO
2,
m/
z 261.0490 and 263.0473 (M + H)
+, found 261.0475 and 263.0453, calculated for C
11H
17BrO
2Na,
m/
z 283.0310 and 285.0290 (M + H)
+, found 283.0304 and 285.0284.
(1S, 2R, 6R, 7S or 1R, 2S, 6S, 7R)-2-Iodo-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (isomer A) and (1S, 2S, 6R, 7R or 1R, 2R, 6S, 7S)-2-Iodo-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (isomer B) (
6a), Iodolactone was prepared according to a known procedure [
32]. Briefly, acid
3a (0.5 g) was dissolved in 25 mL diethyl ether and stirred for 1 h with 25 mL of 0.5 mol sodium bicarbonate solution. Then, 1 g of iodine and 2 g of potassium iodide in 20 mL of water were added to the solution, and the whole mixture was stirred for 24 h. After the reaction was completed, the product was extracted with diethyl ether. After purification of the compound by column chromatography, using a mixture of hexane/acetone 3:1 as eluent, 0.48g (57%) of iodolactone
6a was obtained as a pair of isomers A and B, occurring in the ratio 96%:4%. The spectral data of compound
6a are as follows: Isomer A:
1H NMR (400 MHz, CDCl
3): 1.04 (s, 3H, CH
3-9), 1.09 (s, 3H, CH
3-10), 1.41–1.48 (m, 2H CH
2-4), 1.45 (d,
J = 7.2 Hz, 3H, CH
3-11), 2.03–2.17 (m, 2H, CH
2-4), 2.09–2.14 (m, 2H, CH
2-3), 2.43 (dd,
J = 7.2 and 6.0 Hz, 1H, H-6), 2.71 (q,
J = 7.8 Hz, 1H, H-7), 4.42 (m, H-2), 4.70 (m, 1H, H-1),
13C NMR (100 MHz, CDCl
3): 14.42 (C-11), 25.18 (C-9), 28.58 (C-2), 30.87(C-3), 32.66 (C-10), 37.34 (C-4), 40.09 (C-7), 47.73 (C-6), 54.77 (C-5), 84.06 (C-1), 179.06 (C-8). Isomer B:
1H NMR (400 MHz, CDCl
3): 1.04 (s, 3H, CH
3-9), 1.09 (s, 3H, CH
3-10), 1.41–1.49 (m, 2H, CH
2-4), 1.45 (d,
J = 7.6 Hz, 3H, CH
3-11), 1.92–1.97 (m, 2H, one of CH
2-3), 2.11–2.15 (m, 1H, CH
2-6), 2.20–2.22 (m, 1H, one of CH
2-3), 2.48–2.55 (qm, 1H, H-7), 3.74–3.81 (m, H-2), 4.69–4.72 (m, H-1),
13C NMR (100 MHz, CDCl
3): 17.10 (C-11), 25.18 (C-9), 28.12 (C-2), 29.74 (C-10), 30.87 (C-5), 33.02 (C-3), 37.34 (C-4), 40.01 (C-7), 47.69 (C-6), 82.91 (C-1), 179.06 (C-8). ESIHRMS: calculated for C
11H
17IO
2,
m/
z 309.0352 (M + H)
+, found 309.0352, calculated for C
11H
17IO
2Na,
m/
z 331.0171 (M + H)
+, found 331.0161.
(1R, 7S or 1R, 7R)-Ethyl ester of (4,4,7-trimethylcyclohex-2-en-1-yl)acetic acid (isomer A) and (1R, 7R or 1S, 7S)-Ethyl ester of (4,4,7-trimethylcyclohex-2-en-1-yl)acetic acid (isomer B) (
2b), The known allylic alcohol
1b [
24] (1.5 g) was subjected to Claisen rearrangement with orthopropionate modification analogously as described for ester
1a. Ester
2b (1.6 g) was obtained as a mixture of two diastereoisomers A and B in the ratio 20%:80% in 65% yield. The spectral data of compound
2b are as follows:
1H NMR (400 MHz, CDCl
3): 0.93 (s, 3H, CH
3-9), 0.94 (s, 3H, CH
3-10), 1.09 (d,
J = 6.8 Hz, 3H, CH
3-11A), 1.11 (d,
J = 6.8 Hz, 3H, CH
3-11B), 1.24 (t,
J = 7.6 Hz, 3H, CH
3-12), 1.36–1.42 (m, 2H, one of CH
2-5, one of CH
2-6), 1.47–1.52 (m, 1H, one of CH
2-6), 1.54–1.53 (m, 1H, one of CH
2-5), 2.29–2.37 (m, 2H, H-1 and H-7), 4.12 (q, 2H, CH
2-12), 5.28–5.31 (dm,
J = 10.0 Hz, 1H, H-3), 5.43–5.45 (dm,
J = 8.4 Hz, 1H, H-2),
13C NMR (100 MHz, CDCl
3): 13.63 (C-11A), 13.92 (C-11B), 14.39 (C-13A and C-13B), 22.39 (C-5A), 24.07 (C-5B), 28.77 (C-9A), 29.00 (C-9B), 30.52 (C-10B), 30.71 (C-10A), 31.66 (C-4A), 31.74 (C-4B), 36.27 (C-6B), 36.39 (C-6A), 38.48 (C-1A), 38.51 (C-1B), 44.06 (C-7B), 44.21 (C-7A), 60.22 (C-12A and C-12B), 125.60 (C-3A), 126.80 (C-3B), 139.02 (C-2A), 139.31 (C-2B), 176.07 (C-8B), 176.16 (C-8A), ESIHRMS: calculated for C
13H
22O
2,
m/
z 211.1698 (M + H)
+, found 211.1700.
(1R, 7S or 1R, 7R)-(4,4,7-trimethylcyclohex-2-en-1-yl)acetic acid (isomer A) and (1R, 7R or 1S, 7S)-(4,4,7-trimethylcyclohex-2-en-1-yl)acetic acid (isomer B) (3b), Ester 2b (1.6g) was given an alkaline hydrolysis, analogously as described for acid 2a. Acid 3b (0.7g) was obtained as a mixture of two diastereoisomers A and B in the ratio 22%:78% in 50% yield. The spectral data of compound 3b are as follows: 1H NMR (400 MHz, CDCl3): 0.90 (s, 3H, CH3-10B), 0.94 (s, 3H, CH3-9A), 0.96 (s, 3H, CH3-10A,), 0.96(s, 3H, CH3-9B), 1.12 (d, J = 6.8 Hz, 3H, CH3-11A), 1.16 (d, J = 6.8 Hz, 3H, CH3-11B), 1.38–1.43 (m, 4H, one of CH2-5A, one of CH2-6A, one of CH2-5B, one of CH2-6B), 1.49–1.53 (m, 2H, one of CH2-6A, one of CH2-6B), 1.63–1.68 (m, 2H, one of CH2-5A, one of CH2-5B), 2.37–2.44 (m, 4H, H-1A, H-1B, H-7A and H-7B), 5.33–5.35 (dm, J = 10.4 Hz, 2H, H-3A, H-3B), 5.46–5.49 (dm, J = 10.0 Hz, 2H, H-2A, H-2B), 13C NMR (100 MHz, CDCl3): 13.19 (C-11A), 13.62 (C-11B), 22.05 (C-5A), 24.19 (C-5B), 28.77 (C-9A), 29.00 (C-9B), 30.49 (C-10B), 30.69 (C-10A), 31.65 (C-4A), 31.75 (C-4B), 43.90 (C-6B), 43.00 (C-6A), 38.19 (C-1A), 38.19 (C-1B), 43.90 (C-7B), 44.00 (C-7A), 125.12 (C-3A), 126.63 (C-3B), 139.36 (C-2A), 139.71 (C-2B), 182.31 (C-8B), 182.39 (C-8A), ESIHRMS: calculated for C11H18O2, m/z 183.1385 (M + H)+, found 183.1376.
(1S, 2R, 6R, 7S or 1R, 2S, 6R, 7R)-2-Chloro-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (isomer A) and (1S, 2S, 6R, 7R or 1R, 2R, 6S, 7S)-2-Chloro-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (isomer B) (4b), Acid 3b (0.2g) was subjected to chlorolactonization in the same manner as given for compound 3a. The product 4b was obtained as a mixture of two diastereoisomers A and B in the ratio 22%:78% with a yield of 0.19 g (80%) with the following spectral data: Isomer A: 1H NMR (400 MHz, CDCl3): 1.07 (s, 3H, CH3-9), 1.07 (s, 3H, CH3-10), 1.18 (d, J = 7.2 Hz, 3H, CH3-11), 1.24–1.30 (m, 2H, CH2-4), 1.56–1.70 (m, 2H, CH2-5), 2.60–2.67 (m, 1H, H-6), 2.75 (q, J = 7.2 Hz, 1H, H-7), 4.10 (m, 1H, H-2), 4.57 (dd, J = 3.6 and 3.6 Hz, 1H, H-1), 13C NMR (100 MHz, CDCl3): 9.56 (C-11), 18.95 (C-4), 25.64 (C-9), 30.17 (C-10), 30.57 (C-5), 34.10 (C-3), 35.49 (C-6), 40.97 (C-7), 65.22 (C-2), 80.83 (C-1), 178.31 (C-8). Isomer B: 1H NMR (400 MHz, CDCl3): 1.00 (s, 3H, CH3-9), 1.07 (s, 3H, CH3-10), 1.20 (d, J = 6.4 Hz, 3H, CH3-11), 1.46 (ddd, J = 14.4, 4.4 and 4.4 Hz, 1H, one of CH2-4), 1.55 (ddd, J = 14.4, 4.8. and 2.4 Hz, 1H, one of CH2-4), 1.64–1.69 (m, 1H, one of CH2-5), 1.85–1.94 (m, 1H, one of CH2-5), 2.36–2.48 (m, 2H, H-6, H-7), 3.56 (d, J = 9.6 Hz, 1H, H-2), 4.48 (dd, J = 9.6 and 6.8 Hz, 1H, H-1), 13C NMR (100 MHz, CDCl3): 13.48 (C-11), 18.54 (C-9), 19.89 (C-5), 29.60 (C-10), 34.00 (C-4), 35.74 (C-6), 36.97 (C-3), 43.53 (C-7), 70.83 (C-2), 81.59 (C-1), 178.30 (C-8). ESIHRMS: calculated for C11H17ClO2, m/z 217.0995 and 219.0969 (M + H)+, found 217.0999 and 219.0970, calculated for C11H17ClO2Na, m/z 239.0815 and 241.0788 (M + H)+, found 239.0823 and 241.079.
(1S, 2R, 6R, 7S or 1R, 2S, 6R, 7R)-2-Bromo-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (isomer A) and (1S, 2S, 6R, 7R or 1R, 2R, 6S, 7S)-2-Bromo-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (isomer B) (5b), Acid 3b (0.2 g) underwent bromolactonization analogously as described for compound 3a. The resulting product 5b was formed as a mixture of two diastereoisomers A and B in the ratio 14%:86% with a yield of 0.26 g (90%) with the following spectral data: Isomer A: 1H NMR (600 MHz, CDCl3): 1.14 (s, 3H, CH3-9), 1.18 (s, 3H, CH3-10), 1.22 (d, J = 7.2 Hz, 3H, CH3-11), 1.29–1.34 (m, 2H, one of CH2-4, one of CH2-5), 1.61–1.66 (m, 1H, CH2-4), 1.71–1.76 (m, 1H, CH2-5), 2.74–2.76 (m, 1H, H-6), 2.80 (q, J = 7.2 Hz, 1H, H-7), 4.32 (m, 1H, H-2), 4.78 (dd, J = 3.6 and 3.6 Hz, 1H, H-1), 13C NMR (151 MHz, CDCl3): 9.51 (C-11), 19.03 (C-4), 25.69 (C-10), 31.07 (C-5), 32.48 (C-9), 33.96 (C-3), 36.45 (C-6), 41.17 (C-7), 60.33 (C-2), 81.21 (C-1), 178.31 (C-8). Isomer B: 1H NMR (600 MHz, CDCl3): 1.08 (s, 3H, CH3-9), 1.12 (s, 3H, CH3-10), 1.24 (d, J = 6.8 Hz, 3H, CH3-11), 1.52 (ddd, J = 14.0, 4.8 and 4.4 Hz, 1H, one of CH2-4), 1.63–1.67 (m, 1H, one of CH2-4), 1.70–1.74 (m, 1H, one of CH2-5), 1.92–1.99 (m, 1H, one of CH2-5), 2.36–2.40 (m, 1H, H-6), 2.50 (q, J = 6.8 Hz, 1H, H-7), 3.74 (d, J = 10.0 Hz, 1H, H-2), 4.67 (dd, J = 10.0 and 7.6 Hz, 1H, H-1), 13C NMR (151 MHz, CDCl3): 13.31 (C-11), 19.69 (C-9), 19.92 (C-5), 30.98 (C-10), 33.60 (C-4), 35.41 (C-7), 36.78 (C-3), 43.69 (C-6), 65.34 (C-2), 81.77 (C-1), 178.05 (C-8). ESIHRMS: calculated for C11H17BrO2, m/z 261.0490 and 263.0471 (M + H)+, found 261.0485 and 263.0465, calculated for C11H17BrO2Na, m/z 283.0310 and 285.0290 (M + H)+, found 283.0305 and 285.0285.
(1S, 2R, 6R, 7S or 1R, 2S, 6R, 7R)-2-Iodo-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (isomer A) and (1S, 2S, 6R, 7R or 1R, 2R, 6S, 7S)-2-Iodo-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (isomer B) (6b), Acid 3b (0.3 g) was submitted to iodolactonization in the same way as compound 3a. The product 6b was formed as a mixture of two diastereoisomers A and B in the ratio 23%:77% with a yield of 0.24 g (48%). The spectral data are as follows: Isomer A: 1H NMR (400 MHz, CDCl3): 1.13 (s, 3H, CH3-9), 1.17 (d, J = 6.8 Hz, 3H, CH3-11), 1.22 (s, 3H, CH3-10), 1.24–1.29 (m, 1H, one ofCH2-4), 1.60–1.69 (m, 2H, one of CH2-4 and CH2-5), 2.76 (q, J = 6.8 Hz, 1H, H-7), 2.80–2.85 (m, 1H, H-6), 4.53–4.54 (m, 1H, H-2), 4.94 (dd, J = 4.0 and 3.2 Hz, 1H, H-1), 13C NMR (100 MHz, CDCl3): 9.62 (C-11), 19.35 (C-5), 22.73 (C-3), 24.85 (C-10), 32.07 (C-4), 35.57 (C-6), 36.99 (C-9), 41.69 (C-7), 45.00 (C-2), 83.48 (C-1), 178.67 (C-8). Isomer B: 1H NMR (400 MHz, CDCl3): 1.05 (s, 3H, CH3-9), 1.06 (s, 3H, CH3-10), 1.19 (d, J = 6.8 Hz, 3H, CH3-11), 1.48–1.57 (m, 1H, one of CH2-4), 1.64–1.73 (m, 2H, one of CH2-4 and one of CH2-5), 1.88–1.98 (m, 1H, one of CH2-5), 2.19–2.24 (m, 1H, H-6), 2.52 (q, J = 6.8 Hz, 1H, H-7), 3.84 (d, J = 10.4 Hz, 1H, H-2), 4.77 (dd, J = 10.4 and 7.2 Hz, 1H, H-1), 13C NMR (100 MHz, CDCl3): 13.24 (C-11), 20.21 (C-5), 21.92 (C-10), 31.81 (C-4), 33.84 (C-9), 34.99 (C-7), 36.62 (C-3), 43.43 (C-6), 48.49 (C-2), 83.00 (C-1), 177.92 (C-8). ESIHRMS: calculated for C11H17IO2, m/z 309.0352 (M + H)+, found 309.0352.
4.4. Biotransformation
4.4.1. Microorganisms
Biotransformation reactions were carried out using strains of filamentous fungi from the collection of the Department of Food Chemistry and Biocatalysis of Wrocław University of Life Sciences. There were the strains of the genus Fusarium: F. culmorum AM10, F. avenaceum AM12, F. equiseti AM16, F. semitectum AM20, F. oxysporum AM21, F. solani AM203, and strains from the genus Absidia: A. coerulea AM93, A. glauca AM177, A. cylindrospora AM336. These strains were cultured on Sabouraud Agar (0.5 g of aminobac, 0.5 g of peptone, 4 g of glucose and 1.5 g of agar dissolved in 100 mL of water) at 28 °C and stored at 4 °C after growth.
4.4.2. Screening Biotransformation
Biotransformation was carried out in 300 mL Erlenmeyer flasks containing 100 mL of Sabouraud medium, consisting of 3 g of glucose and 1 g of peptone dissolved in 100 mL of water. After inoculation of the medium with the given microorganism, the mycelium was allowed to grow for three days. After this time, 10 mg of halolactone 4a,b–6a,b dissolved in 1 mL of acetone was added to each flask. Shaken culture was continued for another seven days, and after three, five and seven days, about 30 mL of medium was taken with the mycelium. This mixture was extracted with dichloromethane (15 mL), dried with magnesium sulfate and analyzed by GC using a SGE BP5 column.
4.4.3. Preparative Biotransformation
To 10 Erlenmeyer flasks, each containing 100 mL of Sabouraud medium with overgrown mycelium, 10 mg each (for a total of 100 mg) of bromo-5a or iodolactone 6a dissolved in 10 mL of acetone was added. Shaken culture was carried out for 7 days, after which the combined contents of ten flasks were extracted with dichloromethane (3 × 30 mL). The combined organic fractions were dried with anhydrous magnesium sulfate. After evaporation of the solvent in vacuo, the products were purified by column chromatography using a mixture of hexane/acetone 6:1 as eluent. The spectral data of the compounds obtained are shown below.
(1S, 2S, 6R, 7S or 1R, 2R, 6S, 7R)-2-Hydroxy-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (7a), 1H NMR (400 MHz, CDCl3): 1.01 (s, 6H, CH3-9 and CH3-10), 1.19–1.27 (m, 1H, one of CH2-4), 1.39–1.41 (m, 1H, one of CH2-4), 1.43 (d, J = 7.2 Hz, 3H, CH3-11), 1.61–1.71 (m, 1H, one of CH2-3), 1.76–1.83 (m, 1H, one of CH2-3), 2.15 (s, 1H, OH), 2.18 (dd, J = 6.8 and 4.4 Hz, 1H, H-6), 2.82 (q, J = 7.2 Hz, 1H, H-7), 3.74 (ddd, J = 10.4, 5.2 and 5.2, 1H, H-2), 4.42 (t, J = 4.4 Hz, 1H, H-1), 13C NMR (151 MHz, CDCl3): 11.87 (C-11), 22.78 (C-9), 26.11 (C-3), 31.53 (C-10), 31.92 (C-5), 38.81 (C-4), 42.20 (C-7), 47.07 (C-6), 69.30 (C-2), 80.52 (C-1), 179.27 (C-8), ESIHRMS: calculated for C11H18O3, m/z 199.1334 (M + H)+, found 199.1330, calculated for C11H18O3Na, m/z 221.1154 (M + H)+, found 221.1134.
(1S, 2R 6R, 7R or 1R, 2S, 6S, 7S)-2-Hydroxy-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (8a), 1H NMR (400 MHz, CDCl3): 1.06 (s, 3H, CH3-9), 1.09 (s, 3H, CH3-10), 1.24–1.26 (m, 1H, one of CH2-4), 1.40 (d, J = 7.2 Hz, 3H, CH3-11), 1.45–1.53 (m, 1H, one of CH2-4), 1.56–1.65 (m, 1H, one of CH2-3), 1.86–1.93 (m, 1H, one of CH2-3), 1.99 (s, 1H, OH), 2.44 (dd, J = 8.4 and 7.2 Hz, 1H, H-6), 2.69 (q, J = 8.0 Hz, 1H, H-7), 3.85–3.91 (m, 1H, H-2), 4.35–4.40 (m, 1H, H-1), 13C NMR (151 MHz, CDCl3): 15.19 (C-11), 23.09 (C-5), 26.35 (C-3), 30.38 (C-10), 32.86 (C-4), 34.22 (C-9), 38.35 (C-7), 48.33 (C-6), 71.55 (C-2), 84.28 (C-1), 180.24 (C-8), ESIHRMS: calculated for C11H18O3, m/z 199.1334 (M + H)+, found 199.1330.
(1S, 2R, 6R, 7R or 1R, 2S, 6S, 7S)-2-Chloro-7-hydroxy-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (9a), 1H NMR (400 MHz, CDCl3): 1.10 (s, 3H, CH3-9), 1.22 (s, 3H, CH3-10), 1.40–1.44 (m, 1H, one of CH2-4), 1.59 (d, J = 7.2 Hz, 3H, CH3-11), 1.66 (td, J = 14.0 and 3.6 Hz, 1H, one of CH2-4), 1.92 (qd, J = 14.0 and 4.0 Hz, 1H, one of CH2-3), 2.12–2.17 (m, 1H, one of CH2-3), 2.54 (d, J = 8.4 Hz, 1H, H-6), 2.76 (s, 1H, OH), 3.92 (ddd, J = 12.8, 10.0, 5.2 Hz, 1H, H-2), 4.61 (dd, J = 10.0 and 8.4 Hz, 1H, H-1), 13C NMR (151 MHz, CDCl3): 24.08 (C-11), 26.56 (C-10), 30.08 (C-3), 30.31 (C-9), 32.84 (C-5), 35.06 (C-4), 55.09 (C-6), 59.71 (C-2), 73.88 (C-7), 81.16 (C-1), 179.60 (C-8), ESIHRMS: calculated for C11H17ClO3, m/z 233.0945 and 235.0918 (M + H)+, found 233.0933 and 235.0893.
(1S, 2R, 6R, 7R or 1R, 2S, 6S, 7S)-2-Bromo-7-hydroxy-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (10a), 1H NMR (400 MHz, CDCl3): 1.10 (s, 3H, CH3-9), 1.21 (s, 3H, CH3-10), 1.24–1.29 (m, 1H, one of CH2-4), 1.60 (d, J = 7.2 Hz, 3H, CH3-11), 1.66 (td, J = 14.0 and 3.6 Hz, 1H, one of CH2-4), 2.09 (qd, J = 13.2 and 4.0 Hz, 1H, one of CH2-3), 2.22–2.25 (m, 1H, one of CH2-3), 2.50 (dd, J = 8,4 and 1.2 Hz, 1H, H-6), 2.72 (s, 1H, OH), 4.00 (ddd, J = 12.8, 10.0, 5.6 Hz, 1H, H-2), 4.74 (dd, J = 10.0 and 8.4 Hz, 1H, H-1), 13C NMR (151 MHz, CDCl3): 24.35 (C-11), 26.67 (C-10), 30.07 (C-9), 31.23 (C-3), 32.84 (C-5), 35.03 (C-4), 50.59 (C-2), 55.39 (C-6), 73.86 (C-7), 81.30 (C-1), 179.44 (C-8), ESIHRMS: calculated for C11H17BrO3, m/z 277.0439 and 279.0420 (M + H)+, found 277.0425 and 279.0387.
(1S, 2R, 6R, 7R or 1R, 2S, 6S, 7S)-2-Iodo-7-hydroxy-3,3,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (11a), 1H NMR (400 MHz, CDCl3): 1.10 (s, 3H, CH3-9), 1.20 (s, 3H, CH3-10), 1.24 (m, 1H, one of CH2-4), 1.64 (d, J = 7.2 Hz, 3H, CH3-11), 1.65 (td, J = 14.0 and 4.0 Hz, 1H, one of CH2-4), 2.25 (qd, J = 13.6 and 4.0 Hz, 2H, CH2-3), 2.38 (d, J = 8.4 Hz, 1H, H-6), 2.58 (s, 1H, OH), 3.95 (ddd, J = 12.8, 10.8, 5.6 Hz, 1H, H-2), 4.61 (dd, J = 10.4 and 8.0 Hz, 1H, H-1), 13C NMR (151 MHz, CDCl3): 24.85 (C-11), 26.93 (C-10), 26.98 (C-2), 30.04 (C-9), 32.83 (C-5), 33.50 (C-3), 37.30 (C-4), 55.24 (C-6), 73.88 (C-7), 82.42 (C-1), 179.12 (C-8), ESIHRMS: calculated for C11H17IO3, m/z 325.0301 (M + H)+, found 325.0309.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}