
Switchable Site-Selective Benzanilide C(sp2)-H Bromination via Promoter Regulation

Abstract
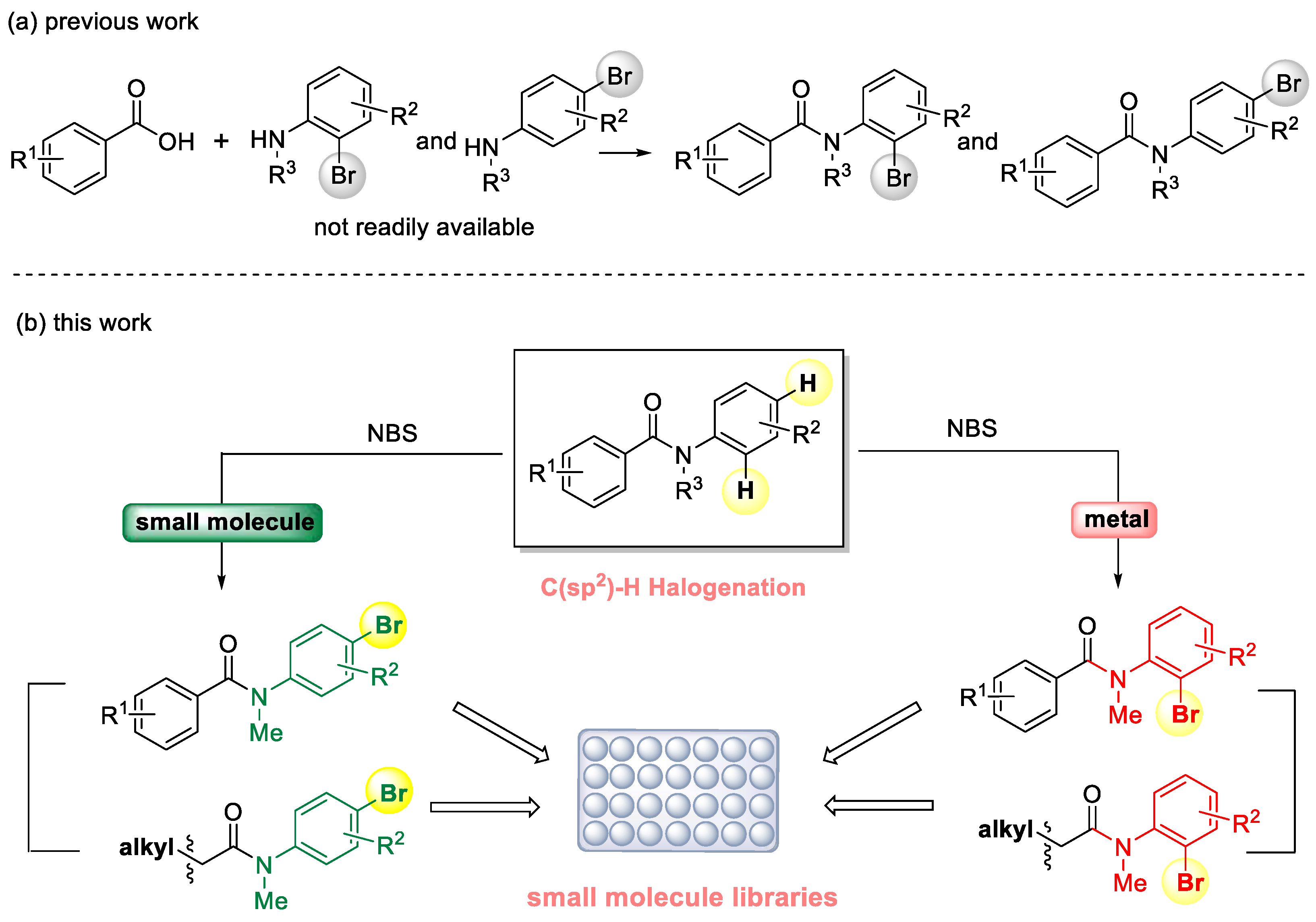
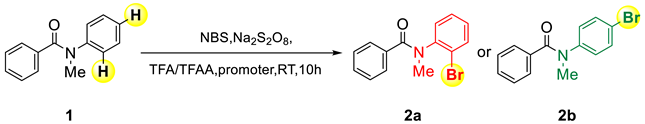
:1. Introduction
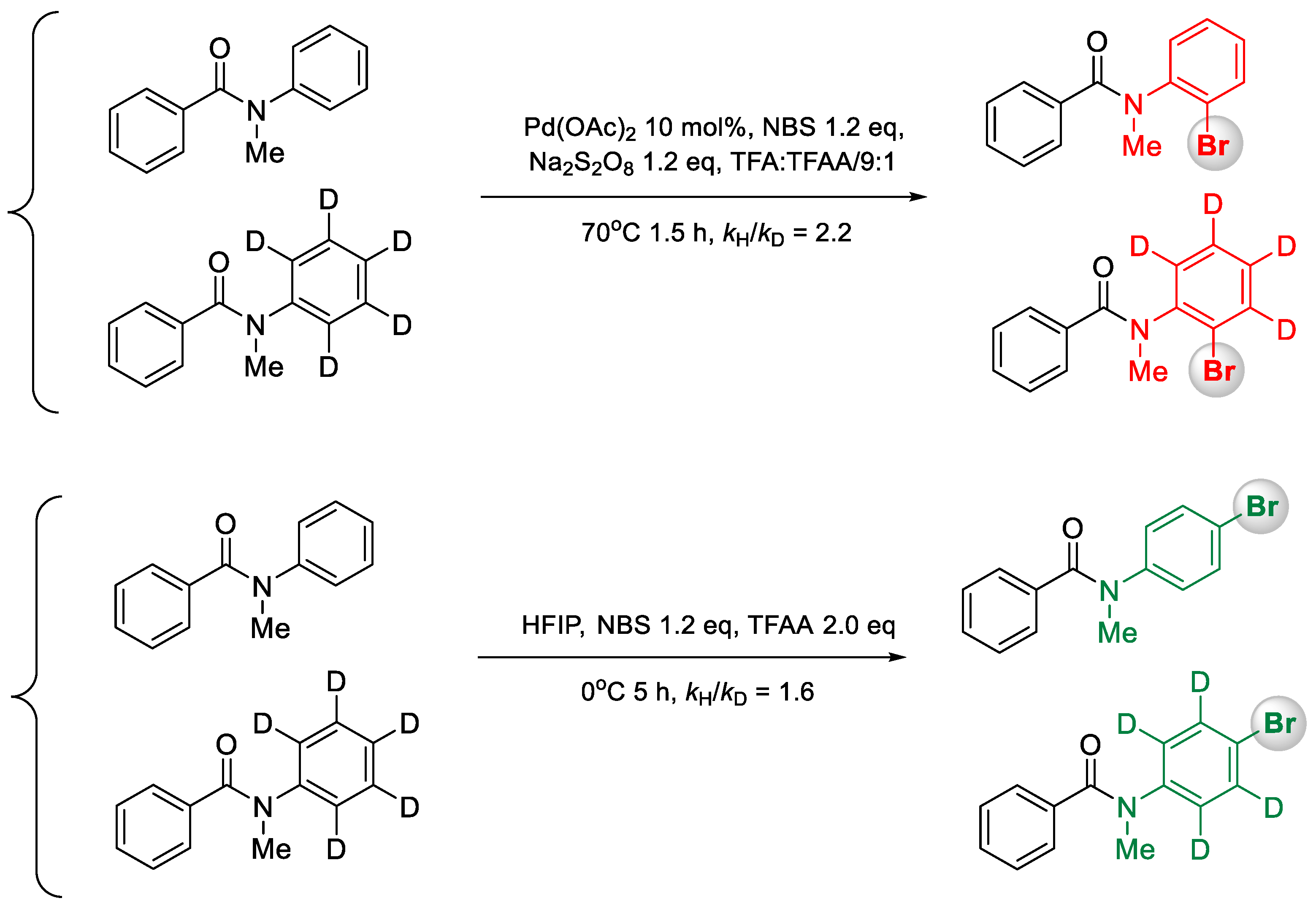
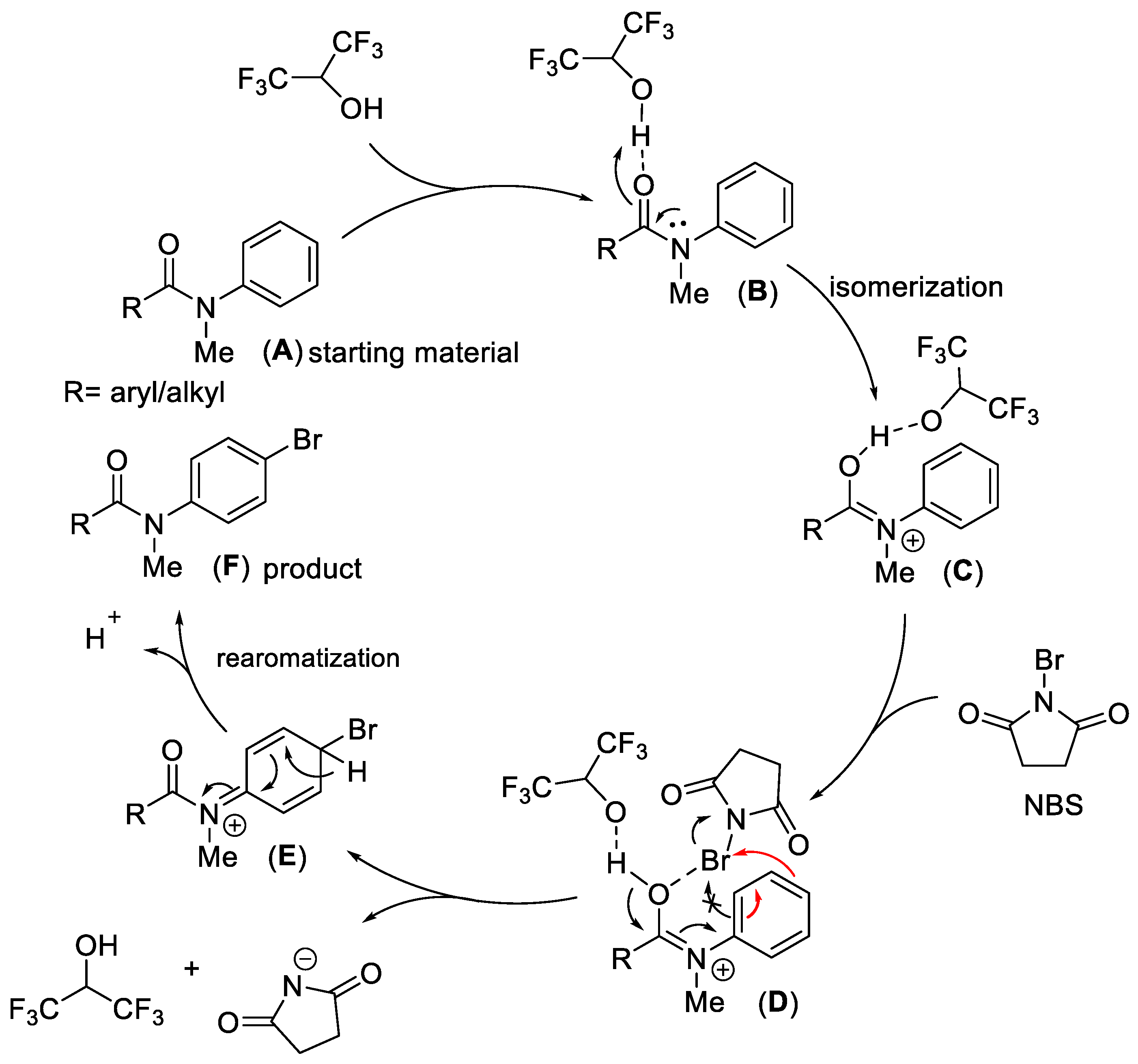
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. General Procedure for N-Methyl-Benzanilides Preparation
3.3. General Procedure for Palladium-Catalyzed Bromination of Benzanilides
3.4. General Procedure for HFIP-Catalyzed Bromination of Benzanilides
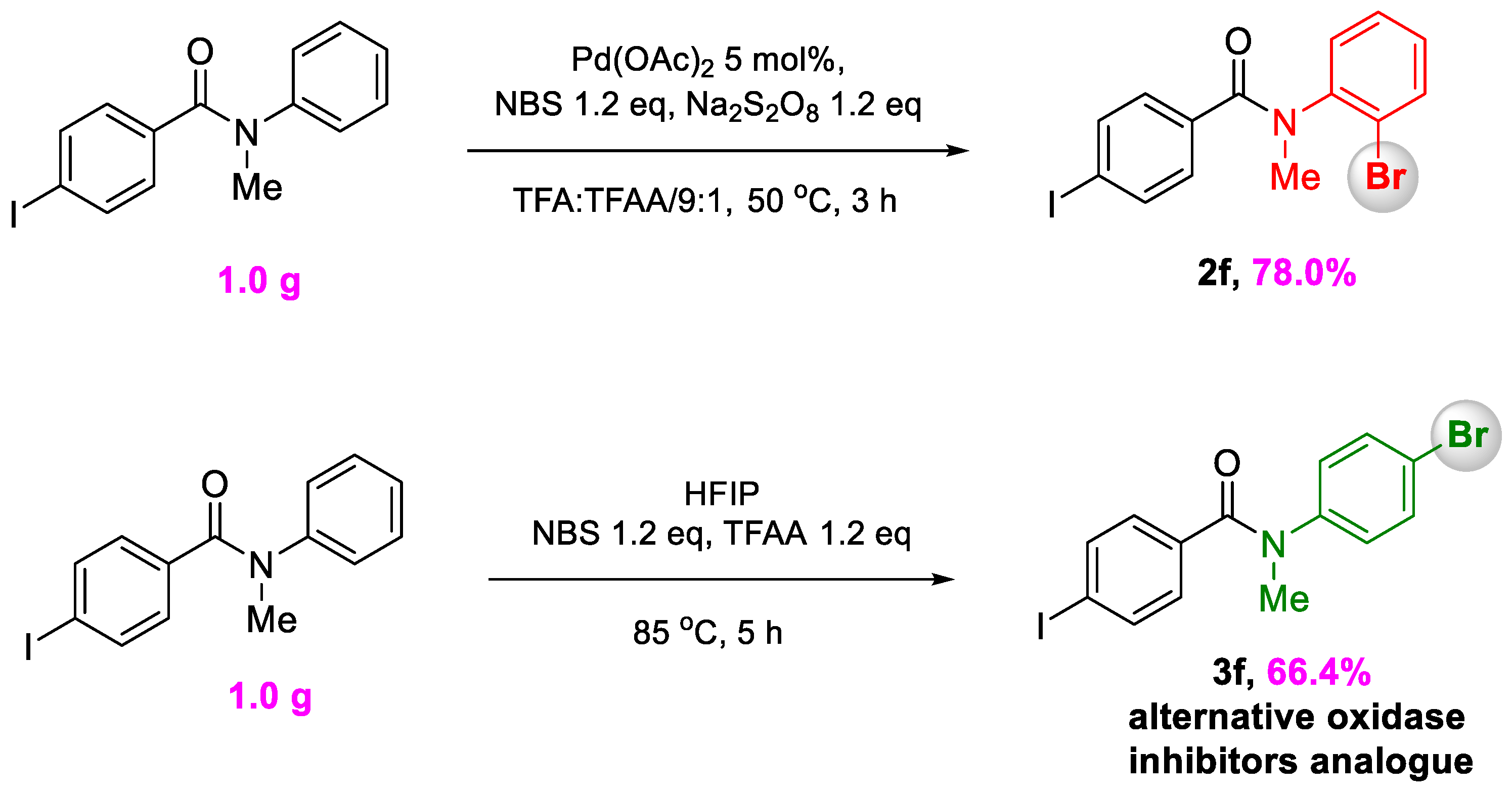
3.5. Procedure for Gram Scale Reaction
3.5.1. N-(2-Bromophenyl)-4-Iodo-N-Methylbenzamide
3.5.2. N-(4-Bromophenyl)-4-Iodo-N-Methylbenzamide
3.6. Characterization of Representative Substrates in Details
3.7. Characterization of Products in Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dudkin, V.; Fraley, M.; Wang, C.; Steen, J. Aminobenzotriazole Derivatives. WO 2010/114726, 7 October 2010. [Google Scholar]
- Wang, Q.; Guo, J.; Wang, Z.; Liu, Y.; Song, H.; Li, Y. Preparation of Trifluoromethylthiotryptamine Derivative and Their Application in Controlling Plant Virus and Fungicides. CN 113024562, 25 June 2021. [Google Scholar]
- Usui, S.; Oyama, K.; Araki, K.; Ushie, M.; Fukuchi, T. Novel 4-Pyridinecarboxamide Derivative and Agricultural and Horticultural Agents Containing Same as Active Ingredient. WO 2016/175017, 3 November 2016. [Google Scholar]
- De Colli, A.A.; Koolik, I.M.; Seminara, A.B.; Hatzios, S.K. A Propeptide-based Biosensor For The Selective Detection of Vibrio Cholerae Using an Environment-sensitive Fluorophore. Cell Chem. Biol. 2022, 29, 1505–1516. [Google Scholar] [CrossRef]
- Ibbeson, B.M.; Laraia, L.; Alza, E.; O’Connor, C.J.; Tan, Y.S.; Davies, H.M.L.; McKenzie, G.; Venkitaraman, A.R.; Spring, D.R. Diversity-oriented Synthesis as a Tool for Identifying New Modulators of Mitosis. Nat. Commun. 2014, 5, 3155. [Google Scholar] [CrossRef]
- Edkins, K.; Tweedy, J.; Fung, S.; Edkins, R.M. Steric Influence on Solvate Formation—A Comparison of Resorcylic Acid and Two Brominated Derivatives. Cryst. Eng. Comm. 2022, 24, 1368–1376. [Google Scholar] [CrossRef]
- Wang, Y.; Nguyen, H.P.; Xue, P.; Xie, Y.; Yi, D.; Lin, F.; Dinh, J.; Viscarra, J.A.; Ibe, N.U.; Duncan, R.E.; et al. ApoL6 Associates with Lipid Droplets and Disrupts Perilipin1-HSL Interaction to Inhibit Lipolysis. Nat. Commun. 2024, 15, 186. [Google Scholar] [CrossRef]
- Liu, S.; Dou, K.; Liu, B.; Pang, M.; Ma, P.A.; Lin, J. Construction of Multiform Hollow-Structured Covalent Organic Frameworks via a Facile and Universal Strategy for Enhanced Sonodynamic Cancer Therapy. Angew. Chem. Int. Ed. Engl. 2023, 62. [Google Scholar] [CrossRef]
- Rahman, A.A.; Butcko, A.J.; Songyekutu, E.; Granneman, J.G.; Mottillo, E.P. Direct Effects of Adipocyte Lipolysis on AMPK through Intracellular Long-chain Acyl-CoA Signaling. Sci. Rep. 2024, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xiong, T.; Du, J.; Tian, R.; Xiao, M.; Guo, L.; Long, S.; Fan, J.; Sun, W.; Shao, K.; et al. Superoxide Radical Photogenerator with Amplification Effect: Surmounting The Achilles’ Heels Of Photodynamic Oncotherapy. J. Am. Chem. Soc. 2019, 141, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Hotra, A.; Ragunathan, P.; Ng, P.S.; Seankongsuk, P.; Harikishore, A.; Sarathy, J.P.; Saw, W.G.; Lakshmanan, U.; Sae-Lao, P.; Kalia, N.P.; et al. Discovery of a Novel Mycobacterial F-ATP Synthase Inhibitor and Its Potency in Combination with Diarylquinolines. Angew. Chem. Int. Ed. Engl. 2020, 59, 13295–13304. [Google Scholar] [CrossRef]
- Luo, R.; Lv, H.; Liao, Q.; Wang, N.; Yang, J.; Li, Y.; Xi, K.; Wu, X.; Ju, H.; Lei, J. Intrareticular Charge Transfer Regulated Electrochemiluminescence of Donor–acceptor Covalent Organic Frameworks. Nat. Commun. 2021, 12, 6808. [Google Scholar] [CrossRef]
- Gerry, C.J.; Schreiber, S.L. Chemical Probes and Drug Leads from Advances in Synthetic Planning and Methodology. Nat. Rev. Drug Discov. 2018, 17, 333–352. [Google Scholar] [CrossRef]
- Schreiber, S.L. Molecular Diversity by Design. Nature 2009, 457, 153–154. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Galloway, W.R.J.D.; Spring, D.R. A Question Of Library Design. Nature 2011, 470, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Mannan, A.M.; Yvone, G.M.; Ross, K.N.; Zhang, Y.-L.; Marton, M.A.; Taylor, B.R.; Crenshaw, A.; Gould, J.Z.; Tamayo, P.; et al. High-throughput Identification of Genotype-specific Cancer Vulnerabilities in Mixtures of Barcoded Tumor Cell Lines. Nat. Biotechnol. 2016, 34, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.-J.; Wong, S.-T.; Hwang, C.-C.; Mou, C.-Y. Highly Efficient and Regioselective Halogenation over Well Dispersed Rhenium-Promoted Mesoporous Zirconia. ACS Catal. 2011, 1, 786–793. [Google Scholar] [CrossRef]
- Ritschel, B.; Poater, J.; Dengel, H.; Bickelhaupt, F.M.; Lichtenberg, C. Double CH Activation of a Masked Cationic Bismuth Amide. Angew. Chem. Int. Ed. Engl. 2018, 57, 3825–3829. [Google Scholar] [CrossRef] [PubMed]
- Hering, T.; Mühldorf, B.; Wolf, R.; König, B. Halogenase-Inspired Oxidative Chlorination Using Flavin Photocatalysis. Angew. Chem. Int. Ed. Engl. 2016, 55, 5342–5345. [Google Scholar] [CrossRef] [PubMed]
- Rogge, T.; Kaplaneris, N.; Chatani, N.; Kim, J.; Chang, S.; Punji, B.; Schafer, L.L.; Musaev, D.G.; Wencel-Delord, J.; Roberts, C.A.; et al. C–H Activation. Nat. Rev. Methods Primes 2021, 1, 43. [Google Scholar] [CrossRef]
- Goswami, N.; Bhattacharya, T.; Maiti, D. Transient Directing Ligands for Selective Metal-catalysed C–H activation. Nat. Rev. Chem. 2021, 5, 646–659. [Google Scholar] [CrossRef]
- Barranco, S.; Zhang, J.; López-Resano, S.; Casnati, A.; Pérez-Temprano, M.H. Transition Metal-catalysed Directed C–H functionalization with Nucleophiles. Nat. Synth. 2022, 1, 841–853. [Google Scholar] [CrossRef]
- Dutta, U.; Maiti, S.; Bhattacharya, T.; Maiti, D. Arene Diversification Through Distal C(sp2)−H Functionalization. Science 2021, 372, eabd5992. [Google Scholar] [CrossRef]
- Bakanas, I.; Lusi, R.F.; Wiesler, S.; Hayward Cooke, J.; Sarpong, R. Strategic Application of C–H Oxidation in Natural Product total Synthesis. Nat. Rev. Chem. 2023, 7, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Lam, N.Y.S.; Lucas, E.L.; Saint-Denis, T.G.; Verma, P.; Chekshin, N.; Yu, J.-Q. Achieving Site-Selectivity for C–H Activation Processes Based on Distance and Geometry: A Carpenter’s Approach. J. Am. Chem. Soc. 2020, 142, 10571–10591. [Google Scholar] [CrossRef] [PubMed]
- Loup, J.; Dhawa, U.; Pesciaioli, F.; Wencel-Delord, J.; Ackermann, L. Enantioselective C−H Activation with Earth-Abundant 3D Transition Metals. Angew. Chem. Int. Ed. Engl. 2019, 58, 12803–12818. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Yeung, Y.Y. Highly Ortho-Selective Chlorination of Anilines Using a Secondary Ammonium Salt Organocatalyst. Angew. Chem. Int. Ed. Engl. 2016, 55, 16101–16105. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.Y.; Shan, G.; Sun, Y.H.; Rao, Y. Regio-and Chemoselective C-H Chlorination/Bromination of Electron-Deficient Arenes by Weak Coordination and Study of Relative Directing-Group Abilities. Angew. Chem. Int. Ed. Engl. 2013, 52, 4440–4444. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Dong, G. Rapid and Modular Access to Multifunctionalized 1,2-Azaborines via Palladium/Norbornene Cooperative Catalysis. J. Am. Chem. Soc. 2024, 146, 9512–9518. [Google Scholar] [CrossRef]
- Liang, Y.; Lin, F.; Adeli, Y.; Jin, R.; Jiao, N. Efficient Electrocatalysis for the Preparation of (Hetero)Aryl Chlorides and Vinyl Chloride With 1,2-Dichloroethane. Angew. Chem. Int. Ed. Engl. 2019, 58, 4566–4570. [Google Scholar] [CrossRef] [PubMed]
- Colomer, I.; Chamberlain, A.E.R.; Haughey, M.B.; Donohoe, T.J. Hexafluoroisopropanol as a Highly Versatile Solvent. Nat. Rev. Chem. 2017, 1, 88. [Google Scholar] [CrossRef]
- Robertson, J.C.; Coote, M.L.; Bissember, A.C. Synthetic Applications of light, Electricity, Mechanical Force and Flow. Nat. Rev. Chem. 2019, 3, 290–304. [Google Scholar] [CrossRef]
- Ito, M.; Kubo, H.; Itani, I.; Morimoto, K.; Dohi, T.; Kita, Y. Organocatalytic C–H/C–H′ Cross-Biaryl Coupling: C-Selective Arylation of Sulfonanilides with Aromatic Hydrocarbons. J. Am. Chem. Soc. 2013, 135, 14078–14081. [Google Scholar] [CrossRef]
- Cai, S.; Tang, H.; Li, B.; Shao, Y.; Zhang, D.; Zheng, H.; Qiao, T.; Chu, X.; He, G.; Xue, X.-S.; et al. Formaldehyde-Mediated Hydride Liberation of Alkylamines for Intermolecular Reactions in Hexafluoroisopropanol. J. Am. Chem. Soc. 2024, 146, 5952–5963. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Yu, J.-Q. Lactonization As A General Route To β-C(sp3)–H Functionalization. Nature 2019, 577, 656–659. [Google Scholar] [CrossRef]
- Herron, A.N.; Liu, D.; Xia, G.; Yu, J.-Q. δ-C–H Mono- And Dihalogenation of Alcohols. J. Am. Chem. Soc. 2020, 142, 2766–2770. [Google Scholar] [CrossRef]
- Chen, Y.-Q.; Singh, S.; Wu, Y.; Wang, Z.; Hao, W.; Verma, P.; Qiao, J.X.; Sunoj, R.B.; Yu, J.-Q. Pd-Catalyzed γ-C(sp3)–H Fluorination of Free Amines. J. Am. Chem. Soc. 2020, 142, 9966–9974. [Google Scholar] [CrossRef] [PubMed]
- Garry, O.L.; Heilmann, M.; Chen, J.; Liang, Y.; Zhang, X.; Ma, X.; Yeung, C.S.; Bennett, D.J.; MacMillan, D.W.C. Rapid Access to 2-Substituted Bicyclo[1.1.1]pentanes. J. Am. Chem. Soc. 2023, 145, 3092–3100. [Google Scholar] [CrossRef]
- Chen, T.Q.; Pedersen, P.S.; Dow, N.W.; Fayad, R.; Hauke, C.E.; Rosko, M.C.; Danilov, E.O.; Blakemore, D.C.; Dechert-Schmitt, A.-M.; Knauber, T.; et al. A Unified Approach to Decarboxylative Halogenation of (Hetero)Aryl Carboxylic Acids. J. Am. Chem. Soc. 2022, 144, 8296–8305. [Google Scholar] [CrossRef]
- Lovett, G.H.; Chen, S.; Xue, X.-S.; Houk, K.N.; MacMillan, D.W.C. Open-Shell Fluorination of Alkyl Bromides: Unexpected Selectivity in A Silyl Radical-Mediated Chain Process. J. Am. Chem. Soc. 2019, 141, 20031–20036. [Google Scholar] [CrossRef]
- Liu, W.; Lavagnino, M.N.; Gould, C.A.; Alcázar, J.; MacMillan, D.W.C. A Biomimetic SH2 Cross-coupling Mechanism for Quaternary sp3-carbon Formation. Science 2021, 374, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, M.K.; Kauppi, A.M.; Olsson, I.-M.; Linusson, A.; Elofsson, M. Design, Synthesis, And Multivariate Quantitative Structure−Activity Relationship of SalicylanilidesPotent Inhibitors of Type III Secretion in Yersinia. J. Med. Chem. 2007, 50, 6177–6188. [Google Scholar] [CrossRef]
- Mihara, J.; Araki, K.; Mori, T.; Murata, T.; Yoneta, Y.; Watanabe, Y.; Shimojo, E.; Ichihara, T.; Ataka, M.; Shibuya, K.; et al. Pesticidal Carboxamides. WO 2011/018170, 17 February 2011. [Google Scholar]
- Yoneda, T.; Yoshida, K.; Tazawa, Y.; Kani, T.; Cho, Y.; Noujima, A.; Murai, Y. N-(4-Pyridyl)Benzamide Compound or Salt Thereof, and Pest Control Agent Containing Compound or Salt Thereof as Active Ingredient. WO 2017/222037, 28 December 2017. [Google Scholar]
- Costa, P.C.S.; Barsottini, M.R.O.; Vieira, M.L.L.; Pires, B.A.; Evangelista, J.S.; Zeri, A.C.M.; Nascimento, A.F.Z.; Silva, J.S.; Carazzolle, M.F.; Pereira, G.A.G.; et al. N-Phenylbenzamide Derivatives as Alternative Oxidase Inhibitors: Synthesis, Molecular Properties, 1H-STD NMR, and QSAR. J. Mol. Struct. 2020, 1208, 127903. [Google Scholar] [CrossRef]
- Chao, J.; Enyedy, I.; Van Vloten, K.; Marcotte, D.; Guertin, K.; Hutchings, R.; Powell, N.; Jones, H.; Bohnert, T.; Peng, C.-C.; et al. Discovery of Biaryl Carboxylamides as Potent RORγ Inverse Agonists. Bioorg. Med. Chem. Lett. 2015, 25, 2991–2997. [Google Scholar] [CrossRef] [PubMed]
- Janicka, M.; Tyihak, E.; Moricz, A.M.; Oscik-Mendyk, B. Crucial Role of Formaldehyde and Its reaction Products in the Antiproliferative Activity of Some Potential Pesticides. JPC-J. Planar. Chromat. 2008, 21, 161–166. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
| entry | promoter | 2a | 2b |
| yield b (%) | yield b (%) | ||
| 1 c | none | <5 | <5 |
| 2 | Pd/C | <5 | <5 |
| 3 | Pd(OAc)2 | 56 | <5 |
| 4 | Pd(TFA)2 | 42 | <5 |
| 5 | PdCl2 | <5 | <5 |
| 6 d | Pd(OAc)2 | 72 | <5 |
| 7 | Co(acac)2 | <5 | <5 |
| 8 | Cu(OAc)2 | <5 | <5 |
| 9 e | HFIP | <5 | 68 |
| 10 | Cp*Co(CO)I2 | <5 | <5 |
| 11 | Ni(TFA)2 | <5 | <5 |
| 12 f | HFIP | <5 | 83 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; He, Q.; Lv, X.; Zhang, N.; Yan, W.; Sun, J.; Zhuang, L. Switchable Site-Selective Benzanilide C(sp2)-H Bromination via Promoter Regulation. Molecules 2024, 29, 2861. https://doi.org/10.3390/molecules29122861
Sun Y, He Q, Lv X, Zhang N, Yan W, Sun J, Zhuang L. Switchable Site-Selective Benzanilide C(sp2)-H Bromination via Promoter Regulation. Molecules. 2024; 29(12):2861. https://doi.org/10.3390/molecules29122861
Chicago/Turabian StyleSun, Yonghui, Qiyu He, Xucheng Lv, Naizhen Zhang, Wei Yan, Jianghui Sun, and Lili Zhuang. 2024. "Switchable Site-Selective Benzanilide C(sp2)-H Bromination via Promoter Regulation" Molecules 29, no. 12: 2861. https://doi.org/10.3390/molecules29122861