
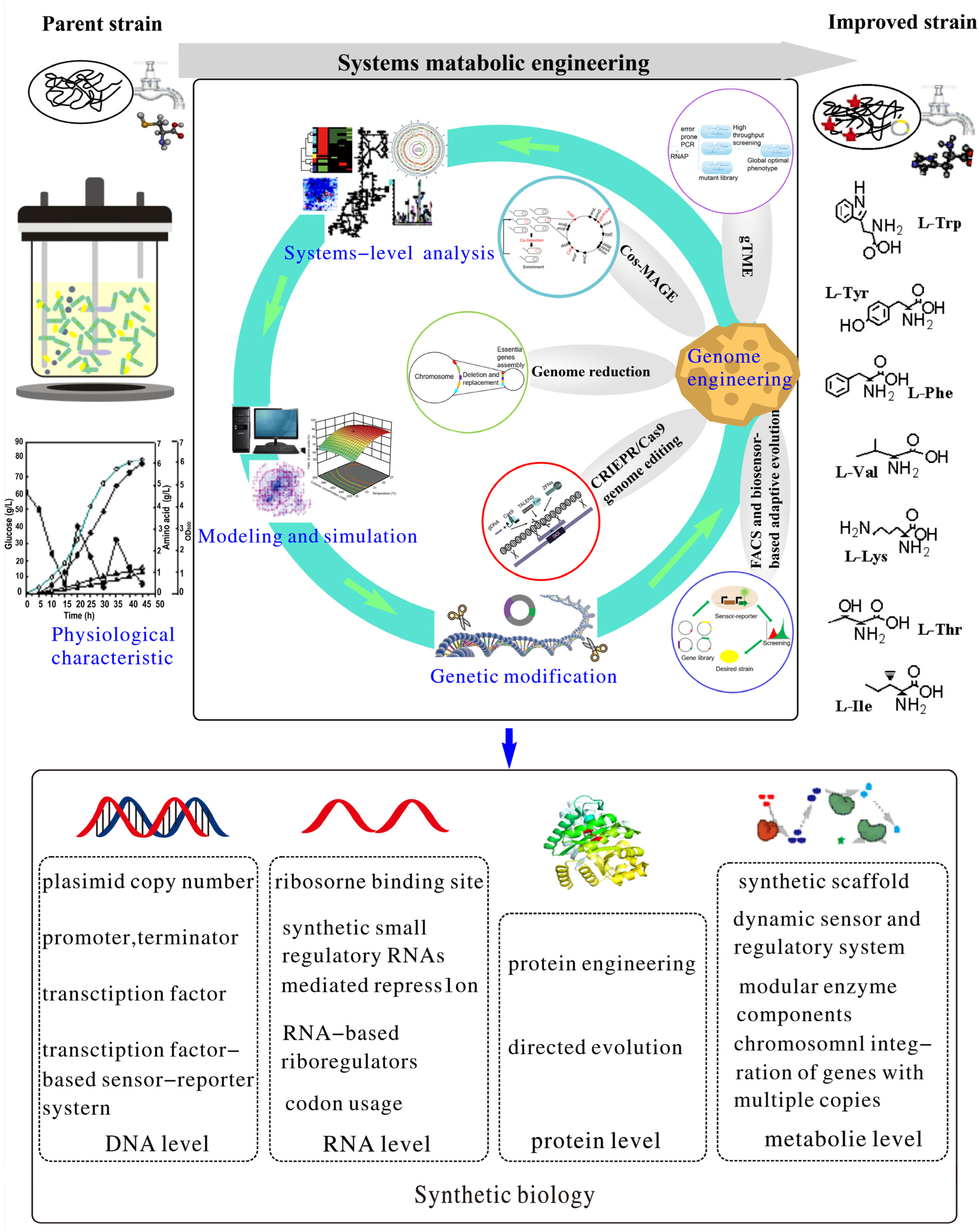
Recent Advances in Metabolic Engineering for the Biosynthesis of Phosphoenol Pyruvate–Oxaloacetate–Pyruvate-Derived Amino Acids

Abstract
:1. Introduction
2. L-Tryptophan
2.1. Engineering the Common Pathway and L-Trp Branch for Enhanced L-Trp Production
2.2. Engineering the POP Node and PPP to Improve Carbon Flux for PEP and E4P Formation
2.3. Combined Transport Engineering and By-Product Elimination
2.4. Combined High-Throughput Screening (HTS) and Multidimensional Regulation
3. L-Tyrosine
3.1. L-Tyr Biosynthetic Pathway Modification, Competition Pathway Elimination, and Cofactor Availability Improvement
3.2. Combined Regulation of the PPP and POP Node
3.3. Global Metabolic Engineering for Enhanced L-Tyr Formation
3.4. Development of Glucose–Xylose–Phenolic (GXP) System for L-Tyr Production from Lignocellulosic Biomass
4. L-Phenylalanine
4.1. Engineering to Improve Carbon Flux for L-Phe Production
4.2. Engineering to Weaken Competing Branches and By-Product Formation
4.3. Engineering to Strengthen Precursor Pathways
4.4. Modular Engineering and Regulator Redesign for Enhanced L-Phe Production
4.5. Adaptive Laboratory Evolution for Enhanced L-Phe Production
5. L-Valine
5.1. Engineering to Strengthen L-Val Biosynthetic Pathways and Eliminate Competition Pathways
5.2. Combined Regulation of the POP Node and Cofactor Supply
5.3. Engineering to Eliminate By-Product Formation
5.4. L-Val Biosynthetic Pathway with Transporter Engineering Enhancement and the Carbon Flux Loss Decrement
6. L-Lysine
6.1. Engineering the L-Lys Biosynthetic Route for Enhanced L-Lys Generation
6.2. Engineering to Regulate the POP Node and Block By-Product Formation
6.3. Engineering to Improve Cofactor NADPH Provision for L-Lys Generation
6.4. Engineering of C. glutamicum for L-Lys Production
6.5. Engineering to Enhance Multiple Carbon Catabolism in C. glutamicum
6.6. Establishment of Artificial Rare Cryptosystem for Mutating and Screening High-Yielding Strains
7. L-Threonine
7.1. L-Thr Biosynthetic Pathway Modification and By-Product Elimination
7.2. Transport Engineering for Enhanced L-Thr Production
7.3. Engineering to Dynamically Regulate the POP Node and Operon
7.4. Systems Metabolic Engineering
8. L-Isoleucine
8.1. Engineering to Enhance L-Ile Biosynthesis and Weaken Competing Branches
8.2. Engineering to Enhance NADPH Supply for L-Ile Production
8.3. Engineering to Improve Secretion for L-Ile Production
9. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cuvas-Limon, R.B.; Nobre, C.; Cruz, M.; Rodriguez-Jasso, R.M.; Ruíz, H.A.; Loredo-Treviño, A.; Texeira, J.A.; Belmares, R. Spontaneously fermented traditional beverages as a source of bioactive compounds: An overview. Crit. Rev. Food Sci. Nutr. 2021, 61, 2984–3006. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, L.; Tian, K.; Kumar, A.; Singh, S.; Prior, B.A.; Wang, Z. Metabolic engineering of Escherichia coli: A sustainable industrial platform for bio-based chemical production. Biotechnol. Adv. 2013, 31, 1200–1223. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.J.; Son, J.; Lim, H.J.; Lim, S.H.; Park, S.J. Valorization of lignocellulosic biomass for polyhydroxyalkanoate production: Status and perspectives. Bioresour. Technol. 2022, 360, 127575. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Kumar, V. Microbial conversion of waste biomass into bioethanol: Current challenges and future prospects. Biomass Convers. Biorefinery 2021, 13, 6419–6456. [Google Scholar] [CrossRef]
- Gu, P.F.; Su, T.Y.; Qi, Q.S. Novel technologies provide more engineering strategies for amino acid-producing microorganisms. Appl. Microbiol. Biotechnol. 2016, 100, 2097–2105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Q.; Ren, X.N.; Liang, X.H.; Wang, Y.Q.; Feng, D.X.; Zhang, Y.J.; Xian, M.; Zou, H.B. Improving the microbial production of amino acids: From conventional approaches to recent trends. Biotechnol. Bioprocess. Eng. 2021, 26, 708–727. [Google Scholar] [CrossRef]
- D’Este, M.; Alvarado-Morales, M.; Angelidaki, I. Amino acids production focusing on fermentation technologies—A review. Biotechnol. Adv. 2018, 36, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zheng, B.; Chen, Z.; Huo, Y.X. Metabolic engineering of Corynebacterium glutamicum for producing branched chain amino acids. Microb. Cell Fact. 2021, 20, 230. [Google Scholar] [CrossRef]
- Shimizu, K.; Matsuoka, Y. Feedback regulation and coordination of the main metabolism for bacterial growth and metabolic engineering for amino acid fermentation. Biotechnol. Adv. 2022, 55, 107887. [Google Scholar] [CrossRef]
- Huergo, L.F.; Araújo, G.A.T.; Santos, A.S.R.; Gerhardt, E.C.M.; Pedrosa, F.O.; Souza, E.M.; Forchhammer, K. The NADP-dependent malic enzyme MaeB is a central metabolic hub controlled by the acetyl-CoA to CoASH ratio. BBA-Proteins Proteom. 2020, 1868, 140462. [Google Scholar] [CrossRef]
- Krause, J.P.; Polen, T.; Youn, J.W.; Emer, D.; Eikmanns, B.J.; Wendisch, V.F. Regulation of the malic enzyme gene malE by the transcriptional regulator MalR in Corynebacterium glutamicum. J. Biotechnol. 2012, 159, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Kortmann, M.; Baumgart, M.; Bott, M. Pyruvate carboxylase from Corynebacterium glutamicum: Purification and characterization. Appl. Microbiol. Biot. 2019, 103, 6571–6580. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Liang, H.Y.; Han, C.; Zhou, P.; Xing, Z.W.; Chen, Q.Q.; Liu, Y.Y.; Xie, G.A.; Xie, R.F. Engineering of global transcription factor FruR to redirect the carbon flow in Escherichia coli for enhancing l-phenylalanine biosynthesis. Microb. Cell Fact. 2022, 21, 222. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M. Towards bacterial strains overproducing L-tryptophan and other aromatics by metabolic engineering. Appl. Microbiol. Biotechnol. 2006, 69, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Lütke-Eversloh, T.; Santos, C.N.; Stephanopoulos, G. Perspectives of biotechnological production of L-tyrosine and its applications. Appl. Microbiol. Biotechnol. 2007, 77, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, G.A. From scratch to value: Engineering Escherichia coli wild type cells to the production of L-phenylalanine and other fine chemicals derived from chorismate. Appl. Microbiol. Biotechnol. 2007, 75, 739–749. [Google Scholar] [CrossRef]
- Park, J.H.; Oh, J.E.; Lee, K.H.; Kim, J.Y.; Lee, S.Y. Rational design of Escherichia coli for L-isoleucine production. ACS Synth. Biol. 2012, 1, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Oldiges, M.; Eikmanns, B.J.; Blombach, B. Application of metabolic engineering for the biotechnological production of L-valine. Appl. Microbiol. Biotechnol. 2014, 98, 5859–5870. [Google Scholar] [CrossRef] [PubMed]
- Master, P.B.Z.; Macedo, R.C.O. Effects of dietary supplementation in sport and exercise: A review of evidence on milk proteins and amino acids. Crit. Rev. Food Sci. Nutr. 2021, 61, 1225–1239. [Google Scholar] [CrossRef]
- Zhang, K.; Li, H.; Cho, K.M.; Liao, J.C. Expanding metabolism for total biosynthesis of the nonnatural amino acid L-homoalanine. Proc. Natl. Acad. Sci. USA 2010, 107, 6234–6239. [Google Scholar] [CrossRef]
- Shih, I.L.; Shen, M.H.; Van, Y.T. Microbial synthesis of poly(epsilon-lysine) and its various applications. Bioresour. Technol. 2006, 97, 1148–1159. [Google Scholar] [CrossRef]
- Becker, J.; Wittmann, C. Bio-based production of chemicals, materials and fuels Corynebacterium glutamicum as versatile cell factory. Curr. Opin. Biotechnol. 2012, 23, 631–640. [Google Scholar] [CrossRef]
- Yang, J.; Yang, S. Comparative analysis of Corynebacterium glutamicum genomes: A new perspective for the industrial production of amino acids. BMC Genom. 2017, 18, 940. [Google Scholar] [CrossRef] [PubMed]
- Tyo, K.E.; Alper, H.S.; Stephanopoulos, G.N. Expanding the metabolic engineering toolbox: More options to engineer cells. Trends Biotechnol. 2007, 25, 132–137. [Google Scholar] [CrossRef]
- Adrio, J.L.; Demain, A.L. Recombinant organisms for production of industrial products. Bioeng. Bugs 2010, 1, 116–131. [Google Scholar] [CrossRef]
- Ferrer, L.; Elsaraf, M.; Mindt, M.; Wendisch, V.F. L-serine biosensor-controlled fermentative production of L-tryptophan derivatives by Corynebacterium glutamicum. Biology 2022, 11, 744. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, S.; Wu, D.; Wu, J.; Chen, J. Effect of mufti-gene knockout of L-tryptophan transport system on L-tryptophan production in Escherichia coli. Chin. J. Biotech. 2011, 27, 1765–1772. [Google Scholar]
- Shen, T.; Liu, Q.; Xie, X.; Xu, Q.; Chen, N. Improved production of tryptophan in genetically engineered Escherichia coli with TktA and PpsA overexpression. J. Biomed. Biotechnol. 2012, 2012, 605219. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, B.B.; Xu, J.Z.; Zhang, W.G. Engineering of shikimate pathway and terminal branch for efficient production of L-tryptophan in Escherichia coli. Int. J. Mol. Sci. 2023, 24, 11866. [Google Scholar] [CrossRef]
- Du, L.; Zhang, Z.; Xu, Q.; Chen, N. New strategy for removing acetic acid as a by-product during L-tryptophan production. Biotechnol. Biotechnol. Eq. 2019, 33, 1471–1480. [Google Scholar] [CrossRef]
- Xiong, B.; Zhu, Y.; Tian, D.; Jiang, S.; Fan, X.; Ma, Q.; Wu, H.; Xie, X. Flux redistribution of central carbon metabolism for efficient production of l-tryptophan in Escherichia coli. Biotechnol. Bioeng. 2021, 118, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Yang, F.; Li, F.; Liang, Q.; Qi, Q. Knocking out analysis of tryptophan permeases in Escherichia coli for improving L-tryptophan production. Appl. Microbiol. Biotechnol. 2013, 97, 6677–6683. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhang, Z.; Xu, Q.; Chen, N. Central metabolic pathway modification to improve L-tryptophan production in Escherichia coli. Bioengineered 2019, 10, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cheng, L.K.; Wang, J.; Liu, Q.; Shen, T.; Chen, N. Genetic engineering of Escherichia coli to enhance production of L-tryptophan. Appl. Microbiol. Biotechnol. 2013, 97, 7587–7596. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Pan, X.W.; Yang, T.J.; You, J.J.; Zhu, R.S.; Yang, T.W.; Zhang, X.; Xu, M.J.; Rao, Z.M. Multidimensional engineering of Escherichia coli for efficient synthesis of L-tryptophan. Bioresour. Technol. 2023, 386, 129475. [Google Scholar] [CrossRef] [PubMed]
- Lütke-Eversloh, T.; Stephanopoulos, G. L-tyrosine production by deregulated strains of Escherichia coli. Appl. Microbiol. Biotechnol. 2007, 75, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, R.; Zolandz, R.R.; Green, D.A.; Kraynie, D.F. L-tyrosine production by recombinant Escherichia coli: Fermentation optimization and recovery. Biotechnol. Bioeng. 2008, 99, 741–752. [Google Scholar] [CrossRef]
- Olson, M.M.; Templeton, L.J.; Suh, W.; Youderian, P.; Sariaslani, F.S.; Gatenby, A.A.; Van Dyk, T.K. Production of tyrosine from sucrose or glucose achieved by rapid genetic changes to phenylalanine-producing Escherichia coli strains. Appl. Microbiol. Biotechnol. 2007, 74, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lin, Y.; Yuan, Q.; Yan, Y. Production of tyrosine through phenylalanine hydroxylation bypasses the intrinsic feedback inhibition in Escherichia coli. J. Ind. Microbiol. Biotechnol. 2015, 42, 655–659. [Google Scholar] [CrossRef]
- Takai, A.; Nishi, R.; Joe, Y.; Ito, H. L-Tyrosine-Producing Bacterium and a Method for Producing L-Tyrosine. U.S. Patent 7,482,140, 4 May 2010. [Google Scholar]
- Kim, B.; Binkley, R.; Kim, H.U.; Lee, S.Y. Metabolic engineering of Escherichia coli for the enhanced production of L-tyrosine. Biotechnol. Bioeng. 2018, 115, 2554–2564. [Google Scholar] [CrossRef]
- Li, G.; Chen, Z.; Chen, N.; Xu, Q. Enhancing the efficiency of L-tyrosine by repeated batch fermentation. Bioengineered 2020, 11, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Ping, J.R.; Wang, L.; Qin, Z.J.; Zhou, Z.M.; Zhou, J.W. Synergetic engineering of Escherichia coli for efficient production of L-tyrosine. Synth. Syst. Biotechnol. 2023, 8, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Na, D.; Yoo, S.M.; Chung, H.; Park, H.; Park, J.H.; Lee, S.Y. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat. Biotechnol. 2013, 31, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liao, X.; Liu, L.; Wang, T.; Du, G.; Chen, J. Enhanced L-phenylalanine production by recombinant Escherichia coli BR-42 (pAP-B03) resistant to bacteriophage BP-1 via a two-stage feeding approach. J. Ind. Microbiol. Biotechnol. 2011, 38, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, Y.; Zhao, S.; Sun, J.; Jin, Z.; Zhang, D. Application of dynamic regulation to increase L-phenylalanine production in Escherichia coli. J. Microbiol. Biotechnol. 2019, 29, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Doroshenko, V.G.; Shakulov, R.S.; Kazakova, S.M.; Kivero, A.D.; Yampolskaya, T.A.; Mashko, S.V. Construction of an L-phenylalanine-producing tyrosine-prototrophic Escherichia coli strain using tyrA ssrA-like tagged alleles. Biotechnol. Lett. 2010, 32, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Ojima, Y.; Komaki, M.; Nishioka, M.; Iwatani, S.; Tsujimoto, N.; Taya, M. Introduction of a stress-responsive gene, yggG, enhances the yield of L-phenylalanine with decreased acetic acid production in a recombinant Escherichia coli. Biotechnol. Lett. 2009, 31, 525–530. [Google Scholar] [CrossRef]
- Wu, Y.Q.; Jiang, P.H.; Fan, C.S.; Wang, J.G.; Shang, L.; Huang, W.D. Co-expression of five genes in E. coli for L-phenylalanine in Brevibacterium flavum. World J. Gastroenterol. 2003, 9, 342–346. [Google Scholar] [CrossRef]
- Báez-Viveros, J.L.; Osuna, J.; Hernández-Chávez, G.; Soberón, X.; Bolívar, F.; Gosset, G. Metabolic engineering and protein directed evolution increase the yield of L-phenylalanine synthesized from glucose in Escherichia coli. Biotechnol. Bioeng. 2004, 87, 516–524. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, J.; Kang, Z.; Du, G.; Chen, J. Rational engineering of multiple module pathways for the production of L-phenylalanine in Corynebacterium glutamicum. J. Ind. Microbiol. Biotechnol. 2015, 42, 787–797. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, Y.; Ding, D.; Wen, J.; Zhu, B.; Zhang, D. Genetic engineering of Escherichia coli to improve L-phenylalanine production. BMC Biotechnol. 2018, 18, 5. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Saini, D.; Guleria, R.; Mukherjee, K.J. Designing an Escherichia coli Strain for Phenylalanine Overproduction by Metabolic Engineering. Mol. Biotechnol. 2017, 59, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.G.; Qiu, C.; Chen, C.H.; Gao, C.; Wei, W.Q.; Song, W.; Wu, J.; Liu, L.M.; Chen, X.L. Metabolic engineering of Escherichia coli for high-level production of l-phenylalanine. J. Agric. Food. Chem. 2024, 72, 11029–11040. [Google Scholar] [CrossRef] [PubMed]
- Elisáková, V.; Pátek, M.; Holátko, J.; Nesvera, J.; Leyval, D.; Goergen, J.L.; Delaunay, S. Feedback-resistant acetohydroxy acid synthase increases valine production in Corynebacterium glutamicum. Appl. Environ. Microbiol. 2005, 71, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, J.; Schwentner, A.; Brunnenkan, B.; Gabris, C.; Grimm, S.; Gerstmeir, R.; Takors, R.; Eikmanns, B.J.; Blombach, B. Platform engineering of Corynebacterium glutamicum with reduced pyruvate dehydrogenase complex activity for improved production of L-lysine, L-valine, and 2-ketoisovalerate. Appl. Environ. Microbiol. 2013, 79, 5566–5575. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Uematsu, K.; Natsuma, Y.; Suda, M.; Hiraga, K.; Jojima, T.; Inui, M.; Yukawa, H. Improvement of the redox balance increases L-valine production by Corynebacterium glutamicum under oxygen deprivation conditions. Appl. Environ. Microbiol. 2012, 78, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Jang, Y.S.; Lee, J.W.; Lee, S.Y. Escherichia coli W as a new platform strain for the enhanced production of L-valine by systems metabolic engineering. Biotechnol. Bioeng. 2011, 108, 1140–1147. [Google Scholar] [CrossRef] [PubMed]
- Schwentner, A.; Feith, A.; Münch, E.; Busche, T.; Rückert, C.; Kalinowski, J.; Takors, R.; Blombach, B. Metabolic engineering to guide evolution—Creating a novel mode for L-valine production with Corynebacterium glutamicum. Metab. Eng. 2018, 47, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Zhao, Y.; Liu, T.; Wang, T.; Han, C.; Mao, Q.; Chen, N.; Li, Y. Effects of heterologous pyruvate carboxylase expression on synthesis of L-threonine in Escherichia coli. Lect. Notes Electr. Eng. 2018, 444, 133–143. [Google Scholar]
- Xu, J.Z.; Han, M.; Ren, X.D.; Zhang, W.G. Modification of aspartokinase III and dihydrodipicolinate synthetase increases the production of L-lysine in Escherichia coli. Biochem. Eng. J. 2016, 114, 79–86. [Google Scholar] [CrossRef]
- Becker, J.; Zelder, O.; Häfner, S.; Schröder, H.; Wittmann, C. From zero to hero-design-based systems metabolic engineering of Corynebacterium glutamicum for L-lysine production. Metab. Eng. 2011, 13, 159–168. [Google Scholar] [CrossRef]
- Yanase, M.; Aikoh, T.; Sawada, K.; Ogura, K.; Hagiwara, T.; Imai, K.; Wada, M.; Yokota, A. Pyruvate kinase deletion as an effective phenotype to enhance lysine production in Corynebacterium glutamicum ATCC13032: Redirecting the carbon flow to a precursor metabolite. J. Biosci. Bioeng. 2016, 122, 160–167. [Google Scholar] [CrossRef]
- Xu, J.Z.; Wu, Z.H.; Gao, S.J.; Zhang, W. Rational modification of tricarboxylic acid cycle for improving L-lysine production in Corynebacterium glutamicum. Microb. Cell Fact. 2018, 17, 105. [Google Scholar] [CrossRef]
- Xu, J.Z.; Han, M.; Zhang, J.L.; Guo, Y.F.; Qian, H.; Zhang, W.G. Improvement of L-lysine production combines with minimization of by-products synthesis in Corynebacterium glutamicum. J. Chem. Technol. Biotechnol. 2014, 89, 1924–1933. [Google Scholar] [CrossRef]
- Becker, J.; Klopprogge, C.; Herold, A.; Zelder, O.; Bolten, C.J.; Wittmann, C. Metabolic flux engineering of L-lysine production in Corynebacterium glutamicum—Over expression and modification of G6P dehydrogenase. J. Biotechnol. 2007, 132, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Takeno, S.; Hori, K.; Ohtani, S.; Mimura, A.; Mitsuhashi, S.; Ikeda, M. L-Lysine production independent of the oxidative pentose phosphate pathway by Corynebacterium glutamicum with the Streptococcus mutans gapN gene. Metab. Eng. 2016, 37, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Z.; Yang, H.K.; Liu, L.M.; Wang, Y.Y.; Zhang, W.G. Rational modification of Corynebacterium glutamicum dihydrodipicolinate reductase to switch the nucleotide-cofactor specificity for increasing L-lysine production. Biotechnol. Bioeng. 2018, 115, 1764–1777. [Google Scholar] [CrossRef]
- Pérez-García, F.; Peters-Wendisch, P.; Wendisch, V.F. Engineering Corynebacterium glutamicum for fast production of L-lysine and L-pipecolic acid. Appl. Microbiol. Biotechnol. 2016, 100, 8075–8090. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Z.; Yu, H.B.; Han, M.; Liu, L.M.; Zhang, W.G. Metabolic engineering of glucose uptake systems in Corynebacterium glutamicum for improving the efficiency of L-lysine production. J. Ind. Microbiol. Biotechnol. 2019, 46, 937–949. [Google Scholar] [CrossRef]
- Tateno, T.; Fukuda, H.; Kondo, A. Production of L-Lysine from starch by Corynebacterium glutamicum displaying alpha-amylase on its cell surface. Appl. Microbiol. Biotechnol. 2007, 74, 1213–1220. [Google Scholar] [CrossRef]
- Liu, H.; Yang, C.P.; Yang, L.; Wang, R.M.; Li, P.W.; Du, B.W.; Li, N.; Wang, J.Q. Screening l-lysine-overproducing Escherichia coli using artificial rare codons and a rare codon-rich marker. Fermentation 2023, 9, 899. [Google Scholar] [CrossRef]
- Wang, S.M.; Chen, X.M.; Jin, X.; Gu, F.; Jiang, W.; Qi, Q.S.; Liang, Q.F. Creating polyploid Escherichia coli and its application in efficient L-threonine production. Adv. Sci. 2023, 10, e2302417. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Wu, Z.; Han, S.; Lin, Y.; Zheng, S.P. Construction of recombinant Corynebacterium glutamicum for L-threonine production. Biotechnol. Bioprocess. Eng. 2012, 17, 16–21. [Google Scholar] [CrossRef]
- Wei, L.; Xu, N.; Wang, Y.; Zhou, W.; Han, G.; Ma, Y.; Liu, J. Promoter library-based module combination (PLMC) technology for optimization of threonine biosynthesis in Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2018, 102, 4117–4130. [Google Scholar] [CrossRef]
- Yang, J.; Fang, Y.; Wang, J.; Wang, C.; Zhao, L.; Wang, X. Deletion of regulator-encoding genes fadR, fabR and iclR to increase L-threonine production in Escherichia coli. Appl. Microbiol. Biotechnol. 2019, 103, 4549–4564. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Fang, Y.; Ding, Z.; Zhang, S.; Wang, X. Developing an L-threonine-producing strain from wild-type Escherichia coli by modifying the glucose uptake, glyoxylate shunt, and L-threonine biosynthetic pathway. Biotechnol. Appl. Biochem. 2019, 66, 962–976. [Google Scholar] [CrossRef]
- Xie, X.; Liang, Y.; Liu, H.; Liu, Y.; Xu, Q.; Zhang, C.; Chen, N. Modification of glycolysis and its effect on the production of L-threonine in Escherichia coli. J. Ind. Microbiol. Biotechnol. 2014, 41, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lu, Y.; Yang, J.; Fang, Y.; Zhu, L.; Ding, Z.; Wang, C.; Ma, W.; Hu, X.; Wang, X. Expression regulation of multiple key genes to improve L-threonine in Escherichia coli. Microb. Cell Fact. 2020, 19, 46. [Google Scholar] [CrossRef]
- Livshits, V.A.; Zakataeva, N.P.; Aleshin, V.V.; Vitushkina, M.V. Identification and characterization of the new gene rhtA involved in threonine and homoserine efflux in Escherichia coli. Res. Microbiol. 2003, 154, 123–135. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, J.; Ma, W.; Yang, J.; Zhang, H.; Zhao, L.; Chen, S.; Zhang, S.; Hu, X.; Li, Y.; et al. Rebalancing microbial carbon distribution for L-threonine maximization using a thermal switch system. Metab. Eng. 2020, 61, 33–46. [Google Scholar] [CrossRef]
- Hao, R.X.; Wang, S.M.; Jin, X.; Yang, X.Y.; Qi, Q.S.; Liang, Q.F. Dynamic and balanced regulation of the thrABC operon gene for efficient synthesis of L-threonine. Front. Bioeng. Biotechnol. 2023, 11, 1118948. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Sung, B.H.; Kim, M.S.; Blattner, F.R.; Yoon, B.H.; Kim, J.H.; Kim, S.C. Metabolic engineering of a reduced-genome strain of Escherichia coli for L-threonine production. Microb. Cell Fact. 2009, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Li, K.; Huan, X.; Wang, X. Expression of NAD(H) kinase and glucose-6-phosphate dehydrogenase improve NADPH supply and L-isoleucine biosynthesis in Corynebacterium glutamicum ssp. lactofermentum. Appl. Biochem. Biotechnol. 2013, 171, 504–521. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zhao, J.; Chen, C.; Hu, X.Q.; Wang, X.Y. Enhancing the carbon flux and NADPH supply to increase L-isoleucine production in Corynebacterium glutamicum. Biotechnol. Bioprocess Eng. 2014, 19, 132–142. [Google Scholar] [CrossRef]
- Vogt, M.; Krumbach, K.; Bang, W.G.; van Ooyen, J.; Noack, S.; Klein, B.; Bott, M.; Eggeling, L. The contest for precursors: Channelling L-isoleucine synthesis in Corynebacterium glutamicum without byproduct formation. Appl. Microbiol. Biotechnol. 2015, 99, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Ye, T.; Liu, F.M.; Zhang, X.J.; Wang, J.P.; Wei, X.B.; Neo, Y.P.; Liu, H.Y.; Fang, H.T. Strategies to enhance l-isoleucine synthesis by modifying the threonine metabolism pathway in Escherichia coli. ACS Omega 2024, 9, 10276–10285. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Xu, L.; Shi, J.; Xu, Q.; Chen, N. Effect of transport proteins on L-isoleucine production with the L-isoleucine-producing strain Corynebacterium glutamicum YILW. J. Ind. Microbiol. Biotechnol. 2012, 39, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Duan, X.; Wu, J. L-Tryptophan production in Escherichia coli improved by weakening the Pta-AckA pathway. PLoS ONE 2016, 11, e0158200. [Google Scholar] [CrossRef]
- Bongaerts, J.; Krämer, M.; Müller, U.; Raeven, L.; Wubbolts, M. Metabolic engineering for microbial production of aromatic amino acids and derived compounds. Metab. Eng. 2001, 3, 289–300. [Google Scholar] [CrossRef]
- Doroshenko, V.; Airich, L.; Vitushkina, M.; Kolokolova, A.; Livshits, V.; Mashko, S. YddG from Escherichia coli promotes export of aromatic amino acids. FEMS Microbiol. Lett. 2007, 275, 312–318. [Google Scholar] [CrossRef]
- Mindt, M.; Ferrer, L.; Bosch, D.; Cankar, K.; Wendisch, V.F. De novo tryptophanase-based indole production by metabolically engineered Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2023, 107, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Schoppel, K.; Trachtmann, N.; Korzin, E.J.; Tzanavari, A.; Sprenger, G.A.; Weuster-Botz, D. Metabolic control analysis enables rational improvement of E. coli L-tryptophan producers but methylglyoxal formation limits glycerol-based production. Microb. Cell Fact. 2022, 21, 201. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Y.; Ding, D.; Cong, L.; Zhang, D. Rational design and analysis of an Escherichia coli strain for high-efficiency tryptophan production. J. Ind. Microbiol. Biotechnol. 2018, 45, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Dodge, T.C.; Gerstner, J.M. Optimization of the glucose feed rate profile for the production of tryptophan from recombinant E. coli. J. Chem. Technol. Biotechnol. 2002, 77, 1238–1245. [Google Scholar] [CrossRef]
- Chen, L.; Chen, M.; Ma, C.; Zeng, A.P. Discovery of feed-forward regulation in L-tryptophan biosynthesis and its use in metabolic engineering of E. coli for efficient tryptophan bioproduction. Metab. Eng. 2018, 47, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.J.; Zou, C.; Zhu, Y.X.; Dai, J.; Chen, S.; Wu, D.; Wu, J.; Chen, J. Development of L-tryptophan production strains by defined genetic modification in Escherichia coli. J. Ind. Microbiol. Biotechnol. 2011, 38, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, R.; Ikeda, M. Hyperproduction of tryptophan in Corynebacterium glutamicum by pathway engineering. Nat. Biotechnol. 1993, 11, 921–925. [Google Scholar] [CrossRef]
- Liu, L.; Chen, S.; Wu, J. Phosphoenolpyruvate:glucose phosphotransferase system modification increases the conversion rate during L-tryptophan production in Escherichia coli. J. Ind. Microbiol. Biotechnol. 2017, 44, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhao, J.; Mao, X. Effect of PTS modifications on L-tryptophan production in Escherichia coli. Chin. J. Biotechnol. 2017, 33, 1877–1882. [Google Scholar]
- Zhao, Z.; Chen, S.; Wu, D.; Wu, J.; Chen, J. Effect of gene knockouts of l-tryptophan uptake system on the production of l-tryptophan in Escherichia coli. Process Biochem. 2012, 47, 340–344. [Google Scholar] [CrossRef]
- Liu, Q.; Cheng, Y.; Xie, X.; Xu, Q.; Chen, N. Modification of tryptophan transport system and its impact on production of L-tryptophan in Escherichia coli. Bioresour. Technol. 2012, 114, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Duan, X.; Wu, J. Modulating the direction of carbon flow in Escherichia coli to improve l-tryptophan production by inactivating the global regulator FruR. J. Biotechnol. 2016, 231, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Bai, F.; Chen, N.; Bai, G. Gene modification of the acetate biosynthesis pathway in Escherichia coli and implementation of the cell recycling technology to increase L-tryptophan production. PLoS ONE 2017, 12, e0179240. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Zhang, B.; Cheng, L.; Wang, J.S.; Zhang, S.L.; Fu, S.J.; Xiao, Y.Q.; Liu, H.Y. Gene modification of Escherichia coli and incorporation of process control to decrease acetate accumulation and increase L-tryptophan production. ANN Microbiol. 2017, 67, 567–576. [Google Scholar] [CrossRef]
- Gao, J.S.; Du, M.H.; Zhao, J.H.; Xu, N.; Du, H.M.; Ju, J.S.; Wei, L.; Liu, J. Design of a genetically encoded biosensor to establish a high-throughput screening platform for L-cysteine overproduction. Metab. Eng. 2022, 73, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.N.; Zhou, S.H.; Deng, Y. Transcription-factor-based biosensor engineering for applications in synthetic biology. ACS Synth. Biol. 2021, 10, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.B.; Zeng, A.P. Exploring lysine riboswitch for metabolic flux control and improvement of L-lysine synthesis in Corynebacterium glutamicum. ACS Synth. Biol. 2015, 4, 729–734. [Google Scholar] [CrossRef]
- Chávez-Béjar, M.I.; Báez-Viveros, J.L.; Martínez, A.; Bolívar, F.; Gosset, G. Biotechnological production of L-tyrosine and derived compounds. Process Biochem. 2012, 47, 1017–1026. [Google Scholar] [CrossRef]
- Chávez-Béjar, M.I.; Lara, A.R.; López, H.; Hernández-Chávez, G.; Martinez, A.; Ramírez, O.T.; Bolívar, F.; Gosset, G. Metabolic engineering of Escherichia coli for L-tyrosine production by expression of genes coding for the chorismate mutase domain of the native chorismate mutase-prephenate dehydratase and a cyclohexadienyl dehydrogenase from Zymomonas mobilis. Appl. Environ. Microbiol. 2008, 74, 3284–3290. [Google Scholar] [CrossRef]
- Lütke-Eversloh, T.; Stephanopoulos, G. Combinatorial pathway analysis for improved L-tyrosine production in Escherichia coli: Identification of enzymatic bottlenecks by systematic gene overexpression. Metab. Eng. 2008, 10, 69–77. [Google Scholar] [CrossRef]
- Gold, N.D.; Gowen, C.M.; Lussier, F.X.; Cautha, S.C.; Mahadevan, R.; Martin, V.J. Metabolic engineering of a tyrosine-overproducing yeast platform using targeted metabolomics. Microb. Cell Fact. 2015, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Min, B.E.; Hwang, H.G.; Seo, S.W.; Jung, G.Y. Pathway optimization by re-design of untranslated regions for L-tyrosine production in Escherichia coli. Sci. Rep. 2015, 5, 13853. [Google Scholar] [PubMed]
- Muñoz, A.J.; Hernández-Chávez, G.; de Anda, R.; Martínez, A.; Bolívar, F.; Gosset, G. Metabolic engineering of Escherichia coli for improving L-3,4-dihydroxyphenylalanine (L-DOPA) synthesis from glucose. J. Ind. Microbiol. Biotechnol. 2011, 38, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Noh, M.H.; Lim, H.G.; Kang, C.W.; Im, D.K.; Oh, M.K.; Jung, G.Y. Precise tuning of the glyoxylate cycle in Escherichia coli for efficient tyrosine production from acetate. Microb. Cell Fact. 2019, 18, 57. [Google Scholar] [CrossRef] [PubMed]
- Juminaga, D.; Baidoo, E.E.; Redding-Johanson, A.M.; Batth, T.S.; Burd, H.; Mukhopadhyay, A.; Petzold, C.J.; Keasling, J.D. Modular engineering of L-tyrosine production in Escherichia coli. Appl. Environ. Microbiol. 2012, 78, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.N.; Xiao, W.; Stephanopoulos, G. Rational, combinatorial, and genomic approaches for engineering L-tyrosine production in Escherichia coli. Proc. Natl. Acad. Sci. USA 2012, 109, 13538–13543. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.P.; Feng, Y.C.; Fu, J.; Guo, H.W.; Guo, Y.; Han, B.X.; Jiang, Z.C.; Kong, L.Z.; Li, C.Z.; Liu, H.C.; et al. Catalytic conversion of lignocellulosic biomass into chemicals and fuels. Green Energy Environ. 2023, 8, 10–114. [Google Scholar] [CrossRef]
- Zhao, M.T.; Wu, X.F.; Tao, Y.K.; Xiao, Y. Full use of lignocellulosic biomass for efficient synthesis of l-tyrosine and its analogues by engineering microbial consortia. Green Chem. 2024, 26, 6760–6773. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, J.; Kang, Z.; Du, G.; Yu, X.; Wang, T.; Chen, J. Enhanced production of L-phenylalanine in Corynebacterium glutamicum due to the introduction of Escherichia coli wild-type gene aroH. J. Ind. Microbiol. Biotechnol. 2013, 40, 643–651. [Google Scholar] [CrossRef]
- Braga, A.; Faria, N. Bioprocess optimization for the production of aromatic compounds with metabolically engineered hosts: Recent developments and future challenges. Front. Bioeng. Biotechnol. 2020, 8, 96. [Google Scholar] [CrossRef]
- Gerigk, M.; Bujnicki, R.; Ganpo-Nkwenkwa, E.; Bongaerts, J.; Sprenger, G.; Takors, R. Process control for enhanced L-phenylalanine production using different recombinant Escherichia coli strains. Biotechnol. Bioeng. 2002, 80, 746–754. [Google Scholar] [CrossRef]
- Ding, D.; Liu, Y.; Xu, Y.; Zheng, P.; Li, H.; Zhang, D.; Sun, J. Improving the production of L-phenylalanine by identifying key enzymes through multi-enzyme reaction system in vitro. Sci. Rep. 2016, 6, 32208. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.P.; Xiao, M.R.; Zhang, L.; Xu, J.; Ding, Z.Y.; Gu, Z.H.; Shi, G.Y. Production of L-phenylalanine from glucose by metabolic engineering of wild type Escherichia coli W3110. Process Biochem. 2013, 48, 413–419. [Google Scholar] [CrossRef]
- Koma, D.; Kishida, T.; Yamanaka, H.; Moriyoshi, K.; Nagamori, E.; Ohmoto, T. Escherichia coli chromosome-based T7-dependent constitutive overexpression system and its application to generating a phenylalanine producing strain. J. Biosci. Bioeng. 2018, 126, 586–595. [Google Scholar] [CrossRef]
- Liu, D.X.; Fan, C.S.; Tao, J.H.; Liang, G.X.; Gao, S.E.; Wang, H.J.; Li, X.; Song, D.X. Integration of E. coli aroG-pheA tandem genes into Corynebacterium glutamicum tyrA locus and its effect on L-phenylalanine biosynthesis. World J. Gastroenterol 2004, 10, 3683–3687. [Google Scholar] [CrossRef]
- Yakandawala, N.; Romeo, T.; Friesen, A.D.; Madhyastha, S. Metabolic engineering of Escherichia coli to enhance phenylalanine production. Appl. Microbiol. Biotechnol. 2008, 78, 283–291. [Google Scholar] [CrossRef]
- Nie, M.Z.; Wang, J.Y.; Chen, Z.Y.; Cao, C.K.; Zhang, K.C. Systematic engineering enables efficient biosynthesis of L-phenylalanine in E. coli from inexpensive aromatic precursors. Microb. Cell Fact. 2024, 23, 12. [Google Scholar] [CrossRef]
- Lennen, R.M.; Lim, H.G.; Jensen, K.; Mohammed, E.T.; Phaneuf, P.V.; Noh, M.H.; Malla, S.; Börner, R.A.; Chekina, K.; Özdemir, E.; et al. Laboratory evolution reveals generaland specific tolerance mechanisms for commodity chemicals. Metab. Eng. 2023, 76, 179–192. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, S.Y. Fermentative production of branched chain amino acids: A focus on metabolic engineering. Appl. Microbiol. Biotechnol. 2010, 85, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zhang, X.; Dai, J.; Wang, Y.; He, W. Engineering of leucine-responsive regulatory protein improves spiramycin and bitespiramycin biosynthesis. Microb. Cell Fact. 2019, 18, 38. [Google Scholar] [CrossRef]
- Bartek, T.; Blombach, B.; Zönnchen, E.; Makus, P.; Lang, S.; Eikmanns, B.J.; Oldiges, M. Importance of NADPH supply for improved L-valine formation in Corynebacterium glutamicum. Biotechnol. Prog. 2010, 26, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Wendisch, V.F. Metabolic engineering advances and prospects for amino acid production. Metab. Eng. 2020, 58, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Ge, X.; Wu, D.; Qian, H.; Zhang, W. Improvement of L-valine production at high temperature in Brevibacterium flavum by overexpressing ilvEBNrC genes. J. Ind. Microbiol. Biotechnol. 2012, 39, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Radmacher, E.; Vaitsikova, A.; Burger, U.; Krumbach, K.; Sahm, H.; Eggeling, L. Linking central metabolism with increased pathway flux: L-valine accumulation by Corynebacterium glutamicum. Appl. Environ. Microbiol. 2002, 68, 2246–2250. [Google Scholar] [CrossRef] [PubMed]
- Holátko, J.; Elisáková, V.; Prouza, M.; Sobotka, M.; Nesvera, J.; Pátek, M. Metabolic engineering of the L-valine biosynthesis pathway in Corynebacterium glutamicum using promoter activity modulation. J. Biotechnol. 2009, 139, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, A.W.; Moo-Young, M.; Chou, C.P. Development of a CRISPR-Cas9 tool kit for comprehensive engineering of Bacillus subtilis. Appl. Environ. Microbiol. 2016, 82, 4876–4895. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, A.W.; Ren, X.; Moo-Young, M.; Chou, C.P. Metabolic engineering of Bacillus subtilis for L-valine overproduction. Biotechnol. Bioeng. 2018, 115, 2778–2792. [Google Scholar] [CrossRef] [PubMed]
- Geraskina, N.V.; Sycheva, E.V.; Samsonov, V.V.; Eremina, N.S.; Hook, C.D.; Serebrianyi, V.A.; Stoynova, N.V. Engineering Escherichia coli for autoinducible production of L-valine: An example of an artificial positive feedback loop in amino acid biosynthesis. PLoS ONE 2019, 14, e0215777. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.W.; Tang, M.; You, J.J.; Hao, Y.A.; Zhang, X.; Yang, T.W.; Rao, Z.M. A movel method to screen strong constitutive promoters in Escherichia coli and Serratia marcescens for industrial applications. Biology 2023, 12, 71. [Google Scholar]
- Wada, M.; Hijikata, N.; Aoki, R.; Takesue, N.; Yokota, A. Enhanced valine production in Corynebacterium glutamicum with defective H+-ATPase and C-terminal truncated acetohydroxyacid synthase. Biosci. Biotechnol. Biochem. 2008, 72, 2959–2965. [Google Scholar] [CrossRef]
- Ma, Y.; Cui, Y.; Du, L.; Liu, X.; Xie, X.; Chen, N. Identification and application of a growth-regulated promoter for improving L-valine production in Corynebacterium glutamicum. Microb. Cell Fact. 2018, 17, 185. [Google Scholar] [CrossRef]
- Blombach, B.; Schreiner, M.E.; Bartek, T.; Oldiges, M.; Eikmanns, B.J. Corynebacterium glutamicum tailored for high-yield L-valine production. Appl. Microbiol. Biotechnol. 2008, 79, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Chen, X.; Zhang, Y.; Qian, H.; Zhang, W. (L)-Valine production with minimization of by-products’ synthesis in Corynebacterium glutamicum and Brevibacterium flavum. Amino Acids 2012, 43, 2301–2311. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, Y.; Hu, J.; Dong, X.; Wang, X. Metabolic engineering of Corynebacterium glutamicum ATCC13869 for L-valine production. Metab. Eng. 2015, 29, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Suda, M.; Uematsu, K.; Natsuma, Y.; Hiraga, K.; Jojima, T.; Inui, M.; Yukawa, H. Engineering of Corynebacterium glutamicum for high-yield L-valine production under oxygen deprivation conditions. Appl. Environ. Microbiol. 2013, 79, 1250–1257. [Google Scholar] [CrossRef]
- Liang, C.; Huo, Y.; Qi, G.; Wei, X.; Wang, Q.; Chen, S. Enhancement of L-valine production in Bacillus licheniformis by blocking three branched pathways. Biotechnol. Lett. 2015, 37, 1243–1248. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, K.H.; Kim, T.Y.; Lee, S.Y. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc. Natl. Acad. Sci. USA 2007, 104, 7797–7802. [Google Scholar] [CrossRef]
- Hao, Y.; Pan, X.; Xing, R.; You, J.; Hu, M.; Liu, Z.; Li, X.; Xu, M.; Rao, Z. High-level production of L-valine in Escherichia coli using multi-modular engineering. Bioresour. Technol. 2022, 359, 127461. [Google Scholar] [CrossRef]
- Mahr, R.; Gätgens, C.; Gätgens, J.; Polen, T.; Kalinowski, J.; Frunzke, J. Biosensor-driven adaptive laboratory evolution of L-valine production in Corynebacterium glutamicum. Metab. Eng. 2015, 32, 184–194. [Google Scholar] [CrossRef]
- Blombach, B.; Schreiner, M.E.; Moch, M.; Oldiges, M.; Eikmanns, B.J. Effect of pyruvate dehydrogenase complex deficiency on L-lysine production with Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2007, 76, 615–623. [Google Scholar] [CrossRef]
- Viswanath, B.; Rajagopal, S.; Basha, P.A.; Rao, D.M.; Begum, P.S.; Rajakumari, D.; Razak, M.A. Enhancing the Activity of Aspartate Kinase for an Overproduction of L-lysine by Corynebacterium glutamicum. Am. J. Biochem. Mol. Biol. 2016, 6, 33–44. [Google Scholar] [CrossRef]
- Wang, J.; Gao, D.; Yu, X.; Li, W.; Qi, Q. Evolution of a chimeric aspartate kinase for L-lysine production using a synthetic RNA device. Appl. Microbiol. Biotechnol. 2015, 99, 8527–8536. [Google Scholar] [CrossRef] [PubMed]
- van Ooyen, J.; Noack, S.; Bott, M.; Reth, A.; Eggeling, L. Improved L-lysine production with Corynebacterium glutamicum and systemic insight into citrate synthase flux and activity. Biotechnol. Bioeng. 2012, 109, 2070–2081. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.B.; Zeng, A.P. Engineering a Lysine-ON riboswitch for metabolic control of lysine production in Corynebacterium glutamicum. ACS Synth. Biol. 2015, 4, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Kind, S.; Becker, J.; Wittmann, C. Increased lysine production by flux coupling of the tricarboxylic acid cycle and the lysine biosynthetic pathway—Metabolic engineering of the availability of succinyl-CoA in Corynebacterium glutamicum. Metab. Eng. 2013, 15, 184–195. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, J.; Liu, D.; Zhang, W. Increased glucose utilization and cell growth of Corynebacterium glutamicum by modifying the glucose-specific phosphotransferase system (PTS(Glc)) genes. Can. J. Microbiol. 2016, 62, 983–992. [Google Scholar] [CrossRef]
- Brautaset, T.; Jakobsen, Ø.M.; Degnes, K.F.; Netzer, R.; Naerdal, I.; Krog, A.; Dillingham, R.; Flickinger, M.C.; Ellingsen, T.E. Bacillus methanolicus pyruvate carboxylase and homoserine dehydrogenase I and II and their roles for L-lysine production from methanol at 50 °C. Appl. Microbiol. Biotechnol. 2010, 87, 951–964. [Google Scholar] [CrossRef]
- Blombach, B.; Hans, S.; Bathe, B.; Eikmanns, B.J. Acetohydroxyacid synthase, a novel target for improvement of L-lysine production by Corynebacterium glutamicum. Appl. Environ. Microbiol. 2009, 75, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Peters-Wendisch, P.G.; Schiel, B.; Wendisch, V.F.; Katsoulidis, E.; Möckel, B.; Sahm, H.; Eikmanns, B.J. Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J. Mol. Microbiol. Biotechnol. 2001, 3, 295–300. [Google Scholar]
- Wang, Z.H.; Moslehi-Jenabian, S.; Solem, C.; Jensen, P.R. Increased expression of pyruvate carboxylase and biotin protein ligase increases lysine production in a biotin prototrophic Corynebacterium glutamicum strain. Eng. Life Sci. 2014, 15, 73–82. [Google Scholar] [CrossRef]
- Kortmann, M.; Mack, C.; Baumgart, M.; Bott, M. Pyruvate carboxylase variants enabling improved lysine production from glucose identified by biosensor-based high-throughput fluorescence-activated cell sorting screening. ACS Synth. Biol. 2019, 8, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Bommareddy, R.R.; Frank, D.; Rappert, S.; Zeng, A.P. Deregulation of feedback inhibition of phosphoenolpyruvate carboxylase for improved lysine production in Corynebacterium glutamicum. Appl. Environ. Microbiol. 2014, 80, 1388–1393. [Google Scholar] [CrossRef]
- Becker, J.; Klopprogge, C.; Schröder, H.; Wittmann, C. Metabolic engineering of the tricarboxylic acid cycle for improved lysine production by Corynebacterium glutamicum. Appl. Environ. Microbiol. 2009, 75, 7866–7869. [Google Scholar] [CrossRef]
- van Ooyen, J.; Emer, D.; Bussmann, M.; Bott, M.; Eikmanns, B.J.; Eggeling, L. Citrate synthase in Corynebacterium glutamicum is encoded by two gltA transcripts which are controlled by RamA, RamB, and GlxR. J. Biotechnol. 2011, 154, 140–148. [Google Scholar] [CrossRef]
- Radmacher, E.; Eggeling, L. The three tricarboxylate synthase activities of Corynebacterium glutamicum and increase of L-lysine synthesis. Appl. Microbiol. Biotechnol. 2007, 76, 587–595. [Google Scholar] [CrossRef]
- Mitsuhashi, S.; Hayashi, M.; Ohnishi, J.; Ikeda, M. Disruption of malate: Quinone oxidoreductase increases L-lysine production by Corynebacterium glutamicum. Biosci. Biotechnol. Biochem. 2006, 70, 2803–2806. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Chai, X.; Lu, X.; Li, Y.; Zhang, Y.; Wang, G.; Zhang, C.; Liu, S.; Zhang, Y.; Ma, J.; et al. Native promoters of Corynebacterium glutamicum and its application in L-lysine production. Biotechnol. Lett. 2018, 40, 383–391. [Google Scholar] [CrossRef]
- Marx, A.; Hans, S.; Möckel, B.; Bathe, B.; de Graaf, A.A.; McCormack, A.C.; Stapleton, C.; Burke, K.; O’Donohue, M.; Dunican, L.K. Metabolic phenotype of phosphoglucose isomerase mutants of Corynebacterium glutamicum. J. Biotechnol. 2003, 104, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Klopprogge, C.; Zelder, O.; Heinzle, E.; Wittmann, C. Amplified expression of fructose 1,6-bisphosphatase in Corynebacterium glutamicum increases in vivo flux through the pentose phosphate pathway and lysine production on different carbon sources. Appl. Environ. Microbiol. 2005, 71, 8587–8596. [Google Scholar] [CrossRef]
- Ohnishi, J.; Katahira, R.; Mitsuhashi, S.; Kakita, S.; Ikeda, M. A novel gnd mutation leading to increased L-lysine production in Corynebacterium glutamicum. FEMS Microbiol. Lett. 2005, 242, 265–274. [Google Scholar] [CrossRef]
- Contador, C.A.; Rizk, M.L.; Asenjo, J.A.; Liao, J.C. Ensemble modeling for strain development of L-lysine-producing Escherichia coli. Metab. Eng. 2009, 11, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Takeno, S.; Murata, R.; Kobayashi, R.; Mitsuhashi, S.; Ikeda, M. Engineering of Corynebacterium glutamicum with an NADPH-generating glycolytic pathway for L-lysine production. Appl. Environ. Microbiol. 2010, 76, 7154–7160. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Z.; Ruan, H.Z.; Chen, X.L.; Zhang, F.; Zhang, W. Equilibrium of the intracellular redox state for improving cell growth and L-lysine yield of Corynebacterium glutamicum by optimal cofactor swapping. Microb. Cell Fact. 2019, 18, 65. [Google Scholar] [CrossRef] [PubMed]
- Kabus, A.; Georgi, T.; Wendisch, V.F.; Bott, M. Expression of the Escherichia coli pntAB genes encoding a membrane-bound transhydrogenase in Corynebacterium glutamicum improves L-lysine formation. Appl. Microbiol. Biotechnol. 2007, 75, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhang, Y.; Liu, D.; Chen, Z. Efficient mining of natural NADH-utilizing dehydrogenases enables systematic cofactor engineering of lysine synthesis pathway of Corynebacterium glutamicum. Metab. Eng. 2019, 52, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, J.; Mitsuhashi, S.; Hayashi, M.; Ando, S.; Yokoi, H.; Ochiai, K.; Ikeda, M. A novel methodology employing Corynebacterium glutamicum genome information to generate a new L-lysine-producing mutant. Appl. Microbiol. Biotechnol. 2002, 58, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Han, M.; Zhang, J.; Guo, Y.; Zhang, W. Metabolic engineering Corynebacterium glutamicum for the L-lysine production by increasing the flux into L-lysine biosynthetic pathway. Amino Acids 2014, 46, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Q.; Zheng, P.; Guo, Y.; Wang, L.; Zhang, T.; Sun, J.; Ma, Y. Evolving the L-lysine high-producing strain of Escherichia coli using a newly developed high-throughput screening method. J. Ind. Microbiol. Biotechnol. 2016, 43, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Adachi, N.; Takahashi, C.; Ono-Murota, N.; Yamaguchi, R.; Tanaka, T.; Kondo, A. Direct L-lysine production from cellobiose by Corynebacterium glutamicum displaying beta-glucosidase on its cell surface. Appl. Microbiol. Biotechnol. 2013, 97, 7165–7172. [Google Scholar] [CrossRef]
- Anusree, M.; Wendisch, V.F.; Nampoothiri, K.M. Co-expression of endoglucanase and β-glucosidase in Corynebacterium glutamicum DM1729 towards direct lysine fermentation from cellulose. Bioresour. Technol. 2016, 213, 239–244. [Google Scholar] [CrossRef]
- Abe, K.; Kuroda, A.; Takeshita, R. Engineering of Escherichia coli to facilitate efficient utilization of isomaltose and panose in industrial glucose feedstock. Appl. Microbiol. Biotechnol. 2017, 101, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Korosh, T.C.; Markley, A.L.; Clark, R.L.; McGinley, L.L.; McMahon, K.D.; Pfleger, B.F. Engineering photosynthetic production of L-lysine. Metab. Eng. 2017, 44, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Malla, S.; van der Helm, E.; Darbani, B.; Wieschalka, S.; Förster, J.; Borodina, I.; Sommer, M.O.A. A novel efficient L-lysine exporter identified by functional metagenomics. Front. Microbiol. 2022, 13, 855736. [Google Scholar] [CrossRef] [PubMed]
- Neuner, A.; Wagner, I.; Sieker, T.; Ulber, R.; Schneider, K.; Peifer, S.; Heinzle, E. Production of L-lysine on different silage juices using genetically engineered Corynebacterium glutamicum. J. Biotechnol. 2013, 163, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, J.; Guo, Y.; Zai, Y.; Zhang, W. Improvement of cell growth and L-lysine production by genetically modified Corynebacterium glutamicum during growth on molasses. J. Ind. Microbiol. Biotechnol. 2013, 40, 1423–1432. [Google Scholar] [CrossRef]
- Matano, C.; Uhde, A.; Youn, J.W.; Maeda, T.; Clermont, L.; Marin, K.; Krämer, R.; Wendisch, V.F.; Seibold, G.M. Engineering of Corynebacterium glutamicum for growth and L-lysine and lycopene production from N-acetyl-glucosamine. Appl. Microbiol. Biotechnol. 2014, 98, 5633–5643. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.L.; Jungmann, L.; Schiefelbein, S.; Peyriga, L.; Cahoreau, E.; Portais, J.C.; Becker, J.; Wittmann, C. Lysine production from the sugar alcohol mannitol: Design of the cell factory Corynebacterium glutamicum SEA-3 through integrated analysis and engineering of metabolic pathway fluxes. Metab. Eng. 2018, 47, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Cazier, A.P.; Blazeck, J. Advances in promoter engineering: Novel applications and predefined transcriptional control. Biotechnol. J. 2021, 16, e2100239. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, D.; van Beek, H.L.; Syberg, F.; Schallmey, M.; Tobola, F.; Cormann, K.U.; Schlicker, C.; Baumann, P.T.; Krumbach, K.; Sokolowsky, S.; et al. Engineering and application of a biosensor with focused ligand specificity. Nat. Commun. 2020, 11, 4851. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Quinn, P.J.; Wang, X. Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for the production of L-threonine. Biotechnol. Adv. 2011, 29, 11–23. [Google Scholar] [CrossRef]
- Lee, S.Y.; Park, J.H. Integration of systems biology with bioprocess engineering: L: -threonine production by systems metabolic engineering of Escherichia coli. Adv. Biochem. Eng. Biotechnol. 2010, 120, 1–19. [Google Scholar] [PubMed]
- Naz, S.; Liu, P.; Farooq, U.; Ma, H. Insight into de-regulation of amino acid feedback inhibition: A focus on structure analysis method. Microb. Cell Fact. 2023, 22, 161. [Google Scholar] [CrossRef]
- Li, Y.; Wei, H.; Wang, T.; Xu, Q.; Zhang, C.; Fan, X.; Ma, Q.; Chen, N.; Xie, X. Current status on metabolic engineering for the production of L-aspartate family amino acids and derivatives. Bioresour. Technol. 2017, 245, 1588–1602. [Google Scholar] [CrossRef]
- Simic, P.; Willuhn, J.; Sahm, H.; Eggeling, L. Identification of glyA (encoding serine hydroxymethyltransferase) and its use together with the exporter ThrE to increase L-threonine accumulation by Corynebacterium glutamicum. Appl. Environ. Microbiol. 2002, 68, 3321–3327. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Fang, Y.; Wang, X.; Zhao, L.; Wang, J.; Li, Y. Increasing L-threonine production in Escherichia coli by engineering the glyoxylate shunt and the L-threonine biosynthesis pathway. Appl. Microbiol. Biotechnol. 2018, 102, 5505–5518. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Zhao, Y.; Hu, J.; Li, Y.; Wang, X. Attenuating L-lysine production by deletion of ddh and lysE and their effect on L-threonine and L-isoleucine production in Corynebacterium glutamicum. Enzyme Microb. Technol. 2016, 93–94, 70–78. [Google Scholar] [CrossRef]
- Lee, J.H.; Jung, S.C.; Bui, L.M.; Kang, K.H.; Song, J.J.; Kim, S.C. Improved production of L-threonine in Escherichia coli by use of a DNA scaffold system. Appl. Environ. Microbiol. 2013, 79, 774–782. [Google Scholar] [CrossRef]
- Liu, S.; Xiao, H.; Zhang, F.; Lu, Z.; Zhang, Y.; Deng, A.; Li, Z.; Yang, C.; Wen, T. A seamless and iterative DNA assembly method named PS-Brick and its assisted metabolic engineering for threonine and 1-propanol production. Biotechnol. Biofuels 2019, 12, 180. [Google Scholar] [CrossRef]
- Wang, J.; Ma, W.; Fang, Y.; Yang, J.; Zhan, J.; Chen, S.; Wang, X. Increasing L-threonine production in Escherichia coli by overexpressing the gene cluster phaCAB. J. Ind. Microbiol. Biotechnol. 2019, 46, 1557–1568. [Google Scholar] [CrossRef]
- Diesveld, R.; Tietze, N.; Fürst, O.; Reth, A.; Bathe, B.; Sahm, H.; Eggeling, L. Activity of exporters of Escherichia coli in Corynebacterium glutamicum, and their use to increase L-threonine production. J. Mol. Microbiol. Biotechnol. 2009, 16, 198–207. [Google Scholar] [CrossRef]
- Kruse, D.; Krämer, R.; Eggeling, L.; Rieping, M.; Pfefferle, W.; Tchieu, J.H.; Chung, Y.J.; Saier, M.H., Jr.; Burkovski, A. Influence of threonine exporters on threonine production in Escherichia coli. Appl. Microbiol. Biotechnol. 2002, 59, 205–210. [Google Scholar] [PubMed]
- Ding, Z.; Fang, Y.; Zhu, L.; Wang, J.; Wang, X. Deletion of arcA, iclR, and tdcC in Escherichia coli to improve L-threonine production. Biotechnol. Appl. Biochem. 2019, 66, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, H.; Xiong, H.; Xie, X.; Chen, N.; Zhao, G.; Caiyin, Q.; Zhu, H.; Qiao, J. Two-stage carbon distribution and cofactor generation for improving L-threonine production of Escherichia coli. Biotechnol. Bioeng. 2019, 116, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Cai, D.; Chen, W.; Chen, H.; Luo, W. Combined metabolic analyses for the biosynthesis pathway of L-threonine in Escherichia coli. Front. Bioeng. Biotechnol. 2022, 10, 1010931. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Park, J.H.; Kim, T.Y.; Kim, H.U.; Lee, S.Y. Systems metabolic engineering of Escherichia coli for L-threonine production. Mol. Syst. Biol. 2007, 3, 149. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xu, J.; Han, M.; Zhang, W. Generation of mutant threonine dehydratase and its effects on isoleucine synthesis in Corynebacterium glutamicum. World J. Microbiol. Biotechnol. 2015, 31, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.J.; Wang, J.L.; Li, Y.; Yin, L.H.; Wang, X.Y. Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) co-produced with L-isoleucine in Corynebacterium glutamicum WM001. Microb. Cell Fact. 2018, 17, 93. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Wendisch, V.F. Exploring the role of sigma factor gene expression on production by Corynebacterium glutamicum: Sigma factor H and FMN as example. Front. Microbiol. 2015, 6, 740. [Google Scholar] [CrossRef]
- Yin, L.; Hu, X.; Xu, D.; Ning, J.; Chen, J.; Wang, X. Co-expression of feedback-resistant threonine dehydratase and acetohydroxy acid synthase increase L-isoleucine production in Corynebacterium glutamicum. Metab. Eng. 2012, 14, 542–550. [Google Scholar] [CrossRef]
- Wu, M.; Sun, Y.; Zhu, M.; Zhu, L.; Lü, J.; Geng, F. Molecular dynamics-based allosteric prediction method to design key residues in threonine dehydrogenase for amino-acid production. ACS Omega 2021, 6, 10975–10983. [Google Scholar] [CrossRef]
- Ma, W.; Wang, J.; Li, Y.; Wang, X. Cysteine synthase A overexpression in Corynebacterium glutamicum enhances L-isoleucine production. Biotechnol. Appl. Biochem. 2019, 66, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wen, B.; Wang, J.; Xu, Q.; Zhang, C.; Chen, N.; Xie, X. Enhancing (L)-isoleucine production by thrABC overexpression combined with alaT deletion in Corynebacterium glutamicum. Appl. Biochem. Biotechnol. 2013, 171, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Huan, X.; Wang, X.; Ning, J. Overexpression of NAD kinases improves the L-isoleucine biosynthesis in Corynebacterium glutamicum ssp. lactofermentum. Enzyme Microb. Technol. 2012, 51, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Wang, J.; Li, Y.; Hu, X.; Shi, F.; Wang, X. Enhancing pentose phosphate pathway in Corynebacterium glutamicum to improve L-isoleucine production. Biotechnol. Appl. Biochem. 2016, 63, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Shi, F.; Hu, X.; Chen, C.; Wang, X. Increasing L-isoleucine production in Corynebacterium glutamicum by overexpressing global regulator Lrp and two-component export system BrnFE. J. Appl. Microbiol. 2013, 114, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, J.; Wang, Y.; Liu, J.; Huang, J.; Chen, N.; Zheng, P.; Sun, J. Efficient multiplex gene repression by CRISPR-dCpf1 in Corynebacterium glutamicum. Front. Bioeng. Biotechnol. 2020, 8, 357. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Kumar, V.; Baweja, M.; Shukla, P. Gene editing and genetic engineering approaches for advanced probiotics: A review. Crit. Rev. Food Sci. Nutr. 2018, 58, 1735–1746. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.L.; Leenay, R.T.; Beisel, C.L. Current and future prospects for CRISPR-based tools in bacteria. Biotechnol. Bioeng. 2016, 113, 930–943. [Google Scholar] [CrossRef]
- Fonfara, I.; Richter, H.; Bratovic, M.; Le Rhun, A.; Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 2016, 532, 517–521. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Lu, Y.J.; Zheng, P.; Sun, J.B.; Ma, Y.H. Development of a CRISPR/Cas9 genome editing toolbox for Corynebacterium glutamicum. Microb. Cell Fact. 2017, 16, 205. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, Y.; Lv, X.; Li, J.; Du, G.; Liu, L. CAMERS-B: CRISPR/Cpf1 assisted multiple-genes editing and regulation system for Bacillus subtilis. Biotechnol. Bioeng. 2020, 117, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Luan, G.; Lu, X. Tailoring cyanobacterial cell factory for improved industrial properties. Biotechnol. Adv. 2018, 36, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhu, T.; Cai, Z.; Li, Y. From cyanochemicals to cyanofactories: A review and perspective. Microb. Cell Fact. 2016, 15, 2. [Google Scholar] [CrossRef] [PubMed]
- Antonovsky, N.; Gleizer, S.; Noor, E.; Zohar, Y.; Herz, E.; Barenholz, U.; Zelcbuch, L.; Amram, S.; Wides, A.; Tepper, N.; et al. Sugar Synthesis from CO2 in Escherichia coli. Cell 2016, 166, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Liu, G.; Zhai, X.; Zhou, J.; Cai, Z.; Li, Y. Quantitative analysis of an engineered CO2-fixing Escherichia coli reveals great potential of heterotrophic CO2 fixation. Biotechnol. Biofuels 2015, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Tseng, I.T.; Chen, Y.L.; Chen, C.H.; Shen, Z.X.; Yang, C.H.; Li, S.Y. Exceeding the theoretical fermentation yield in mixotrophic Rubisco-based engineered Escherichia coli. Metab. Eng. 2018, 47, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Schwander, T.; Schada von Borzyskowski, L.; Burgener, S.; Cortina, N.S.; Erb, T.J. A synthetic pathway for the fixation of carbon dioxide in vitro. Science 2016, 354, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Li, X.; Duchoud, F.; Chuang, D.S.; Liao, J.C. Augmenting the Calvin-Benson-Bassham cycle by a synthetic malyl-CoA-glycerate carbon fixation pathway. Nat. Commun. 2018, 9, 2008. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Li, Z.; Ma, D.; Ye, C.; Zhang, L.P.; Gao, C.; Liu, L.M.; Chen, X.L. Light-driven CO2 sequestration in Escherichia coli to achieve theoretical yield of chemicals. Nat. Catal. 2021, 4, 395–406. [Google Scholar] [CrossRef]
- Hao, Y.N.; Pan, X.W.; You, J.J.; Li, G.M.; Xu, M.J.; Rao, Z.M. Microbial production of branched chain amino acids: Advances and perspectives. Bioresour. Technol. 2024, 397, 130502. [Google Scholar] [CrossRef]


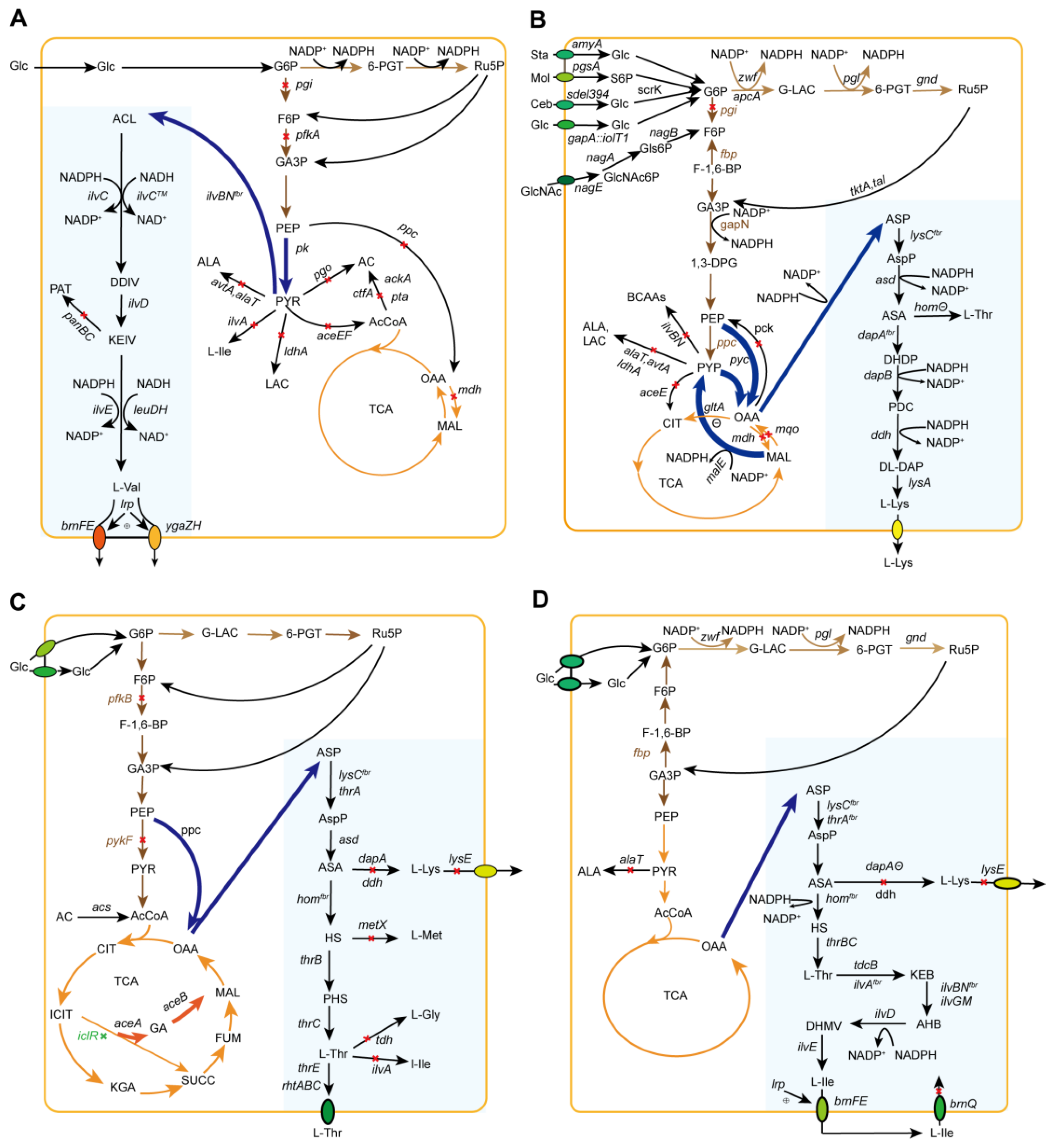

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Engineering Strategy | Culture Method | Titer (g/L) | Yield (g/g) | Productivity (g/L/h) | References |
|---|---|---|---|---|---|---|
| L-tryptophan | ||||||
| C. glutamicum KY9218 (pKW9901) | ↑aroGfbr, ↑trpEfbrDfbrCBA, ↑serA | Fed-batch | 50 | 0.23 | 0.63 | [25] |
| C. glutamicum KY9218 (pIK9960) | ↑tktA, ↑aroGfbr, ↑trpEfbrDfbrBA, ↑serA | Fed-batch | 58 | ns | ns | [26] |
| E. coli FB-04 (pSV03) | ↑aroFfbr, ↑trpEfbrD, ΔtrpR, ΔtnaA, ΔpheA, ΔtyrA | Batch culture | 13.1 | 0.1 | ns | [27] |
| E. coli TRTH1013 | Δtna, ΔtrpR, ↑tktA, ↑ppsA, ↑aroGfbr, ↑trpEfbrDCBA, ↑serA | Fed-batch | 40.2 | 0.17 | ns | [28] |
| E. coli T13 | ycjv::Ptrc-aroEK, mbhA::Ptrc-glnAL159I,E304A, yeeP::PserB-serB, PserC-serC, ΔTnaB | Fed-batch | 53.65 | 0.238 | ns | [29] |
| E. coli TRTH03 | ΔtnaA, Δmtr, ΔpykA, Δppc, ↑trpEfbr, ↑aroGfbr, ↑pck, ↑citT, ↑acnBA, ↑icd, ↑pyc | Fed-batch | 49 | 0.19 | ns | [30] |
| E. coli SX11 | yghX::Ptrc-xfpk, ptsG::PM1-12-glf, ycjv::PM1-12-glk, ΔpykF, ylbE::Ppck-pck, mbhA::Ptrc-pycP458S | Fed-batch | 41.7 | 0.227 | 1.04 | [31] |
| E. coli GPT1017 | Δmtr, ΔtnaB, ΔaroP, ↑tktA, ↑aroGfbr, ↑trpEfbr | Fed-batch | 16.3 | ns | 0.25 | [32] |
| E. coli TRTH03 | Para-acs::araB; Para-aceB-mdh-pck::yghx | Fed-batch | 54.6 | ns | ns | [33] |
| E. coli MG1655-Y | Δpta, Δfmtr, ↑yddG, ↑tktA, ↑ppsA | Fed-batch | 48.7 | 0.22 | 1.39 | [34] |
| E. coli Trp30 | ΔtrpR, ΔtnaA, P2-trpES40FDCBA, ΔackA, Δpta, ΔpoxB | Fed-batch | 42.5 | 0.178 | 0.89 | [35] |
| L-tyrosine | ||||||
| E. coli T2 | ↑tktA, ↑pps, ↑aroGfbr, ↑tyrAfbr, ΔftyrR | Fed-batch | 9.7 | 0.1 | ns | [36] |
| E. coli DPD4193 | ↑aroGfbr, ΔpheAL, ↑tyrAfbr, ↑cscB, ↑cscK, ↑cscA | Fed-batch | 55 | ns | 1.22 | [37,38] |
| E. coli JH7 | ↑PLlacO-1-phhA-phhB-folM-T1, ↑PLlacO-1-mtrA-folX-T1-PLlacO-1-aroL-ppsA-tktA-aroGfbr | Shake flask | 0.401 | ns | ns | [39] |
| E. coli W3110 | ΔtyrR, ΔtyrA, ΔfpheA, ↑aroGfbr, ↑tyrAfbr, ↑aroL | Shake flask | 6.3 | 0.16 | ns | [40] |
| E. coli BL21(DE3) | ↑Ptac-aroGfbr-aroL, Ptrc-tyrC, ΔtyrP | Fed-batch | 43.14 | 0.11 | ns | [41] |
| C. glutamicum KY10865 (pKY1) | ↑aroGfbr, ↑pheACM | Fed-batch | 26 | ns | ns | [42] |
| E. coli HGD (M9) | ΔtyrR, ΔpheA, ΔtrpE, ΔaroP, ΔtyrP, dadX-cvrA::Pj231119-yddG, tyrP::Pj231119-tktA, trpE::Pj231119-ppsA, ykgH-betA::Pj231119-udhA, yeeJ-yeeL::Pj231119-pntAB, ΔpoxB, pAP with aroGfbr, tyrAfbr, fpk, pta, p15A ori with PRPL | Fed-batch | 92.5 | 0.266 | ns | [43] |
| E. coli S17-1 | ↓tyrR, ↓csrA, ↑pgi, ↑ppc | Fed-batch | 21.9 | ns | ns | [44] |
| L-phenylalanine | ||||||
| E. coli BR-42 (pAP-B03) | ↑pheAfbr, ↑aroF | Two-stage process | 57.63 | ns | 1.15 | [45] |
| E. coli xllp1 | ↑PtyrP-aroK | Fed-batch | 61.3 | 0.22 | 1.27 | [46] |
| E. coli DV269 (TyrA-LAA) | ↓tyrA | Batch culture | 7.2 | 0.14 | 0.3 | [47] |
| E. Coli AJ12741 (pHYGG) | ↑yddG | Shake flask | 6.4 | 0.16 | ns | [48] |
| B. flavum 311 (pJN5) | ↑ppsA, ↑pckA, ↑aroG, ↑pheA, ↑tyrB | Shake flask | 5.39 | ns | ns | [49] |
| E. coli PB12-ev2 | ↑aroGfbr, ↑pheAev2, ↑tktA, ΔptsHI, galP | Shake flask | ns | 0.33 | 40 a | [50] |
| C. glutamicum ΔptsI::iolT2-ppgkΔaroP ΔaceEΔldh (pSUTL, pSDTL) | ↑aroFfbr, ↑aroE, ↑ppsA, ↑tktA, ↑pheAfbr, ↑aroA, ↑tyrB, ↑aroL, ↑iolT2-ppgk, ΔfpstI, ΔfaroP, ΔfldhA, ΔfaceE | Fed-batch | 15.76 | ns | 0.197 | [51] |
| E. coli Xllp08 | ΔptsH, ↑galp, ↑glk, TyrRT495I, ↑aroDfbr | Fed-batch | 72.9 | 0.26 | 1.4 | [52] |
| E. coli WF123456+fis | ↑aroKB, ↑aroGfbr, ↑tktA, ΔtyrR, ↑fis | Shake flask | 0.9 | 0.083 | 0.032 | [53] |
| E. coli PHE05 | ↑aroK1, ↑aroL1, ↑pheA1, ↑aroA, ↑aroC, ↑tyrB, adaptive evolution | Fed-batch | 80.48 | 0.27 | 1.68 | [54] |
| Strain | Engineering Strategy | Culture Method | Titer (g/L) | Yield (g/g) | Productivity (g/L/h) | References |
|---|---|---|---|---|---|---|
| L-valine | ||||||
| C. glutamicum ΔilvAΔpanB ilvNM13 (pECKAilvBNC) | ↑ilvBNC, ilvNM13, ΔilvA, ΔpanB | Batch culture | 15.2 | 0.38 | 0.32 | [55] |
| C. glutamicum aceE A16ΔpqoΔppc (pJC4ilvBNCE) | ↓aceE, ↑ilvBNCE, Δppc, Δpqo | Fed-batch | 86.5 | 0.23 | 1.6 | [56] |
| C. glutamicum ΔLDH BNGECTMDLD | ↑ilvBNfbr, ↑ilvCTM, ↑ilvD, ↑LeuDH | Anaerobic, fed-batch | 227.3 | 0.41 | ns | [57] |
| E. coli W ΔilvA (pTrc184ygaZHlrp, pKBRilvBNfbrCED) | ΔilvA, ↑ilvBNfbrCED, ↑lrp, ↑ygaZH | Fed-batch | 60.7 | ns | 2.06 | [58] |
| C. glutamicumΔppcΔpycICDG407S | Δppc, Δpyc, ICDG407S, ilvBNCE | Shake flask | 8.9 | 0.22 | ns | [59] |
| Bacillus subtilis AW015-5 | ↑ilvBHfbrC, ↑ilvD, ↑ybge,↑ ywaA, ↓bcd, ↓pdhA, ΔleuA, ΔilvA, | Shake flask | 4.61 | ns | ns | [60] |
| L-lysine | ||||||
| E. coli LATR11 (pWG-DCSMASMBHc.gLP) | ↑ppc, ↑lysCT344M, ↑asd, ↑dapAH56K, ↑dapB, ↑lysA, ↑ddh | Fed-batch | 125.6 | 0.59 | 3.14 | [61] |
| C. glutamicum LYS-12 | ↑ddh, ↑lysA, ↑lysCT311I, ↑dapB, homV59A, Δpck, ↑pycP458S, ↓icd, ↑fbp, ↑zwf-opcA-pgl-gnd | Fed-batch | 120 | 0.55 | 4 | [62] |
| C. glutamicum DRL2 | Δpyk, ppcD299N, gltAS252C | Shake flask | 15.7 | ns | 0.56 | [63] |
| C. glutamicum JL-6 9Ptac-M gdh | ppc::pck, pyc::odx, ΔP1gltA, Ptac-M::Pgdh | Fed-batch | 181.5 | ns | 3.78 | [64] |
| C. glutamicum Lys9 | ΔaceE, pycC1372T/G1A, ΔalaT, ΔavtA, ΔldhA, Δpck, Δmdh, ↑lysCC932T, ↑pntAB | Fed-batch | 76.8 | 0.42 | 2.69 | [65] |
| C. glutamicum ATCC13032 Psodfbp-zwf243 | ↑zwfA243T, ↑fbp | Shake flask | ns | 0.13 | ns | [66] |
| C. glutamicum RE2Aiol/pCAK311 | gapN::gapA, rhoR696C, ΔgapB, gapA::iolT1 | Shake flask | 9.3 | ns | 0.29 | [67] |
| C. glutamicum JL-6 | Ec-dapBC115G,G116C::dapB | Fed-batch | 117.3 | 0.44 | 2.93 | [68] |
| C. glutamicum GRLys1 | ↑iolT2,↑glcK, ΔsugR, ΔldhA | Shake flask | 4.81 | ns | 0.22 | [69] |
| C. glutamicum ZL-92 | ↑ptsIH, ΔsugR, ↑iolT1, ↑iolT2, ↑ppgK | Fed-batch | 201.6 | 0.65 | 5.04 | [70] |
| C. glutamicum Δhom::HPA | ↑amyA, ↑pgsA | Batch culture | 6.0 | 0.19 | ns | [71] |
| E. coli QD01 ΔtRNA L2 | establishment of artificial rare cryptosystem for mutating and screening | Shake flask | 14.8 | ns | ns | [72] |
| L-threonine | ||||||
| E. coli TH-103Z | artificial construction of polyploid Escherichia coli | Fed-batch | 160.3 | ns | ns | [73] |
| C. glutamicum R102ΔmetXΔdapA(pEX-Box) | ΔmetX, ΔdapA, ↑hom-thrB, ↑thrE | Shake flask | 3.5 | 0.045 | 0.049 | [74] |
| C. glutamicum D | ΔilvA, ΔmetX, ΔtdcB, ↑lysfbr, ↑homfbrM, ↑asd, ↑thrB, ↑thrC | Shake flask | 12 | ns | ns | [75] |
| E. coli TWF044 | ΔfadR, ΔfabR, ΔlacI, fadBA::lacZ, Ptac-trc:Pacs, Ptac-thrAfbrBC-rhtC::lacA, Ptac-ppnk-aspC-ppc::lacZ | Fed-batch | 103.9 | 0.72 | 2.16 | [76] |
| E. coli WMZ016/pFW01-thrA*BC-rhtC | Δcrr, ΔiclR, Ptrc::PgltA, ↑thrAfbrBC-rhtC | Shake flask | 18.0 | 0.35 | ns | [77] |
| E. coli THRD ΔpfkBΔpykF | ↑pfkB, ↑pykF | Fed-batch | 111.4 | 0.37 | 3.98 | [78] |
| E. coli TWF083 | RBSthrL-thrL-RBSthrA::iclR, PcysH-RBSs7-aspC::lacI, ↓arcA, fadR, cpxR, gadE, pykF dynamic regulated by thrR | Fed-batch | 116.6 | 0.49 | 2.43 | [79] |
| E. coli MG422 rhtA23 (pVIC40) | ↑thrAfbrBC, ↑rhtA | Shake flask | 36.3 | 0.73 | 0.79 | [80] |
| E. coli TWF113/pFT24rpa1 | ΔpoxB, ΔpflB, ΔldhA, ΔadhE, ΔtdcC, ΔavtA, ΔalaA, ΔalaC, cIts-PR-PLrhtC-pyc, tetR-PLtetO-1-alaA | Fed-batch | ns | 124.03% | ns | [81] |
| E. coli (P2.1-2901 ΔptsG) | dynamical regulation of the expression of the thrABC operon, ΔptsG | Shake flask | 40.06 | ns | ns | [82] |
| E. coli MDS205 | ↑thrAfbrBC, Δtdh, rhtA23::tdcC, rhtA23::sstT | Batch culture | 40.1 | 0.4 | 1.34 | [83] |
| L-isoleucine | ||||||
| E. coli TLCD | ↑ilvGMEDAfbr, ↑thrAfbrBC, ↑lysCfbr | Shake flask | 12.3 | 0.23 | 0.51 | [17] |
| C. glutamicum JHI3-156 | ↑ppnk, ↑zwf | Shake flask | 4.1 | 0.029 | 0.057 | [84] |
| C. glutamicum IWJ001 | ↑ilvBN1, ↑ilvA1, ↑ppnk1 | Fed-batch | 32.3 | ns | ns | [85] |
| C. glutamicum K2P55 | ↑homfbr, ↑ilvAfbr, ↓dapA | Fed-batch | 14.3 | 0.14 | 0.18 | [86] |
| E. coli NXU102 | ΔleuA/Met−+ Lys−/Δtdh/ΔltaE/ΔyiaY | Shake flask | 7.48 | ns | ns | [87] |
| C. glutamicum YILWΔbrnQpXMJ19brnFE | ΔbrnQ, ↑brnFE | Fed-batch | 29 | 0.24 | 0.48 | [88] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, L.; Zhou, Y.; Ding, N.; Fang, Y. Recent Advances in Metabolic Engineering for the Biosynthesis of Phosphoenol Pyruvate–Oxaloacetate–Pyruvate-Derived Amino Acids. Molecules 2024, 29, 2893. https://doi.org/10.3390/molecules29122893
Yin L, Zhou Y, Ding N, Fang Y. Recent Advances in Metabolic Engineering for the Biosynthesis of Phosphoenol Pyruvate–Oxaloacetate–Pyruvate-Derived Amino Acids. Molecules. 2024; 29(12):2893. https://doi.org/10.3390/molecules29122893
Chicago/Turabian StyleYin, Lianghong, Yanan Zhou, Nana Ding, and Yu Fang. 2024. "Recent Advances in Metabolic Engineering for the Biosynthesis of Phosphoenol Pyruvate–Oxaloacetate–Pyruvate-Derived Amino Acids" Molecules 29, no. 12: 2893. https://doi.org/10.3390/molecules29122893
APA StyleYin, L., Zhou, Y., Ding, N., & Fang, Y. (2024). Recent Advances in Metabolic Engineering for the Biosynthesis of Phosphoenol Pyruvate–Oxaloacetate–Pyruvate-Derived Amino Acids. Molecules, 29(12), 2893. https://doi.org/10.3390/molecules29122893